A Survey of Reported Disease-Related Mutations in the MRE11-RAD50-NBS1 Complex


Abstract
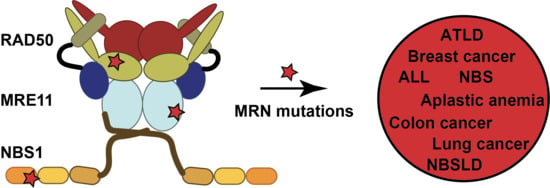
1. Introduction
2. DNA DSBs and the Role of MRN in Repair
3. Correlation of MRE11 with Disease
3.1. MRE11 in Brief
3.2. Disease-Associated MRE11 Variants

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation in MRE11 | Disease/Cancer Type | Location in Gene or Structure | Effect on Structure/Function | Ref. |
|---|---|---|---|---|
| Δ5-7 MRE11 | colon cancer | deletion of exons 5-7; deletes the third and fourth phosphoesterase motifs | Loss of 3′-to-5′ exonuclease activity impairs HR. Weak interactions with RAD50 and NBS1. Suppressed ATM activation. | [76] |
| E42K, E42D, and E42A | ovarian, uterine, and small cell lung cancers | nuclease domain | Inhibits Tel1/ATM activation. May disrupt an ionic interaction stabilizing the interface of the nuclease and capping domains. | [77] |
| S104C | Breast cancer | nuclease domain, NBS1 interaction region 2 | Reduced NBS1 binding. | [57,78] |
| D113G | NBSLD | nuclease domain, NBS1 interaction region 2 | May affect MRE11 dimer geometry/stability and NBS1 binding. | [58,79] |
| N117S | ATLD3/4 | nuclease domain, NBS1 interaction region 2 | May affect MRE11 dimer stability. Reduced NBS1 binding. | [12,80] |
| A177V | lung cancer | nuclease domain | Impairs DSB repair in yeast. Packing of this buried residue might be disrupted. | [77] |
| R202G | breast cancer | nuclease domain | Could affect the local stability around NBS1 binding region 1. Impaired NBS1 interaction. | [57,82] |
| W210C | ATLD7/8 | nuclease domain | Affects MRN protein expression resulting in dysfunctional MRN complex. | [58,83] |
| W243R | ATLD17/18 | nuclease domain | Impaired interactions with RAD50 and NBS1. Disrupts MRE11 dimerization. Causes defects in ATM activation. | [73,74,81] |
| F237C | breast cancer | nuclease domain | Unknown | [85] |
| H302Y | breast cancer | nuclease domain | Unknown | [85] |
| R305W | ovarian cancer | linker between nuclease and capping domains | Possibly affects structural organization of protein. | [57,86] |
| D368Y | lung and nasopharyn-geal cancers | capping domain | Strongly impairs DSB repair. Possibly disrupts the fold of MRE11. | [77] |
| L473F | colorectal cancers | helix-loop-helix | Impaired interaction with RAD50. | [84] |
| T481 Kcompound heterozyg-ous with R527Stop | ATLD5/6 | helix-loop-helix | Decreased expression levels of MRN. Increased sensitivity to IR. Attenuated ATM activation. | [87] |
| R503H | breast carcinoma | helix-loop-helix | Unknown | [78] |
| R572Q | lymphoma | GAR motif | Disrupts interactions with RAD50 and NBS1. Defects in ATR activation. | [74,78] |
| R633Stop | ATLD1/2; breast cancer | truncation near C-terminus | Does not localize to sites of damage. | [12,74,80,82] |
4. Correlation of RAD50 with Disease
4.1. RAD50 in Brief
4.2. Disease-Associated RAD50 Variants
| Mutation in RAD50 | Disease/Cancer Type | Location in the Structure | Effect on Structure/Function | Ref. |
|---|---|---|---|---|
| D69N, D69Y, and D69G | uterine, colorectal, bladder, and lung cancers; myelodysplastic syndrome | Walker A | Impaired Tel1/ATM activation and decreased ATP hydrolysis. Could affect “open” to “closed” global transition dynamics. | [77] |
