The Janus Face of p53-Targeting Ubiquitin Ligases


Abstract
:1. The Wild-Type p53 Signaling Network in Tumor Suppression
1.1. Wild-Type p53 Suppresses Tumorigenesis and Cancer Progression
1.2. Regulation of Wild-Type p53 Activity
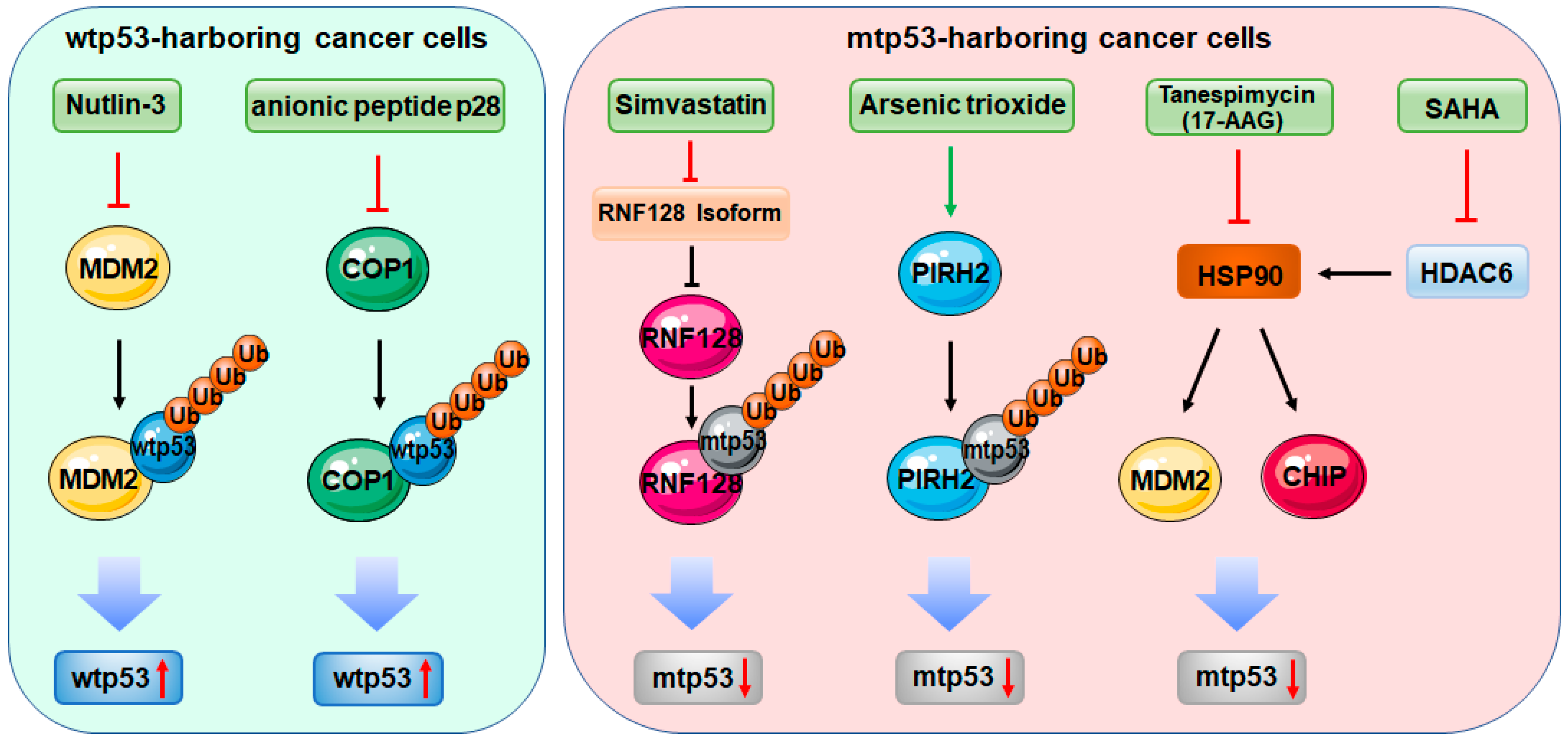
2. p53 Mutation Endorses Cancer Development
3. Divergent Roles of p53-Targeting E3 Ligases in Cancer
3.1. RING-Finger E3 Ligases
3.2. Tripartite Motif Protein Family
3.3. Others
4. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, A.J. The many faces of p53: Something for everyone. J. Mol. Cell Biol. 2019, 11, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Brady, C.A.; Attardi, L.D. p53 at a glance. J. Cell Sci. 2010, 123 Pt 15, 2527–2532. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, S.E.; Ceder, S.; Bykov, V.J.N.; Wiman, K.G. p53 as a hub in cellular redox regulation and therapeutic target in cancer. J. Mol. Cell Biol. 2019, 11, 330–341. [Google Scholar] [CrossRef] [Green Version]
- Vazquez, A.; Bond, E.E.; Levine, A.J.; Bond, G.L. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat. Rev. Drug Discov. 2008, 7, 979–987. [Google Scholar] [CrossRef]
- Zhou, X.; Liao, W.J.; Liao, J.M.; Liao, P.; Lu, H. Ribosomal proteins: Functions beyond the ribosome. J. Mol. Cell Biol. 2015, 7, 92–104. [Google Scholar] [CrossRef] [Green Version]
- Sanz, G.; Singh, M.; Peuget, S.; Selivanova, G. Inhibition of p53 inhibitors: Progress, challenges and perspectives. J. Mol. Cell Biol. 2019, 11, 586–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladds, M.; Lain, S. Small molecule activators of the p53 response. J. Mol. Cell Biol. 2019, 11, 245–254. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Prives, C. Relevance of the p53-MDM2 axis to aging. Cell Death Differ. 2018, 25, 169–179. [Google Scholar] [CrossRef]
- White, E. Autophagy and p53. Cold Spring Harb. Perspect. Med. 2016, 6, a026120. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z. Tumor suppressor p53 and metabolism. J. Mol. Cell Biol. 2019, 11, 284–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Lin, M.; Wu, R.; Wang, X.; Yang, B.; Levine, A.J.; Hu, W.; Feng, Z. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc. Natl. Acad. Sci. USA 2011, 108, 16259–16264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [Green Version]
- Moon, S.H.; Huang, C.H.; Houlihan, S.L.; Regunath, K.; Freed-Pastor, W.A.; Morris, J.P.T.; Tschaharganeh, D.F.; Kastenhuber, E.R.; Barsotti, A.M.; Culp-Hill, R.; et al. p53 Represses the Mevalonate Pathway to Mediate Tumor Suppression. Cell 2019, 176, 564–580.e19. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Hou, T.; Gao, T.; Lu, X.; Yang, Q.; Zhu, Q.; Li, Z.; Liu, C.; Mu, G.; Liu, G.; et al. p53 cooperates with SIRT6 to regulate cardiolipin de novo biosynthesis. Cell Death Dis. 2018, 9, 941. [Google Scholar] [CrossRef]
- Li, L.; Mao, Y.; Zhao, L.; Wu, J.; Zhao, M.; Du, W.; Yu, L.; Jiang, P. p53 regulation of ammonia metabolism through urea cycle controls polyamine biosynthesis. Nature 2019, 567, 253–256. [Google Scholar] [CrossRef]
- Morris, J.P.T.; Yashinskie, J.J.; Koche, R.; Chandwani, R.; Tian, S.; Chen, C.C.; Baslan, T.; Marinkovic, Z.S.; Sanchez-Rivera, F.J.; Leach, S.D.; et al. Alpha-Ketoglutarate links p53 to cell fate during tumour suppression. Nature 2019, 573, 595–599. [Google Scholar] [CrossRef]
- Tajan, M.; Hock, A.K.; Blagih, J.; Robertson, N.A.; Labuschagne, C.F.; Kruiswijk, F.; Humpton, T.J.; Adams, P.D.; Vousden, K.H. A Role for p53 in the Adaptation to Glutamine Starvation through the Expression of SLC1A3. Cell Metab. 2018, 28, 721–736.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maddocks, O.D.; Berkers, C.R.; Mason, S.M.; Zheng, L.; Blyth, K.; Gottlieb, E.; Vousden, K.H. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 2013, 493, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Kroemer, G. Cytoplasmic functions of the tumour suppressor p53. Nature 2009, 458, 1127–1130. [Google Scholar] [CrossRef] [PubMed]
