A Novel Small-Molecule Inhibitor of Endosomal TLRs Reduces Inflammation and Alleviates Autoimmune Disease Symptoms in Murine Models

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Experimental Section
2.1. Ligand Dataset for QSAR Modeling and Virtual Screening
2.2. Cell Culture and Reagents
2.3. Cell Viability Assay
2.4. Detection of TNF-α Secretion
2.5. Measurement of Protein Expression Levels Using Western Blotting
2.6. Confocal Microscopy Analysis
2.7. In Vivo Study Using a Mouse Model of Psoriasis
2.8. In Vivo Study Using a Mouse Model of SLE
2.9. Quantitative Real-Time PCR (qRT-PCR)
2.10. Evaluation of TAC5 Binding to TLR7 Using the Surface Plasmon Resonance (SPR) Assay
2.11. Molecular Docking
2.12. Molecular Dynamics Simulations
2.13. Statistical Analysis
3. Results
3.1. Identification of Probable Inhibitors of Endosomal TLRs
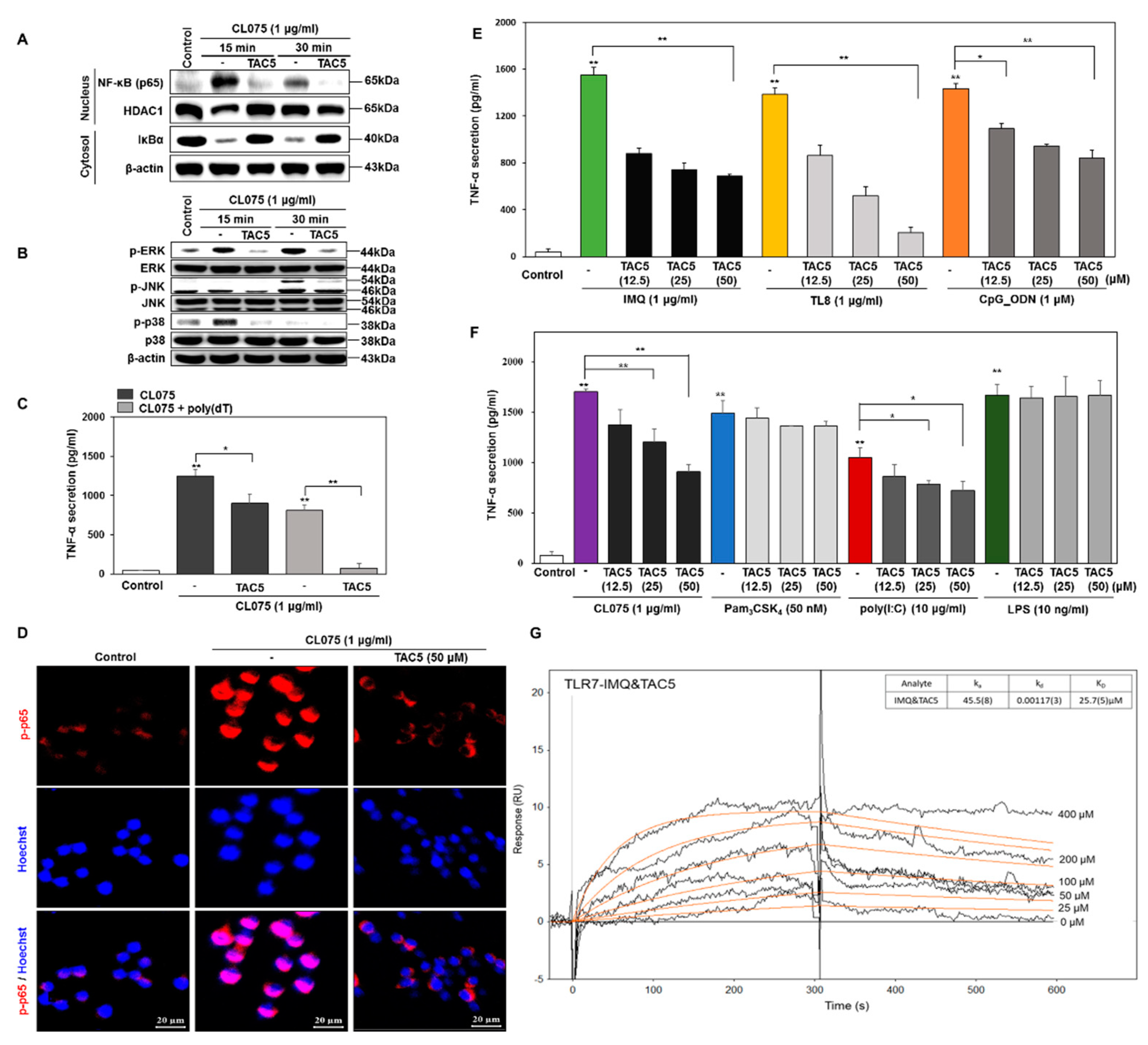
3.2. Effect of TAC5 on CL075-Induced TLR7 and TLR8 Signaling
3.3. TNF-α Inhibition Profile of a 3-Amino-Derivative of TAC5 (TAC5-a)
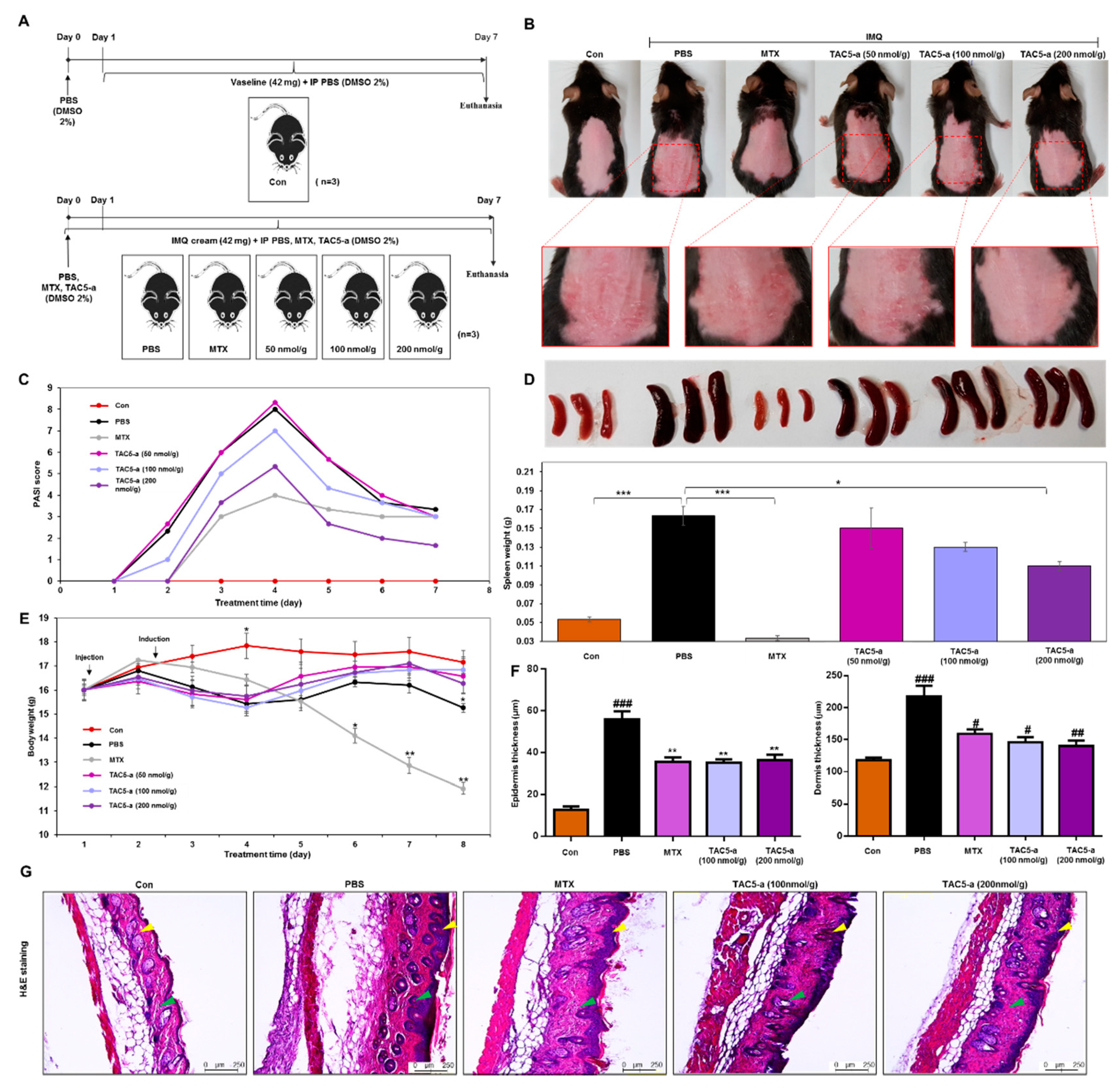
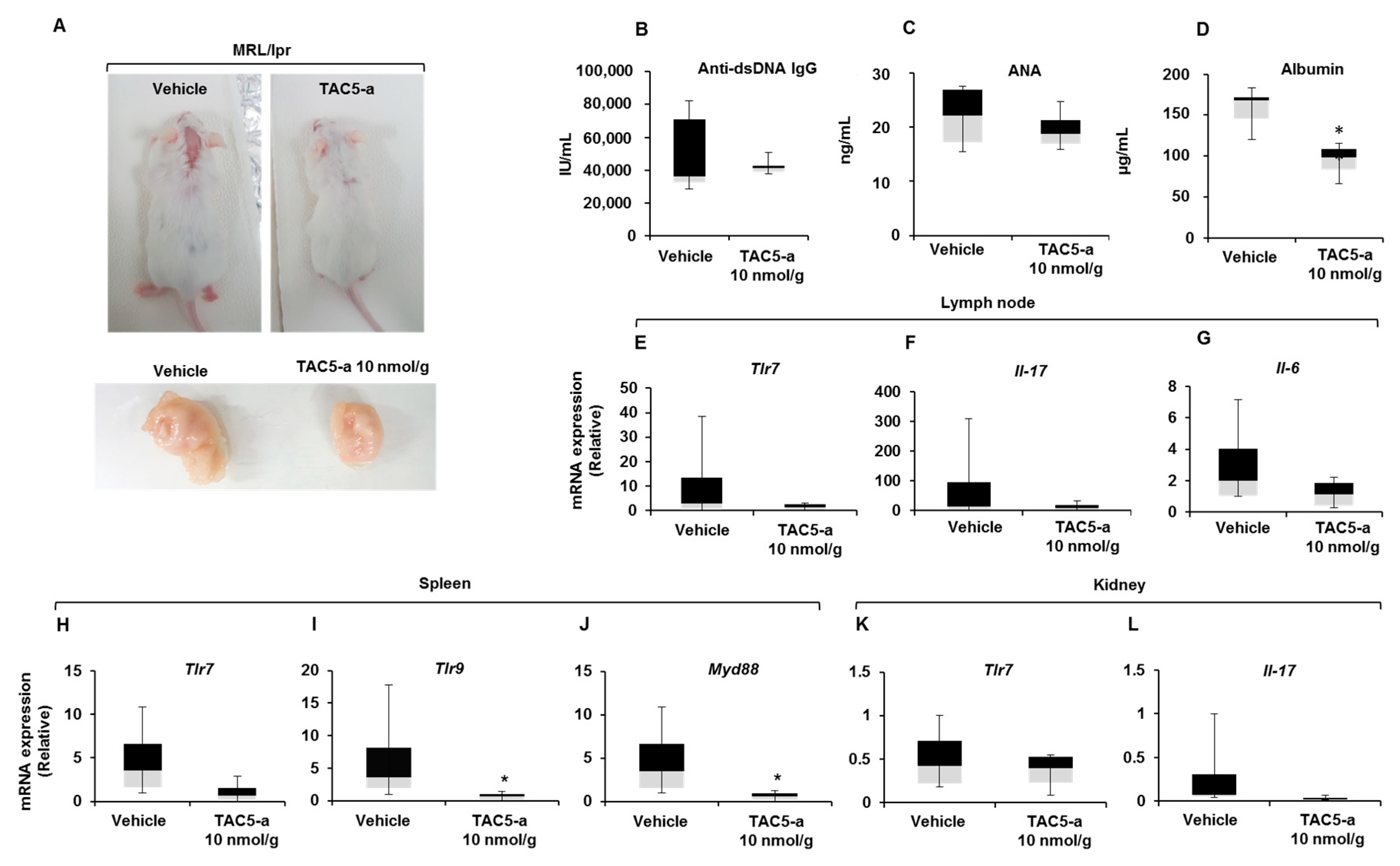
3.4. Effect of TAC5-a Treatment on Mouse Models of Psoriasis and SLE
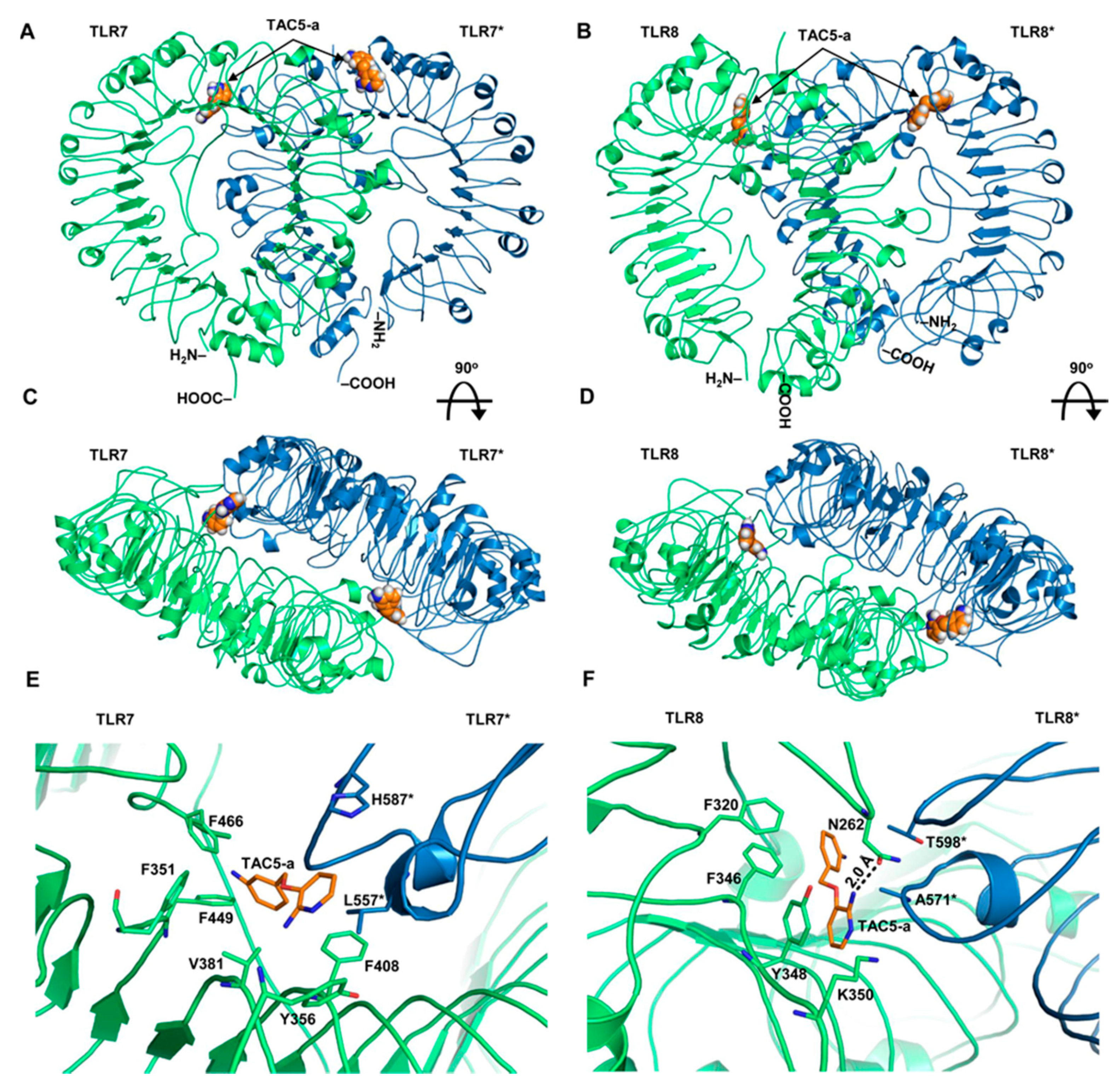
3.5. Prediction of the Binding Modes of TAC5-a on TLR7/8
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, B.; Nicolas, E.; Michaut, L.; Reichhart, J.M.; Hoffmann, J.A. Pillars article: The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell. 1996. 86: 973–983. J. Immunol. 2012, 188, 5210–5220. [Google Scholar] [PubMed]
- Williams, M.J.; Rodriguez, A.; Kimbrell, D.A.; Eldon, E.D. The 18-wheeler mutation reveals complex antibacterial gene regulation in Drosophila host defense. EMBO J. 1997, 16, 6120–6130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patra, M.C.; Shah, M.; Choi, S. Toll-like receptor-induced cytokines as immunotherapeutic targets in cancers and autoimmune diseases. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Gay, N.J.; Symmons, M.F.; Gangloff, M.; Bryant, C.E. Assembly and localization of Toll-like receptor signalling complexes. Nat. Rev. Immunol. 2014, 14, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Takeuchi, O.; Sato, S.; Horiuchi, T.; Hoshino, K.; Takeda, K.; Dong, Z.; Modlin, R.L.; Akira, S. Cutting edge: Role of Toll-like receptor 1 in mediating immune response to microbial lipoproteins. J. Immunol. 2002, 169, 10–14. [Google Scholar] [CrossRef]