| R850C | endometrioid, breast/ovarian cancers | coiled coil region | Unknown | [95,96] |
| R1093Stop compound heterozyg-ous with Stop1313Y | NBSLD | truncated at C-terminal sub-domain | RAD50 deficiency. Increased radiosensitivity, chromosomal instability, impaired activation of ATM, impaired G1/S cellcycle-checkpoint activation. | [14] |
| R1214H and R1214C | pancreatic cancer | extended signature helix/basic switch | Impaired ATP hydrolysis activity, NBD association, and allostery within MRN. | [77,97,98,99] |
| A1229D | Burkitt Lymphoma | Walker B | Unknown | [100] |
| E1232K | lung cancer | Walker B | Defective in ATP hydrolysis. | [47,77] |
| N1236D | Burkitt Lymphoma | D-loop | Unknown | [100] |
| L1237F and L1237V | bladder and colorectal cancer; breast cancer | D-loop | Increased ATP hydrolysis. Destabilization of “closed” MRN complex. Loss of ATM signaling and Mre11 exonuclease activity. | [77,91,101] |
| D1238N | breast cancer | D-loop | Severe DSB repair defect. Increased ATP hydrolysis. Destabilization of “closed” MRN complex. Loss of ATM signaling and Mre11 exonuclease activity. | [91,101] |
| R1256C and R1256H | bladder/uterine cancers | between D-loop and His-loop | Impaired Tel1/ATM activation. | [77] |
| Q1259K | endometrial carcinoma | between D-loop and His-loop | Unknown | [101] |
| Q1263H | endometrial carcinoma | between D-loop and His-loop | Unknown | [95,96] |
5. Correlations of NBS1 with Disease
5.1. NBS1 in Brief
5.2. Disease-Associated NBS1 Variants
| Mutation in NBS1 | Disease/Cancer Type | Location in the Structure | Effect on Structure/Function | Ref. |
|---|---|---|---|---|
| 657del5 | NBS; breast, prostate, and colorectal cancers; medulloblastoma; lymphoblastic leukemia; and non-Hodgkin lymphoma | transition between BRCT1 and BRCT2 | Truncates NBS1 after BRCT1 and expresses a secondary C-terminal fragment starting near BRCT2. Disrupts tandem BRCT domains and proper phosphoprotein interaction. | [22,110] |
| V26I | medulloblastoma | FHA | Possibly disrupts phosphoprotein binding. | [62,116] |
| I41M | hepatocellular carcinoma | FHA | Possibly disrupts phosphoprotein binding | [61,117] |
| L57M/H711Y double mutation | medulloblastoma | FHA; C-terminus | L57M may disrupt the FHA/BRCT1 interface and destabilize the protein. H711Y may disrupt RNF20, MRE11, and/or ATM binding. | [61,116] |
| T90S | intrahepatic cholangiocarcinoma | FHA | May disrupt FHA domain structure and/or phosphoprotein binding site. Decreased nuclear localization of MRE11. | [117] |
| S93L | acute lymphoblastic leukemia (ALL) | FHA | May disrupt FHA domain structure and/or phosphoprotein binding site. | [119] |
| D95N | ALL; breast, larynx, and prostate cancers | FHA | May disrupt FHA domain structure and/or phosphoprotein binding site. | [119,120,121,122] |
| T148I/ P427L double mutation | medulloblastoma | BRCT1; Intrinsically disordered region | T148I may disrupt the hydrophobic cluster where it is located and nearby phosphoserine binding cleft. | [116] |
| L150F | breast cancer | BRCT1 | Possible disruption of a hydrophobic cluster and phosphoserine binding cleft. Increases chromosomal instability. | [61,86,92] |