- Mihara, M.; Erster, S.; Zaika, A.; Petrenko, O.; Chittenden, T.; Pancoska, P.; Moll, U.M. p53 has a direct apoptogenic role at the mitochondria. Mol. Cell 2003, 11, 577–590. [Google Scholar] [CrossRef]
- Leu, J.I.; Dumont, P.; Hafey, M.; Murphy, M.E.; George, D.L. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat. Cell Biol. 2004, 6, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Bouchier-Hayes, L.; Kuwana, T.; Newmeyer, D.D.; Green, D.R. PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science 2005, 309, 1732–1735. [Google Scholar] [CrossRef]
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef] [Green Version]
- Oliner, J.D.; Saiki, A.Y.; Caenepeel, S. The Role of MDM2 Amplification and Overexpression in Tumorigenesis. Cold Spring Harb. Perspect. Med. 2016, 6, a026336. [Google Scholar] [CrossRef]
- Danovi, D.; Meulmeester, E.; Pasini, D.; Migliorini, D.; Capra, M.; Frenk, R.; de Graaf, P.; Francoz, S.; Gasparini, P.; Gobbi, A.; et al. Amplification of Mdmx (or Mdm4) directly contributes to tumor formation by inhibiting p53 tumor suppressor activity. Mol. Cell. Biol. 2004, 24, 5835–5843. [Google Scholar] [CrossRef] [Green Version]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef]
- Kubbutat, M.H.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, S.Y.; Adler, V.; Buschmann, T.; Wu, X.; Ronai, Z. Mdm2 association with p53 targets its ubiquitination. Oncogene 1998, 17, 2543–2547. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Brooks, C.L.; Wu-Baer, F.; Chen, D.; Baer, R.; Gu, W. Mono- versus polyubiquitination: Differential control of p53 fate by Mdm2. Science 2003, 302, 1972–1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Momand, J.; Zambetti, G.P.; Olson, D.C.; George, D.; Levine, A.J. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992, 69, 1237–1245. [Google Scholar] [CrossRef]
- Oliner, J.D.; Kinzler, K.W.; Meltzer, P.S.; George, D.L.; Vogelstein, B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature 1992, 358, 80–83. [Google Scholar] [CrossRef]
- Oliner, J.D.; Pietenpol, J.A.; Thiagalingam, S.; Gyuris, J.; Kinzler, K.W.; Vogelstein, B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature 1993, 362, 857–860. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Bayle, J.H.; Olson, D.; Levine, A.J. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993, 7, 1126–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ofir-Rosenfeld, Y.; Boggs, K.; Michael, D.; Kastan, M.B.; Oren, M. Mdm2 regulates p53 mRNA translation through inhibitory interactions with ribosomal protein L26. Mol. Cell 2008, 32, 180–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shvarts, A.; Steegenga, W.T.; Riteco, N.; van Laar, T.; Dekker, P.; Bazuine, M.; van Ham, R.C.; van der Houven van Oordt, W.; Hateboer, G.; van der Eb, A.J.; et al. MDMX: A novel p53-binding protein with some functional properties of MDM2. Embo J. 1996, 15, 5349–5357. [Google Scholar] [CrossRef] [PubMed]
- Tanimura, S.; Ohtsuka, S.; Mitsui, K.; Shirouzu, K.; Yoshimura, A.; Ohtsubo, M. MDM2 interacts with MDMX through their RING finger domains. FEBS Lett. 1999, 447, 5–9. [Google Scholar] [CrossRef]
- Badciong, J.C.; Haas, A.L. MdmX is a RING finger ubiquitin ligase capable of synergistically enhancing Mdm2 ubiquitination. J. Biol. Chem. 2002, 277, 49668–49675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.N.; Roe, A.E.; Donehower, L.A.; Bradley, A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 1995, 378, 206–208. [Google Scholar] [CrossRef] [PubMed]