- Takeuchi, O.; Kawai, T.; Muhlradt, P.F.; Morr, M.; Radolf, J.D.; Zychlinsky, A.; Takeda, K.; Akira, S. Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int. Immunol. 2001, 13, 933–940. [Google Scholar] [CrossRef]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.Y.; Van Huffel, C.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in Tlr4 gene. Science 1998, 282, 2085–2088. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, F.; Smith, K.D.; Ozinsky, A.; Hawn, T.R.; Yi, E.C.; Goodlett, D.R.; Eng, J.K.; Akira, S.; Underhill, D.M.; Aderem, A. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 2001, 410, 1099–1103. [Google Scholar] [CrossRef]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Diebold, S.S.; Kaisho, T.; Hemmi, H.; Akira, S.; Reis e Sousa, C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 2004, 303, 1529–1531. [Google Scholar] [CrossRef]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; et al. A Toll-like receptor recognizes bacterial DNA. Nature 2000, 408, 740–745. [Google Scholar] [CrossRef]
- Lee, S.M.; Kok, K.H.; Jaume, M.; Cheung, T.K.; Yip, T.F.; Lai, J.C.; Guan, Y.; Webster, R.G.; Jin, D.Y.; Peiris, J.S. Toll-like receptor 10 is involved in induction of innate immune responses to influenza virus infection. Proc. Natl. Acad. Sci. USA 2014, 111, 3793–3798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanji, H.; Ohto, U.; Shibata, T.; Miyake, K.; Shimizu, T. Structural reorganization of the Toll-like receptor 8 dimer induced by agonistic ligands. Science 2013, 339, 1426–1429. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [Green Version]
- Brencicova, E.; Diebold, S.S. Nucleic acids and endosomal pattern recognition: How to tell friend from foe? Front. Cell Infect. Microbiol. 2013, 3, 37. [Google Scholar] [CrossRef] [Green Version]
- Jensen, S.; Thomsen, A.R. Sensing of RNA viruses: A review of innate immune receptors involved in recognizing RNA virus invasion. J. Virol. 2012, 86, 2900–2910. [Google Scholar] [CrossRef] [Green Version]