| I171V | ALL; breast, larynx, and colorectal cancers; head and neck tumors; aplastic anemia | BRCT1 | Possible disruption of a hydrophobic cluster and phosphoserine binding cleft. Increased sensitivity to IR and MMS and lower frequency of HR repair. Loss of association with MDC1. | [62,119,123,124,125,126] |
| E185Q | leukemia and lung cancers; urinary system cancer | BRCT1 | Possibly affects the interaction with BRCA1. May cause an increase in tumor aggression. | [120,127,128,129] |
| V210F | ALL and Non-Hodgkin lymphoma | BRCT1/BRCT2 linker | Hydrophobic residue could disrupt phosphoprotein binding and/or protein stability. | [62,119,130] |
| R215W | ALL; Hodgkin and Non-Hodgkin lymphomas; melanoma; prostate, breast, and colorectal cancers | BRCT1/BRCT2 linker | Disruption of salt bridge destabilizes structure. Decreased co-localization with γ-H2AX at sites of DNA damage and decreased repair efficiency. | [61,62,108,124,125,131] |
| D272N | hepatocellular carcinoma | BRCT2 | Could disrupt ATM phosphorylation of serine 278. | [111,117] |
| A308T | medulloblastoma | BRCT2 | May disrupt the structure of BRCT2 or interface between BRCT1/BRCT2. | [62,116] |
| G311R | medulloblastoma | BRCT2 | May disrupt the structure of BRCT2 or interface between BRCT1/BRCT2. | [62,116] |
| V348D | hepatocellular carcinoma | intrinsically disordered region | Could disrupt ATM phosphorylation of serine 343. | [117] |
| T402A | glioblastoma | intrinsically disordered region | Could disrupt ATM phosphorylation of serine 397. | [115] |
| S406F | glioblastoma | intrinsically disordered region | Could disrupt ATM phosphorylation of serine 397. May introduce order to the intrinsically disordered domain. | [115] |
| S415R | hepatocellular carcinoma | intrinsically disordered region | Could interrupt the sequence space of the intrinsically disordered domain, altering the surrounding structure. | [117] |
| M424V | glioblastoma | intrinsically disordered region | Unknown | [115] |
| T463I/ Q616H double mutation | glioblastoma | intrinsically disordered region | T463I could disrupt nuclear localization of MRE11; Q616H may affect ATM phosphorylation of serine 615. | [115] |
| T485M | glioblastoma | intrinsically disordered region | Unknown | [115] |
| F603L | hepatocellular carcinoma | C-terminal region | Could disrupt RAD18 binding. | [117] |
| S633T | hepatocellular carcinoma | C-terminal region | Could disrupt MRE11 binding. Deficient in nuclear localization of MRE11. | [117] |
| S638P | intrahepatic cholangiocarcinoma | C-terminal region | Could disrupt MRE11 binding. Deficient in nuclear localization of MRE11. | [117] |
| Y679H | renal cell carcinoma | C-terminal region | Could disrupt MRE11 binding and nuclear localization of MRE11. | [135] |
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Romero-Laorden, N.; Castro, E. Inherited mutations in DNA repair genes and cancer risk. Curr. Probl. Cancer 2017, 41, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.K.; Anker, J.F.; Carneiro, B.A.; Chandra, S.; Kaplan, J.; Kalyan, A.; Santa-Maria, C.A.; Platanias, L.C.; Giles, F.J. Genomic landscape of DNA repair genes in cancer. Oncotarget 2016, 7, 23312–23321. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; DuBoff, M.; Jayakumaran, G.; Kris, M.G.; Ladanyi, M.; Robson, M.E.; Mandelker, D.; Zauderer, M.G. Novel Germline Mutations in DNA Damage Repair in Patients with Malignant Pleural Mesotheliomas. J. Thorac. Oncol. 2020, 15, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 2018, 23, 239–254.e6. [Google Scholar] [CrossRef] [PubMed]