- Montes de Oca Luna, R.; Wagner, D.S.; Lozano, G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 1995, 378, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Parant, J.; Chavez-Reyes, A.; Little, N.A.; Yan, W.; Reinke, V.; Jochemsen, A.G.; Lozano, G. Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat. Genet. 2001, 29, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Hao, Q.; Liao, P.; Luo, S.; Zhang, M.; Hu, G.; Liu, H.; Zhang, Y.; Cao, B.; Baddoo, M.; et al. Nerve growth factor receptor negates the tumor suppressor p53 as a feedback regulator. eLife 2016, 5, e15099. [Google Scholar] [CrossRef] [Green Version]
- Chao, T.; Zhou, X.; Cao, B.; Liao, P.; Liu, H.; Chen, Y.; Park, H.W.; Zeng, S.X.; Lu, H. Pleckstrin homology domain-containing protein PHLDB3 supports cancer growth via a negative feedback loop involving p53. Nat. Commun. 2016, 7, 13755. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Cao, B.; Lu, H. Negative auto-regulators trap p53 in their web. J. Mol. Cell Biol. 2017, 9, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Hao, Q.; Cho, W.C. Battle against cancer: An everlasting saga of p53. Int. J. Mol. Sci. 2014, 15, 22109–22127. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Tavana, O.; Gu, W. p53 modifications: Exquisite decorations of the powerful guardian. J. Mol. Cell Biol. 2019, 11, 564–577. [Google Scholar] [CrossRef] [Green Version]
- Shieh, S.Y.; Ikeda, M.; Taya, Y.; Prives, C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 1997, 91, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Shieh, S.Y.; Ahn, J.; Tamai, K.; Taya, Y.; Prives, C. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev. 2000, 14, 289–300. [Google Scholar]
- Kruse, J.P.; Gu, W. Modes of p53 regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, W.; Roeder, R.G. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 1997, 90, 595–606. [Google Scholar] [CrossRef] [Green Version]
- Sykes, S.M.; Mellert, H.S.; Holbert, M.A.; Li, K.; Marmorstein, R.; Lane, W.S.; McMahon, S.B. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol. Cell 2006, 24, 841–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Luo, J.; Zhang, W.; Gu, W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol. Cell 2006, 24, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Zhao, W.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation is indispensable for p53 activation. Cell 2008, 133, 612–626. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Lu, H. Signaling to p53: Ribosomal proteins find their way. Cancer Cell 2009, 16, 369–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marechal, V.; Elenbaas, B.; Piette, J.; Nicolas, J.C.; Levine, A.J. The ribosomal L5 protein is associated with mdm-2 and mdm-2-p53 complexes. Mol. Cell. Biol. 1994, 14, 7414–7420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Liao, J.M.; Liao, W.J.; Lu, H. Scission of the p53-MDM2 Loop by Ribosomal Proteins. Genes Cancer 2012, 3, 298–310. [Google Scholar] [CrossRef]
- Zhou, X.; Hao, Q.; Liao, J.; Zhang, Q.; Lu, H. Ribosomal protein S14 unties the MDM2-p53 loop upon ribosomal stress. Oncogene 2013, 32, 388–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, Q.; Wang, J.; Chen, Y.; Wang, S.; Cao, M.; Lu, H.; Zhou, X. Dual regulation of p53 by the ribosome maturation factor SBDS. Cell Death Dis. 2020, 11, 197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bywater, M.J.; Poortinga, G.; Sanij, E.; Hein, N.; Peck, A.; Cullinane, C.; Wall, M.; Cluse, L.; Drygin, D.; Anderes, K.; et al. Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of p53. Cancer Cell 2012, 22, 51–65. [Google Scholar] [CrossRef] [Green Version]