- Barrat, F.J.; Meeker, T.; Gregorio, J.; Chan, J.H.; Uematsu, S.; Akira, S.; Chang, B.; Duramad, O.; Coffman, R.L. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J. Exp. Med. 2005, 202, 1131–1139. [Google Scholar] [CrossRef] [Green Version]
- Marshak-Rothstein, A. Toll-like receptors in systemic autoimmune disease. Nat. Rev. Immunol. 2006, 6, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Kasai, M.; Tomizawa, H.; Aoki, M.; Eiho, K.; Isobe, Y.; Asano, S. Dihydropyrrolo[2,3-d]pyrimidines: Selective Toll-Like receptor 9 antagonists from scaffold morphing efforts. ACS Med. Chem. Lett. 2014, 5, 1235–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, K.; Wang, X.; Yin, H. Small-molecule inhibitors of the TLR3/dsRNA complex. J. Am. Chem. Soc. 2011, 133, 3764–3767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, N.M.; Kimbrell, M.R.; Malladi, S.S.; David, S.A. Regioisomerism-dependent TLR7 agonism and antagonism in an imidazoquinoline. Bioorganic Med. Chem. Lett. 2009, 19, 2211–2214. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Hu, Z.; Tanji, H.; Jiang, S.; Das, N.; Li, J.; Sakaniwa, K.; Jin, J.; Bian, Y.; Ohto, U.; et al. Small-molecule inhibition of TLR8 through stabilization of its resting state. Nat. Chem. Biol. 2018, 14, 58–64. [Google Scholar] [CrossRef] [Green Version]
- Hennessy, E.J.; Parker, A.E.; O′Neill, L.A. Targeting Toll-like receptors: Emerging therapeutics? Nature Reviews. Drug Discovery 2010, 9, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Kawai, T.; Akira, S. Dissecting negative regulation of Toll-like receptor signaling. Trends Immunol. 2012, 33, 449–458. [Google Scholar] [CrossRef]
- Walter, A.; Schafer, M.; Cecconi, V.; Matter, C.; Urosevic-Maiwald, M.; Belloni, B.; Schonewolf, N.; Dummer, R.; Bloch, W.; Werner, S.; et al. Aldara activates TLR7-independent immune defence. Nat. Commun. 2013, 4, 1560. [Google Scholar] [CrossRef]
- Hemmi, H.; Kaisho, T.; Takeuchi, O.; Sato, S.; Sanjo, H.; Hoshino, K.; Horiuchi, T.; Tomizawa, H.; Takeda, K.; Akira, S. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat. Immunol. 2002, 3, 196–200. [Google Scholar] [CrossRef]