- Vineis, P.; Manuguerra, M.; Kavvoura, F.K.; Guarrera, S.; Allione, A.; Rosa, F.; Di Gregorio, A.; Polidoro, S.; Saletta, F.; Ioannidis, J.P.A.; et al. A field synopsis on low-penetrance variants in DNA repair genes and cancer susceptibility. J. Natl. Cancer Inst. 2009, 101, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Rupnik, A.; Lowndes, N.F.; Grenon, M. MRN and the race to the break. Chromosoma 2010, 119, 115–135. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.J.; Lees-Miller, S.P.; Tainer, J.A. Mre11-Rad50-Nbs1 conformations and the control of sensing, signaling, and effector responses at DNA double-strand breaks. DNA Repair (Amst) 2010, 9, 1299–1306. [Google Scholar] [CrossRef]
- Oh, J.; Symington, L.S. Role of the Mre11 complex in preserving genome integrity. Genes (Basel) 2018, 9, 589. [Google Scholar] [CrossRef]
- Xiao, Y.; Weaver, D.T. Conditional gene targeted deletion by Cre recombinase demonstrates the requirement for the double-strand break repair Mre11 protein in murine embryonic stem cells. Nucleic Acids Res. 1997, 25, 2985–2991. [Google Scholar] [CrossRef]
- Luo, G.; Yao, M.S.; Bender, C.F.; Mills, M.; Bladl, A.R.; Bradley, A.; Petrini, J.H.J. Disruption of mRad50 causes embryonic stem cell lethality, abnormal embryonic development, and sensitivity to ionizing radiation. Proc. Natl. Acad. Sci. USA 1999, 96, 7376–7381. [Google Scholar] [CrossRef]
- Zhu, J.; Petersen, S.; Tessarollo, L.; Nussenzweig, A. Targeted disruption of the Nijmegen breakage syndrome gene NBS1 leads to early embryonic lethality in mice. Curr. Biol. 2001, 11, 105–109. [Google Scholar] [CrossRef]
- Stewart, G.S.; Maser, R.S.; Stankovic, T.; Bressan, D.A.; Kaplan, M.I.; Jaspers, N.G.; Raams, A.; Byrd, P.J.; Petrini, J.H.J.; Taylor, A.M. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell 1999, 99, 577–587. [Google Scholar] [CrossRef]
- Chrzanowska, K.H.; Gregorek, H.; Dembowska-Bagińska, B.; Kalina, M.A.; Digweed, M. Nijmegen breakage syndrome (NBS). Orphanet J. Rare Dis. 2012, 7, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Waltes, R.; Kalb, R.; Gatei, M.; Kijas, A.W.; Stumm, M.; Sobeck, A.; Wieland, B.; Varon, R.; Lerenthal, Y.; Lavin, M.F.; et al. Human RAD50 Deficiency in a Nijmegen Breakage Syndrome-like Disorder. Am. J. Hum. Genet. 2009, 84, 605–616. [Google Scholar] [CrossRef]
- Koczkowska, M.; Krawczynska, N.; Stukan, M.; Kuzniacka, A.; Brozek, I.; Sniadecki, M.; Debniak, J.; Wydra, D.; Biernat, W.; Kozlowski, P.; et al. Spectrum and prevalence of pathogenic variants in ovarian cancer susceptibility genes in a group of 333 patients. Cancers (Basel) 2018, 10, 442. [Google Scholar] [CrossRef] [PubMed]