- Devlin, J.R.; Hannan, K.M.; Hein, N.; Cullinane, C.; Kusnadi, E.; Ng, P.Y.; George, A.J.; Shortt, J.; Bywater, M.J.; Poortinga, G.; et al. Combination Therapy Targeting Ribosome Biogenesis and mRNA Translation Synergistically Extends Survival in MYC-Driven Lymphoma. Cancer Discov. 2016, 6, 59–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patch, A.M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.Z.; Ma, D.; Suo, C.; Shi, J.; Xue, M.; Hu, X.; Xiao, Y.; Yu, K.D.; Liu, Y.R.; Yu, Y.; et al. Genomic and Transcriptomic Landscape of Triple-Negative Breast Cancers: Subtypes and Treatment Strategies. Cancer Cell 2019, 35, 428–440.e5. [Google Scholar] [CrossRef] [Green Version]
- Leroy, B.; Anderson, M.; Soussi, T. TP53 mutations in human cancer: Database reassessment and prospects for the next decade. Hum. Mutat. 2014, 35, 672–688. [Google Scholar] [CrossRef]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [Green Version]
- Chan, W.M.; Siu, W.Y.; Lau, A.; Poon, R.Y. How many mutant p53 molecules are needed to inactivate a tetramer? Mol. Cell. Biol. 2004, 24, 3536–3551. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Reumers, J.; Couceiro, J.R.; De Smet, F.; Gallardo, R.; Rudyak, S.; Cornelis, A.; Rozenski, J.; Zwolinska, A.; Marine, J.C.; et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat. Chem. Biol. 2011, 7, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Hao, Q.; Lu, H. Mutant p53 in cancer therapy-the barrier or the path. J. Mol. Cell Biol. 2019, 11, 293–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adorno, M.; Cordenonsi, M.; Montagner, M.; Dupont, S.; Wong, C.; Hann, B.; Solari, A.; Bobisse, S.; Rondina, M.B.; Guzzardo, V.; et al. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell 2009, 137, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Xu, J.; Liu, J.; Amjad, A.; Zhang, K.; Liu, Q.; Zhou, L.; Xiao, J.; Li, X. Mutant p53 promotes tumor cell malignancy by both positive and negative regulation of the transforming growth factor beta (TGF-beta) pathway. J. Biol. Chem. 2015, 290, 11729–11740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerod, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef] [Green Version]
- Liao, P.; Zeng, S.X.; Zhou, X.; Chen, T.; Zhou, F.; Cao, B.; Jung, J.H.; Del Sal, G.; Luo, S.; Lu, H. Mutant p53 Gains Its Function via c-Myc Activation upon CDK4 Phosphorylation at Serine 249 and Consequent PIN1 Binding. Mol. Cell 2017, 68, 1134–1146 e6. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Liao, P.; Zeng, S.X.; Lu, H. It takes a team: A gain-of-function story of p53-R249S. J. Mol. Cell Biol. 2019, 11, 277–283.e6. [Google Scholar] [CrossRef]
- Ameyar-Zazoua, M.; Larochette, N.; Dorothee, G.; Daugas, E.; Haddada, H.; Gouloumet, V.; Metivier, D.; Stancou, R.; Mami-Chouaib, F.; Kroemer, G.; et al. Wild-type p53 induced sensitization of mutant p53 TNF-resistant cells: Role of caspase-8 and mitochondria. Cancer Gene Ther. 2002, 9, 219–227. [Google Scholar] [CrossRef] [Green Version]
- Chee, J.L.; Saidin, S.; Lane, D.P.; Leong, S.M.; Noll, J.E.; Neilsen, P.M.; Phua, Y.T.; Gabra, H.; Lim, T.M. Wild-type and mutant p53 mediate cisplatin resistance through interaction and inhibition of active caspase-9. Cell Cycle 2013, 12, 278–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, A.K.; Pietsch, E.C.; Dumont, P.; Tao, J.; Murphy, M.E. Wild-type and mutant p53 proteins interact with mitochondrial caspase-3. Cancer Biol. Ther. 2011, 11, 740–745. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Hollstein, M.; Xu, Y. p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat. Cell Biol. 2007, 9, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Lang, G.A.; Iwakuma, T.; Suh, Y.A.; Liu, G.; Rao, V.A.; Parant, J.M.; Valentin-Vega, Y.A.; Terzian, T.; Caldwell, L.C.; Strong, L.C.; et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004, 119, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olive, K.P.; Tuveson, D.A.; Ruhe, Z.C.; Yin, B.; Willis, N.A.; Bronson, R.T.; Crowley, D.; Jacks, T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 2004, 119, 847–860. [Google Scholar] [CrossRef] [Green Version]