- Achek, A.; Shah, M.; Seo, J.Y.; Kwon, H.K.; Gui, X.; Shin, H.J.; Cho, E.Y.; Lee, B.S.; Kim, D.J.; Lee, S.H.; et al. Linear and rationally designed stapled peptides abrogate TLR4 pathway and relieve inflammatory symptoms in rheumatoid arthritis rat model. J. Med. Chem. 2019, 62, 6495–6511. [Google Scholar] [CrossRef]
- Kwon, H.K.; Patra, M.C.; Shin, H.J.; Gui, X.; Achek, A.; Panneerselvam, S.; Kim, D.J.; Song, S.J.; Hong, R.; Kim, K.S.; et al. A cell-penetrating peptide blocks Toll-like receptor-mediated downstream signaling and ameliorates autoimmune and inflammatory diseases in mice. Exp. Mol. Med. 2019, 51, 50. [Google Scholar] [CrossRef] [Green Version]
- Kandimalla, E.R.; Bhagat, L.; Wang, D.; Yu, D.; Sullivan, T.; La Monica, N.; Agrawal, S. Design, synthesis and biological evaluation of novel antagonist compounds of Toll-like receptors 7, 8 and 9. Nucleic Acids Res. 2013, 41, 3947–3961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, W.; Zhu, F.G.; Bhagat, L.; Yu, D.; Tang, J.X.; Kandimalla, E.R.; La Monica, N.; Agrawal, S. A Toll-like receptor 7, 8, and 9 antagonist inhibits Th1 and Th17 responses and inflammasome activation in a model of IL-23-induced psoriasis. J. Invest. Dermatol 2013, 133, 1777–1784. [Google Scholar] [CrossRef] [Green Version]
- Walker, T.; Grulke, C.M.; Pozefsky, D.; Tropsha, A. Chembench: A cheminformatics workbench. Bioinformatics 2010, 26, 3000–3001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capuzzi, S.J.; Kim, I.S.; Lam, W.I.; Thornton, T.E.; Muratov, E.N.; Pozefsky, D.; Tropsha, A. Chembench: A publicly accessible, integrated cheminformatics portal. J. Chem. Inf. Model. 2017, 57, 105–108. [Google Scholar] [CrossRef] [Green Version]
- MOE. Molecular Operating Environment (MOE), 2013.08; Chemical Computing Group ULC: Montreal, QC, Canada, 2017; Volume 2013. [Google Scholar]