- Brandt, S.; Samartzis, E.P.; Zimmermann, A.K.; Fink, D.; Moch, H.; Noske, A.; Dedes, K.J. Lack of MRE11-RAD50-NBS1 (MRN) complex detection occurs frequently in low-grade epithelial ovarian cancer. BMC Cancer 2017, 17, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, Y.; Zhou, K.; Zhou, C.; Zhou, R.; Cheng, C.; Wei, Q.; Lu, D.; Zhou, L. Genetic variations in the homologous recombination repair pathway genes modify risk of glioma. J. Neurooncol. 2016, 126, 11–17. [Google Scholar] [CrossRef]
- Damiola, F.; Pertesi, M.; Oliver, J.; Le Calvez-Kelm, F.; Voegele, C.; Young, E.L.; Robinot, N.; Forey, N.; Durand, G.; Vallée, M.P.; et al. Rare key functional domain missense substitutions in MRE11A, RAD50, and NBNcontribute to breast cancer susceptibility: Results from a Breast Cancer Family Registry case-control mutation-screening study. Breast Cancer Res. 2014, 16, R58. [Google Scholar] [CrossRef]
- Amemiya, Y.; Bacopulos, S.; Al-Shawarby, M.; Al-Tamimi, D.; Naser, W.; Ahmed, A.; Khalifa, M.; Slodkowska, E.; Seth, A. A comparative analysis of breast and ovarian cancer-related gene mutations in Canadian and Saudi Arabian patients with breast cancer. Anticancer Res. 2015, 35, 2601–2610. [Google Scholar]
- Uzunoglu, H.; Korak, T.; Ergul, E.; Uren, N.; Sazci, A.; Utkan, N.Z.; Kargi, E.; Triyaki, Ç.; Yirmibesoglu, O. Association of the nibrin gene (NBN) variants with breast cancer. Biomed. Rep. 2016, 4, 369–373. [Google Scholar] [CrossRef]
- Khan, R.T.; Siddique, A.; Shahid, N.; Khokher, S.; Fatima, W. Breast cancer risk associated with genes encoding DNA repair MRN complex: A study from Punjab, Pakistan. Breast Cancer 2018, 25, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Zeng, Y.; Liu, Z.; Wei, W. Significant association between Nijmegen breakage syndrome 1 657del5 polymorphism and breast cancer risk. Tumor Biol. 2013, 34, 2753–2757. [Google Scholar] [CrossRef] [PubMed]
- Couch, F.J.; Shimelis, H.; Hu, C.; Hart, S.N.; Polley, E.C.; Na, J.; Hallberg, E.; Moore, R.; Thomas, A.; Lilyquist, J.; et al. Associations between cancer predisposition testing panel genes and breast cancer. JAMA Oncol. 2017, 3, 1190–1196. [Google Scholar] [CrossRef] [PubMed]
- Situ, Y.; Chung, L.; Lee, C.S.; Ho, V. MRN (MRE11-RAD50-NBS1) Complex in Human Cancer and Prognostic Implications in Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 816. [Google Scholar] [CrossRef]
- Ho, V.; Chung, L.; Singh, A.; Lea, V.; Revoltar, M.; Lim, S.; Tut, T.-G.; Ng, W.; Lee, M.; de Souza, P.; et al. Early Postoperative Low Expression of RAD50 in Rectal Cancer Patients Associates with Disease-Free Survival. Cancers (Basel) 2017, 9, 163. [Google Scholar] [CrossRef] [PubMed]
- Ho, V.; Chung, L.; Singh, A.; Lea, V.; Abubakar, A.; Lim, S.H.; Ng, W.; Lee, M.; de Souza, P.; Shin, J.S.; et al. Overexpression of the MRE11-RAD50-NBS1 (MRN) complex in rectal cancer correlates with poor response to neoadjuvant radiotherapy and prognosis. BMC Cancer 2018, 18, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Su, T.; Yang, L.; Zhang, C.; He, Y. High expression of MRE11 correlates with poor prognosis in gastric carcinoma. Diagn. Pathol. 2019, 14, 1–9. [Google Scholar] [CrossRef]
- Cannan, W.J.; Pederson, D.S. Mechanisms and Consequences of Double-Strand DNA Break Formation in Chromatin. J. Cell. Physiol. 2016, 231, 3–14. [Google Scholar] [CrossRef]