- Terzian, T.; Suh, Y.A.; Iwakuma, T.; Post, S.M.; Neumann, M.; Lang, G.A.; Van Pelt, C.S.; Lozano, G. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev. 2008, 22, 1337–1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; Chen, L.; Li, C.; Lu, W.; Chen, J. Inhibition of MDM2 by hsp90 contributes to mutant p53 stabilization. J. Biol. Chem. 2001, 276, 40583–40590. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Marchenko, N.D.; Schulz, R.; Fischer, V.; Velasco-Hernandez, T.; Talos, F.; Moll, U.M. Functional inactivation of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Mol. Cancer Res. MCR 2011, 9, 577–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.C.; Chan, J.Y.; Chiu, Y.L.; Liu, S.T.; Lozano, G.; Wang, S.L.; Ho, C.L.; Huang, S.M. Grail as a molecular determinant for the functions of the tumor suppressor p53 in tumorigenesis. Cell Death Differ. 2013, 20, 732–743. [Google Scholar] [CrossRef] [PubMed]
- Ray, D.; Ray, P.; Ferrer-Torres, D.; Wang, Z.; Nancarrow, D.; Yoon, H.W.; San Martinho, M.; Hinton, T.; Owens, S.; Thomas, D.; et al. Isoforms of RNF128 Regulate the Stability of Mutant P53 in Barrett’s Esophageal Cells. Gastroenterology 2020, 158, 583–597.e1. [Google Scholar] [CrossRef]
- Leng, R.P.; Lin, Y.; Ma, W.; Wu, H.; Lemmers, B.; Chung, S.; Parant, J.M.; Lozano, G.; Hakem, R.; Benchimol, S. Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell 2003, 112, 779–791. [Google Scholar] [CrossRef] [Green Version]
- Yan, W.; Jung, Y.S.; Zhang, Y.; Chen, X. Arsenic trioxide reactivates proteasome-dependent degradation of mutant p53 protein in cancer cells in part via enhanced expression of Pirh2 E3 ligase. PLoS ONE 2014, 9, e103497. [Google Scholar] [CrossRef] [PubMed]
- Dornan, D.; Wertz, I.; Shimizu, H.; Arnott, D.; Frantz, G.D.; Dowd, P.; O’Rourke, K.; Koeppen, H.; Dixit, V.M. The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature 2004, 429, 86–92. [Google Scholar] [CrossRef]
- Yamada, T.; Das Gupta, T.K.; Beattie, C.W. p28, an anionic cell-penetrating peptide, increases the activity of wild type and mutated p53 without altering its conformation. Mol. Pharm. 2013, 10, 3375–3383. [Google Scholar] [CrossRef]
- Nguyen, D.T.T.; Richter, D.; Michel, G.; Mitschka, S.; Kolanus, W.; Cuevas, E.; Wulczyn, F.G. The ubiquitin ligase LIN41/TRIM71 targets p53 to antagonize cell death and differentiation pathways during stem cell differentiation. Cell Death Differ. 2017, 24, 1063–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Hao, Q.; Wang, J.; Li, J.; Huang, C.; Zhang, Y.; Wu, X.; Lu, H.; Zhou, X. Ubiquitin ligase TRIM71 suppresses ovarian tumorigenesis by degrading mutant p53. Cell Death Dis. 2019, 10, 737. [Google Scholar] [CrossRef] [PubMed]
- Allton, K.; Jain, A.K.; Herz, H.M.; Tsai, W.W.; Jung, S.Y.; Qin, J.; Bergmann, A.; Johnson, R.L.; Barton, M.C. Trim24 targets endogenous p53 for degradation. Proc. Natl. Acad. Sci. USA 2009, 106, 11612–11616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivlin, N.; Katz, S.; Doody, M.; Sheffer, M.; Horesh, S.; Molchadsky, A.; Koifman, G.; Shetzer, Y.; Goldfinger, N.; Rotter, V.; et al. Rescue of embryonic stem cells from cellular transformation by proteomic stabilization of mutant p53 and conversion into WT conformation. Proc. Natl. Acad. Sci. USA 2014, 111, 7006–7011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearson, M.; Carbone, R.; Sebastiani, C.; Cioce, M.; Fagioli, M.; Saito, S.; Higashimoto, Y.; Appella, E.; Minucci, S.; Pandolfi, P.P.; et al. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature 2000, 406, 207–210. [Google Scholar] [CrossRef]
- Hofmann, T.G.; Moller, A.; Sirma, H.; Zentgraf, H.; Taya, Y.; Droge, W.; Will, H.; Schmitz, M.L. Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2. Nat. Cell Biol. 2002, 4, 1–10. [Google Scholar] [CrossRef]