- Chen, C.Y. TCM Database@Taiwan: The world’s largest traditional Chinese medicine database for drug screening in silico. PLoS ONE 2011, 6, e15939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irwin, J.J.; Shoichet, B.K. ZINC--a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Ohto, U.; Shibata, T.; Krayukhina, E.; Taoka, M.; Yamauchi, Y.; Tanji, H.; Isobe, T.; Uchiyama, S.; Miyake, K.; et al. Structural analysis reveals that Toll-like receptor 7 is a dual receptor for guanosine and single-stranded RNA. Immunity 2016, 45, 737–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Gallo Cassarino, T.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, W252–W258. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Schuttelkopf, A.W.; van Aalten, D.M. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Gorden, K.K.; Qiu, X.; Battiste, J.J.; Wightman, P.P.; Vasilakos, J.P.; Alkan, S.S. Oligodeoxynucleotides differentially modulate activation of TLR7 and TLR8 by imidazoquinolines. J. Immunol. 2006, 177, 8164–8170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurk, M.; Kritzler, A.; Schulte, B.; Tluk, S.; Schetter, C.; Krieg, A.M.; Vollmer, J. Modulating responsiveness of human TLR7 and 8 to small molecule ligands with T-rich phosphorothiate oligodeoxynucleotides. Eur. J. Immunol. 2006, 36, 1815–1826. [Google Scholar] [CrossRef]
- Demaria, O.; Pagni, P.P.; Traub, S.; de Gassart, A.; Branzk, N.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; Flavell, R.A.; Alexopoulou, L. TLR8 deficiency leads to autoimmunity in mice. J. Clin. Investig. 2010, 120, 3651–3662. [Google Scholar] [CrossRef]
- Vincent, F.B.; Morand, E.F.; Schneider, P.; Mackay, F. The BAFF/APRIL system in SLE pathogenesis. Nat. Rev. Rheumatol. 2014, 10, 365–373. [Google Scholar] [CrossRef]
- Krieg, A.M.; Vollmer, J. Toll-like receptors 7, 8, and 9: Linking innate immunity to autoimmunity. Immunol. Rev. 2007, 220, 251–269. [Google Scholar] [CrossRef]
- Terhorst, D.; Chelbi, R.; Wohn, C.; Malosse, C.; Tamoutounour, S.; Jorquera, A.; Bajenoff, M.; Dalod, M.; Malissen, B.; Henri, S. Dynamics and transcriptomics of skin dendritic cells and macrophages in an imiquimod-induced, biphasic mouse model of psoriasis. J. Immunol. 2015, 195, 4953–4961. [Google Scholar] [CrossRef] [Green Version]
- Matsushima, N.; Tanaka, T.; Enkhbayar, P.; Mikami, T.; Taga, M.; Yamada, K.; Kuroki, Y. Comparative sequence analysis of leucine-rich repeats (LRRs) within vertebrate toll-like receptors. BMC Genom. 2007, 8, 124. [Google Scholar] [CrossRef] [Green Version]