- Keeney, S.; Neale, M.J. Initiation of meiotic recombination by formation of DNA double-strand breaks: Mechanism and regulation. Biochem. Soc. Trans. 2006, 34, 523–525. [Google Scholar] [CrossRef]
- Dudley, D.D.; Chaudhuri, J.; Bassing, C.H.; Alt, F.W. Mechanism and control of V(D)J recombination versus class switch recombination: Similarities and differences. Adv. Immunol. 2005. [Google Scholar] [CrossRef]
- Han, J.; Huang, J. DNA double-strand break repair pathway choice: The fork in the road. Genome Instab. Dis. 2019. [Google Scholar] [CrossRef]
- Lieber, M.R. The Mechanism of Double-Strand DNA Break Repair by the Nonhomologous DNA End-Joining Pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed]
- Boulton, S.J.; Jackson, S.P. Components of the Ku-dependent non-homologous end-joining pathway are involved in telomeric length maintenance and telomeric silencing. EMBO J. 1998, 17, 1819–1828. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Trujillo, K.; Ramos, W.; Sung, P.; Tomkinson, A.E. Promotion of Dnl4-Catalyzed DNA End-Joining by the Rad50/Mre11/Xrs2 and Hdf1/Hdf2 Complexes. Mol. Cell 2001, 8, 1105–1115. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Shinohara, A.; Shinohara, M. Forkhead-associated domain of yeast Xrs2, a homolog of human Nbs1, promotes nonhomologous end joining through interaction with a ligase IV partner protein, Lif1. Genetics 2008, 179, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Rass, E.; Grabarz, A.; Plo, I.; Gautier, J.; Bertrand, P.; Lopez, B.S. Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat. Struct. Mol. Biol. 2009, 16, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Xie, A.; Kwok, A.; Scully, R. Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat. Struct. Mol. Biol. 2009, 16, 814–818. [Google Scholar] [CrossRef]
- Myler, L.R.; Gallardo, I.F.; Soniat, M.M.; Deshpande, R.A.; Gonzalez, X.B.; Kim, Y.; Paull, T.T.; Finkelstein, I.J. Single-Molecule Imaging Reveals How Mre11-Rad50-Nbs1 Initiates DNA Break Repair. Mol. Cell 2017, 67, 891–898.e4. [Google Scholar] [CrossRef]
- Reginato, G.; Cannavo, E.; Cejka, P. Physiological protein blocks direct the Mre11-Rad50-Xrs2 and Sae2 nuclease complex to initiate DNA end resection. Genes Dev. 2017, 31, 2325–2330. [Google Scholar] [CrossRef]
- Langerak, P.; Mejia-Ramirez, E.; Limbo, O.; Russell, P. Release of Ku and MRN from DNA ends by Mre11 nuclease activity and Ctp1 is required for homologous recombination repair of double-strand breaks. PLoS Genet. 2011, 7. [Google Scholar] [CrossRef]
- Cassani, C.; Vertemara, J.; Bassani, M.; Marsella, A.; Tisi, R.; Zampella, G.; Longhese, M.P. The ATP-bound conformation of the Mre11–Rad50 complex is essential for Tel1/ATM activation. Nucleic Acids Res. 2019, 47, 3550–3567. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Mand, M.R.; Deshpande, R.A.; Kinoshita, E.; Yang, S.-H.; Wyman, C.; Paull, T.T. Ataxia Telangiectasia-Mutated (ATM) Kinase Activity Is Regulated by ATP-driven Conformational Changes in the Mre11/Rad50/Nbs1 (MRN) Complex. J. Biol. Chem. 2013, 288, 12840–12851. [Google Scholar] [CrossRef] [PubMed]
- Syed, A.; Tainer, J.A. The MRE11–RAD50–NBS1 Complex Conducts the Orchestration of Damage Signaling and Outcomes to Stress in DNA Replication and Repair. Annu. Rev. Biochem. 2018, 87, 263–294. [Google Scholar] [CrossRef] [PubMed]