- Louria-Hayon, I.; Grossman, T.; Sionov, R.V.; Alsheich, O.; Pandolfi, P.P.; Haupt, Y. The promyelocytic leukemia protein protects p53 from Mdm2-mediated inhibition and degradation. J. Biol. Chem. 2003, 278, 33134–33141. [Google Scholar] [CrossRef] [Green Version]
- Alsheich-Bartok, O.; Haupt, S.; Alkalay-Snir, I.; Saito, S.; Appella, E.; Haupt, Y. PML enhances the regulation of p53 by CK1 in response to DNA damage. Oncogene 2008, 27, 3653–3661. [Google Scholar] [CrossRef] [Green Version]
- Rokudai, S.; Laptenko, O.; Arnal, S.M.; Taya, Y.; Kitabayashi, I.; Prives, C. MOZ increases p53 acetylation and premature senescence through its complex formation with PML. Proc. Natl. Acad. Sci. USA 2013, 110, 3895–3900. [Google Scholar] [CrossRef] [Green Version]
- Haupt, S.; di Agostino, S.; Mizrahi, I.; Alsheich-Bartok, O.; Voorhoeve, M.; Damalas, A.; Blandino, G.; Haupt, Y. Promyelocytic leukemia protein is required for gain of function by mutant p53. Cancer Res. 2009, 69, 4818–4826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Zhu, Y.; Hu, W.; Feng, Z. TRIM32 is a novel negative regulator of p53. Mol. Cell. Oncol. 2015, 2, e970951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Zhang, C.; Wang, X.L.; Ly, P.; Belyi, V.; Xu-Monette, Z.Y.; Young, K.H.; Hu, W.; Feng, Z. E3 ubiquitin ligase TRIM32 negatively regulates tumor suppressor p53 to promote tumorigenesis. Cell Death Differ. 2014, 21, 1792–1804. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Huang, N.J.; Chen, C.; Tang, W.; Kornbluth, S. Ubiquitylation of p53 by the APC/C inhibitor Trim39. Proc. Natl. Acad. Sci. USA 2012, 109, 20931–20936. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Ji, Z.; Wang, Y.; Li, J.; Cao, H.; Zhu, H.H.; Gao, W.Q. TRIM59 is up-regulated in gastric tumors, promoting ubiquitination and degradation of p53. Gastroenterology 2014, 147, 1043–1054. [Google Scholar] [CrossRef]
- Chen, Y.; Guo, Y.; Yang, H.; Shi, G.; Xu, G.; Shi, J.; Yin, N.; Chen, D. TRIM66 overexpresssion contributes to osteosarcoma carcinogenesis and indicates poor survival outcome. Oncotarget 2015, 6, 23708–23719. [Google Scholar] [CrossRef] [Green Version]
- Reddy, B.A.; van der Knaap, J.A.; Bot, A.G.; Mohd-Sarip, A.; Dekkers, D.H.; Timmermans, M.A.; Martens, J.W.; Demmers, J.A.; Verrijzer, C.P. Nucleotide biosynthetic enzyme GMP synthase is a TRIM21-controlled relay of p53 stabilization. Mol. Cell 2014, 53, 458–470. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Elabd, S.; Hammer, S.; Solozobova, V.; Yan, H.; Bartel, F.; Inoue, S.; Henrich, T.; Wittbrodt, J.; Loosli, F.; et al. TRIM25 has a dual function in the p53/Mdm2 circuit. Oncogene 2015, 34, 5729–5738. [Google Scholar] [CrossRef]
- Wang, C.; Ivanov, A.; Chen, L.; Fredericks, W.J.; Seto, E.; Rauscher, F.J., 3rd; Chen, J. MDM2 interaction with nuclear corepressor KAP1 contributes to p53 inactivation. Embo J. 2005, 24, 3279–3290. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.; Villagra, A.; Peng, L.; Coppola, D.; Glozak, M.; Sotomayor, E.M.; Chen, J.; Lane, W.S.; Seto, E. The ATDC (TRIM29) protein binds p53 and antagonizes p53-mediated functions. Mol. Cell. Biol. 2010, 30, 3004–3015. [Google Scholar] [CrossRef] [Green Version]
- Caratozzolo, M.F.; Valletti, A.; Gigante, M.; Aiello, I.; Mastropasqua, F.; Marzano, F.; Ditonno, P.; Carrieri, G.; Simonnet, H.; D’Erchia, A.M.; et al. TRIM8 anti-proliferative action against chemo-resistant renal cell carcinoma. Oncotarget 2014, 5, 7446–7457. [Google Scholar] [CrossRef] [PubMed]
- Joo, H.M.; Kim, J.Y.; Jeong, J.B.; Seong, K.M.; Nam, S.Y.; Yang, K.H.; Kim, C.S.; Kim, H.S.; Jeong, M.; An, S.; et al. Ret finger protein 2 enhances ionizing radiation-induced apoptosis via degradation of AKT and MDM2. Eur. J. Cell Biol. 2011, 90, 420–431. [Google Scholar] [CrossRef] [PubMed]