- Roach, J.C.; Glusman, G.; Rowen, L.; Kaur, A.; Purcell, M.K.; Smith, K.D.; Hood, L.E.; Aderem, A. The evolution of vertebrate Toll-like receptors. Proc. Natl. Acad. Sci. USA 2005, 102, 9577–9582. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Flynn, A.D. Drugging membrane protein interactions. Annu. Rev. Biomed. Eng. 2016, 18, 51–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill, A.L.; Frederickson, M.; Cleasby, A.; Woodhead, S.J.; Carr, M.G.; Woodhead, A.J.; Walker, M.T.; Congreve, M.S.; Devine, L.A.; Tisi, D.; et al. Identification of novel p38alpha MAP kinase inhibitors using fragment-based lead generation. J. Med. Chem. 2005, 48, 414–426. [Google Scholar] [CrossRef]
- Shukla, N.M.; Malladi, S.S.; Day, V.; David, S.A. Preliminary evaluation of a 3H imidazoquinoline library as dual TLR7/TLR8 antagonists. Bioorganic Med. Chem. 2011, 19, 3801–3811. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Dewan, V.; Yin, H. Discovery of small molecules as Multi-Toll-like receptor agonists with proinflammatory and anticancer activities. J. Med. Chem. 2017, 60, 5029–5044. [Google Scholar] [CrossRef] [PubMed]
- Salunke, D.B.; Yoo, E.; Shukla, N.M.; Balakrishna, R.; Malladi, S.S.; Serafin, K.J.; Day, V.W.; Wang, X.; David, S.A. Structure-activity relationships in human Toll-like receptor 8-active 2,3-diamino-furo[2,3-c]pyridines. J. Med. Chem. 2012, 55, 8137–8151. [Google Scholar] [CrossRef] [Green Version]
- Shukla, N.M.; Malladi, S.S.; Mutz, C.A.; Balakrishna, R.; David, S.A. Structure-activity relationships in human toll-like receptor 7-active imidazoquinoline analogues. J. Med. Chem. 2010, 53, 4450–4465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nestle, F.O.; Kaplan, D.H.; Barker, J. Psoriasis. N. Engl. J. Med. 2009, 361, 496–509. [Google Scholar] [CrossRef] [PubMed]
- Flutter, B.; Nestle, F.O. TLRs to cytokines: Mechanistic insights from the imiquimod mouse model of psoriasis. Eur. J. Immunol. 2013, 43, 3138–3146. [Google Scholar] [CrossRef]
- Ronnblom, L. Potential role of IFNalpha in adult lupus. Arthritis Res. Ther. 2010, 12 (Suppl. 1), S3. [Google Scholar] [CrossRef] [Green Version]