- Esser, C.; Scheffner, M.; Hohfeld, J. The chaperone-associated ubiquitin ligase CHIP is able to target p53 for proteasomal degradation. J. Biol. Chem. 2005, 280, 27443–27448. [Google Scholar] [CrossRef] [Green Version]
- Thirunavukarasou, A.; Singh, P.; Govindarajalu, G.; Bandi, V.; Baluchamy, S. E3 ubiquitin ligase Cullin4B mediated polyubiquitination of p53 for its degradation. Mol. Cell. Biochem. 2014, 390, 93–100. [Google Scholar] [CrossRef]
- Andrews, P.; He, Y.J.; Xiong, Y. Cytoplasmic localized ubiquitin ligase cullin 7 binds to p53 and promotes cell growth by antagonizing p53 function. Oncogene 2006, 25, 4534–4548. [Google Scholar] [CrossRef] [Green Version]
- Nikolaev, A.Y.; Li, M.; Puskas, N.; Qin, J.; Gu, W. Parc: A cytoplasmic anchor for p53. Cell 2003, 112, 29–40. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Kon, N.; Li, M.; Zhang, W.; Qin, J.; Gu, W. ARF-BP1/Mule is a critical mediator of the ARF tumor suppressor. Cell 2005, 121, 1071–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tovar, C.; Graves, B.; Packman, K.; Filipovic, Z.; Higgins, B.; Xia, M.; Tardell, C.; Garrido, R.; Lee, E.; Kolinsky, K.; et al. MDM2 small-molecule antagonist RG7112 activates p53 signaling and regresses human tumors in preclinical cancer models. Cancer Res. 2013, 73, 2587–2597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, L.; Liew, H.P.; Camus, S.; Goh, A.M.; Chee, L.L.; Lunny, D.P.; Lane, E.B.; Lane, D.P. Ionizing radiation induces a dramatic persistence of p53 protein accumulation and DNA damage signaling in mutant p53 zebrafish. Oncogene 2013, 32, 4009–4016. [Google Scholar] [CrossRef] [Green Version]
- Anandasabapathy, N.; Ford, G.S.; Bloom, D.; Holness, C.; Paragas, V.; Seroogy, C.; Skrenta, H.; Hollenhorst, M.; Fathman, C.G.; Soares, L. GRAIL: An E3 ubiquitin ligase that inhibits cytokine gene transcription is expressed in anergic CD4+ T cells. Immunity 2003, 18, 535–547. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Hsieh, P.; Lin, H.; Liu, T.; Wu, H.; Chen, C.; Chen, Y. Grail is involved in adipocyte differentiation and diet-induced obesity. Cell Death Dis. 2018, 9, 525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, G.; Liu, B.; Li, Z.; Wu, H.; Wang, P.; Zhao, K.; Jiang, G.; Zhang, L.; Gao, C. E3 ubiquitin ligase RNF128 promotes innate antiviral immunity through K63-linked ubiquitination of TBK1. Nat. Immunol. 2016, 17, 1342–1351. [Google Scholar] [CrossRef] [PubMed]
- Migliorini, D.; Bogaerts, S.; Defever, D.; Vyas, R.; Denecker, G.; Radaelli, E.; Zwolinska, A.; Depaepe, V.; Hochepied, T.; Skarnes, W.C.; et al. Cop1 constitutively regulates c-Jun protein stability and functions as a tumor suppressor in mice. J. Clin. Investig. 2011, 121, 1329–1343. [Google Scholar] [CrossRef] [Green Version]
- Hakem, A.; Bohgaki, M.; Lemmers, B.; Tai, E.; Salmena, L.; Matysiak-Zablocki, E.; Jung, Y.S.; Karaskova, J.; Kaustov, L.; Duan, S.; et al. Role of Pirh2 in mediating the regulation of p53 and c-Myc. PLoS Genet. 2011, 7, e1002360. [Google Scholar] [CrossRef] [PubMed]
- Vitari, A.C.; Leong, K.G.; Newton, K.; Yee, C.; O’Rourke, K.; Liu, J.; Phu, L.; Vij, R.; Ferrando, R.; Couto, S.S.; et al. COP1 is a tumour suppressor that causes degradation of ETS transcription factors. Nature 2011, 474, 403–406. [Google Scholar] [CrossRef]
- Khetchoumian, K.; Teletin, M.; Tisserand, J.; Mark, M.; Herquel, B.; Ignat, M.; Zucman-Rossi, J.; Cammas, F.; Lerouge, T.; Thibault, C.; et al. Loss of Trim24 (Tif1alpha) gene function confers oncogenic activity to retinoic acid receptor alpha. Nat. Genet. 2007, 39, 1500–1506. [Google Scholar] [CrossRef]
- Tsai, W.W.; Wang, Z.; Yiu, T.T.; Akdemir, K.C.; Xia, W.; Winter, S.; Tsai, C.Y.; Shi, X.; Schwarzer, D.; Plunkett, W.; et al. TRIM24 links a non-canonical histone signature to breast cancer. Nature 2010, 468, 927–932. [Google Scholar] [CrossRef] [Green Version]
- Groner, A.C.; Cato, L.; de Tribolet-Hardy, J.; Bernasocchi, T.; Janouskova, H.; Melchers, D.; Houtman, R.; Cato, A.C.B.; Tschopp, P.; Gu, L.; et al. TRIM24 Is an Oncogenic Transcriptional Activator in Prostate Cancer. Cancer Cell 2016, 29, 846–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, D.; Li, Y.; Zhang, W.; Alvarez, A.A.; Song, L.; Tang, J.; Gao, W.Q.; Hu, B.; Cheng, S.Y.; Feng, H. TRIM24 is an oncogenic transcriptional co-activator of STAT3 in glioblastoma. Nat. Commun. 2017, 8, 1454. [Google Scholar] [CrossRef]
- Elabd, S.; Meroni, G.; Blattner, C. TRIMming p53’s anticancer activity. Oncogene 2016, 35, 5577–5584. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.S.; Jin, Y.; Gallegos, J.R.; Lu, H. Balance of Yin and Yang: Ubiquitylation-mediated regulation of p53 and c-Myc. Neoplasia 2006, 8, 630–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Marchenko, N.D.; Moll, U.M. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011, 18, 1904–1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Tan, M.; Liu, X.; Xiong, X.; Sun, Y. Inactivation of ribosomal protein S27-like confers radiosensitivity via the Mdm2-p53 and Mdm2-MRN-ATM axes. Cell Death Dis. 2018, 9, 145. [Google Scholar] [CrossRef] [PubMed]
- Wienken, M.; Moll, U.M.; Dobbelstein, M. Mdm2 as a chromatin modifier. J. Mol. Cell Biol. 2017, 9, 74–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| E3 Ligases | Mechanism | Reference |
|---|---|---|
| RING-finger E3 ligases | ||
| MDM2 | Induces ubiquitination and degradation of wtp53; suppresses wtp53 transcriptional activity | [30,31,34,35,36,37] |
| Promotes mtp53 degradation | [83,84,85] | |
| RNF128 | Induces ubiquitination and degradation of wtp53 | [86] |
| Promotes mtp53 degradation | [87] | |
| PIRH2 | Induces ubiquitination and degradation of wtp53 | [88] |
| Promotes mtp53 degradation | [89] | |
| COP1 | Induces ubiquitination and degradation of wtp53 | [90] |
| Promotes mtp53 degradation | [91] | |
| TRIM protein family | ||
| TRIM71 | Induces ubiquitination and degradation of wtp53 | [92] |
| Promotes mtp53 ubiquitination and degradation | [93] | |
| TRIM24 | Induces ubiquitination and degradation of wtp53 | [94] |
| Converts mtp53-R172H to a wild-type conformation | [95] | |
| TRIM19 | Enhances wtp53 acetylation and phosphorylation, and protects it from MDM2-mediated degradation | [96,97,98,99,100] |
| Binds to and enhances transcriptional activity of mtp53 | [101] | |
| TRIM32, TRIM39, TRIM59, TRIM66 | Induces ubiquitination and degradation of wtp53 | [102,103,104,105,106] |
| TRIM21, TRIM25, TRIM28 | Promotes MDM2-mediated p53 ubiquitination and degradation | [107,108,109] |
| TRIM29 | Relocates wtp53 from the nucleus to the cytoplasm | [110] |
| TRIM8, TRIM13 | Stabilizes and activates wtp53 by degrading MDM2 | [111,112] |
| Others | ||
| CHIP | Induces degradation of both wt and mtp53 | [84,85,113] |
| Cullin 4B | Mediates poly-ubiquitination and degradation of wtp53 | [114] |
| Cullin 7 | Mediates mono- or di-ubiquitination of wtp53 | [115] |
| Cullin 9 | Sequesters wtp53 in the cytoplasm | [116] |
| HUWE1 | Mediates mono- or poly-ubiquitination of wtp53 | [117] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hao, Q.; Chen, Y.; Zhou, X. The Janus Face of p53-Targeting Ubiquitin Ligases. Cells 2020, 9, 1656. https://doi.org/10.3390/cells9071656
Hao Q, Chen Y, Zhou X. The Janus Face of p53-Targeting Ubiquitin Ligases. Cells. 2020; 9(7):1656. https://doi.org/10.3390/cells9071656
Chicago/Turabian StyleHao, Qian, Yajie Chen, and Xiang Zhou. 2020. "The Janus Face of p53-Targeting Ubiquitin Ligases" Cells 9, no. 7: 1656. https://doi.org/10.3390/cells9071656
APA StyleHao, Q., Chen, Y., & Zhou, X. (2020). The Janus Face of p53-Targeting Ubiquitin Ligases. Cells, 9(7), 1656. https://doi.org/10.3390/cells9071656