- Lovgren, T.; Eloranta, M.L.; Bave, U.; Alm, G.V.; Ronnblom, L. Induction of interferon-alpha production in plasmacytoid dendritic cells by immune complexes containing nucleic acid released by necrotic or late apoptotic cells and lupus IgG. Arthritis Rheum. 2004, 50, 1861–1872. [Google Scholar] [CrossRef]
- Saxena, R.; Mahajan, T.; Mohan, C. Lupus nephritis: Current update. Arthritis Res. Ther. 2011, 13, 240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miossec, P.; Kolls, J.K. Targeting IL-17 and TH17 cells in chronic inflammation. Nat. Rev. Drug Discov. 2012, 11, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Kolls, J.K.; Linden, A. Interleukin-17 family members and inflammation. Immunity 2004, 21, 467–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurk, M.; Heil, F.; Vollmer, J.; Schetter, C.; Krieg, A.M.; Wagner, H.; Lipford, G.; Bauer, S. Human TLR7 or TLR8 independently confer responsiveness to the antiviral compound R-848. Nat. Immunol. 2002, 3, 499. [Google Scholar] [CrossRef] [PubMed]
- Guiducci, C.; Gong, M.; Cepika, A.M.; Xu, Z.; Tripodo, C.; Bennett, L.; Crain, C.; Quartier, P.; Cush, J.J.; Pascual, V.; et al. RNA recognition by human TLR8 can lead to autoimmune inflammation. J. Exp. Med. 2013, 210, 2903–2919. [Google Scholar] [CrossRef]
- Kugelberg, E. Innate immunity: Making mice more human the TLR8 way. Nat. Rev. Immunol. 2014, 14, 6. [Google Scholar] [CrossRef]
- Patra, M.C.; Choi, S. Recent progress in the development of Toll-like receptor (TLR) antagonists. Expert Opin. Ther. Pat. 2016, 26, 719–730. [Google Scholar] [CrossRef]
- Schmitt, F.C.F.; Freund, I.; Weigand, M.A.; Helm, M.; Dalpke, A.H.; Eigenbrod, T. Identification of an optimized 2′-O-methylated trinucleotide RNA motif inhibiting Toll-like receptors 7 and 8. RNA 2017, 23, 1344–1351. [Google Scholar] [CrossRef] [Green Version]
- Balak, D.M.; van Doorn, M.B.; Arbeit, R.D.; Rijneveld, R.; Klaassen, E.; Sullivan, T.; Brevard, J.; Thio, H.B.; Prens, E.P.; Burggraaf, J.; et al. IMO-8400, a toll-like receptor 7, 8, and 9 antagonist, demonstrates clinical activity in a phase 2a, randomized, placebo-controlled trial in patients with moderate-to-severe plaque psoriasis. Clin. Immunol. 2017, 174, 63–72. [Google Scholar] [CrossRef]
- Juliano, R.L. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016, 44, 6518–6548. [Google Scholar] [CrossRef]
- Hanagata, N. Structure-dependent immunostimulatory effect of CpG oligodeoxynucleotides and their delivery system. Int. J. Nanomed. 2012, 7, 2181–2195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mocsai, A.; Kovacs, L.; Gergely, P. What is the future of targeted therapy in rheumatology: Biologics or small molecules? BMC Med. 2014, 12, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Machine Learning Method | Descriptors | Prediction CCR | Accuracy | Sensitivity | Specificity |
|---|---|---|---|---|---|
| k-Nearest Neighbor | MOE 2D | 0.711 ± 0.031 | 0.748 | 0.875 | 0.547 |
| k-Nearest Neighbor | DragonX-H | 0.733 ± 0.083 | 0.766 | 0.881 | 0.585 |
| Random Forest | MOE 2D | 0.737 ± 0.057 | 0.777 | 0.632 | 0.869 |
| Random Forest | DragonX-H | 0.717 ± 0.029 | 0.732 | 0.635 | 0.809 |
| Support Vector Machine | MOE 2D | 0.773 ± 0.031 | 0.762 | 0.721 | 0.794 |
| Support Vector Machine | DragonX-H | 0.705 ± 0.043 | 0.745 | 0.594 | 0.839 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patra, M.C.; Achek, A.; Kim, G.-Y.; Panneerselvam, S.; Shin, H.-J.; Baek, W.-Y.; Lee, W.H.; Sung, J.; Jeong, U.; Cho, E.-Y.; et al. A Novel Small-Molecule Inhibitor of Endosomal TLRs Reduces Inflammation and Alleviates Autoimmune Disease Symptoms in Murine Models. Cells 2020, 9, 1648. https://doi.org/10.3390/cells9071648
Patra MC, Achek A, Kim G-Y, Panneerselvam S, Shin H-J, Baek W-Y, Lee WH, Sung J, Jeong U, Cho E-Y, et al. A Novel Small-Molecule Inhibitor of Endosomal TLRs Reduces Inflammation and Alleviates Autoimmune Disease Symptoms in Murine Models. Cells. 2020; 9(7):1648. https://doi.org/10.3390/cells9071648
Chicago/Turabian StylePatra, Mahesh Chandra, Asma Achek, Gi-Young Kim, Suresh Panneerselvam, Hyeon-Jun Shin, Wook-Yong Baek, Wang Hee Lee, June Sung, Uisuk Jeong, Eun-Young Cho, and et al. 2020. "A Novel Small-Molecule Inhibitor of Endosomal TLRs Reduces Inflammation and Alleviates Autoimmune Disease Symptoms in Murine Models" Cells 9, no. 7: 1648. https://doi.org/10.3390/cells9071648