- Lammens, K.; Bemeleit, D.J.; Möckel, C.; Clausing, E.; Schele, A.; Hartung, S.; Schiller, C.B.; Lucas, M.; Angermüller, C.; Söding, J.; et al. The Mre11:Rad50 structure shows an ATP-dependent molecular clamp in DNA double-strand break repair. Cell 2011, 145, 54–66. [Google Scholar] [CrossRef]
- Möckel, C.; Lammens, K.; Schele, A.; Hopfner, K.-P.; Pemberton, T. ATP driven structural changes of the bacterial Mre11:Rad50 catalytic head complex. Nucleic Acids Res. 2012, 40, 914–927. [Google Scholar] [CrossRef]
- Lim, H.S.; Kim, J.S.; Park, Y.B.; Gwon, G.H.; Cho, Y. Crystal structure of the Mre11-Rad50-ATPγS complex: Understanding the interplay between Mre11 and Rad50. Genes Dev. 2011, 25, 1091–1104. [Google Scholar] [CrossRef]
- Rojowska, A.; Lammens, K.; Seifert, F.U.; Direnberger, C.; Feldmann, H.; Hopfner, K.-P. Structure of the Rad50 DNA double-strand break repair protein in complex with DNA. EMBO J. 2014, 33, 2847–2859. [Google Scholar] [CrossRef]
- Herdendorf, T.J.; Albrecht, D.W.; Benkovic, S.J.; Nelson, S.W. Biochemical characterization of bacteriophage T4 Mre11-Rad50 complex. J. Biol. Chem. 2011, 286, 2382–2392. [Google Scholar] [CrossRef]
- Hopfner, K.-P.; Karcher, A.; Shin, D.S.; Craig, L.; Arthur, L.M.; Carney, J.P.; Tainer, J.A. Structural biology of Rad50 ATPase: ATP-driven conformational control in DNA double-strand break repair and the ABC-ATPase superfamily. Cell 2000, 101, 789–800. [Google Scholar] [CrossRef]
- Hopfner, K.-P.; Karcher, A.; Craig, L.; Woo, T.T.; Carney, J.P.; Tainer, J.A. Structural biochemistry and interaction architecture of the DNA double-strand break repair Mre11 nuclease and Rad50-ATPase. Cell 2001, 105, 473–485. [Google Scholar] [CrossRef]
- Hopfner, K.-P.; Craig, L.; Moncalian, G.; Zinkel, R.A.; Usui, T.; Owen, B.A.L.L.; Karcher, A.; Henderson, B.; Bodmer, J.-L.; McMurray, C.T.; et al. The Rad50 zinc-hook is a structure joining Mre11 complexes in DNA recombination and repair. Nature 2002, 418, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.S.; Moncalian, G.; Williams, J.S.; Yamada, Y.; Limbo, O.; Shin, D.S.; Groocock, L.M.; Cahill, D.; Hitomi, C.; Guenther, G.; et al. Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell 2008, 135, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.J.; Williams, R.S.; Williams, J.S.; Moncalian, G.; Arvai, A.S.; Limbo, O.; Guenther, G.; SilDas, S.; Hammel, M.; Russell, P.; et al. ABC ATPase signature helices in Rad50 link nucleotide state to Mre11 interface for DNA repair. Nat. Struct. Mol. Biol. 2011, 18, 423–431. [Google Scholar] [CrossRef]
- Sung, S.; Li, F.; Park, Y.B.; Kim, J.S.; Kim, A.; Song, O.-K.; Kim, J.; Che, J.; Lee, S.E.; Cho, Y. DNA end recognition by the Mre11 nuclease dimer: Insights into resection and repair of damaged DNA. EMBO J. 2014, 33, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sung, S.; Kim, Y.; Li, F.; Gwon, G.; Jo, A.; Kim, T.; Song, O.-K.; Lee, S.E.; Cho, Y.; et al. ATP-dependent DNA binding, unwinding, and resection by the Mre11/Rad50 complex. EMBO J. 2016, 35, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Käshammer, L.; Saathoff, J.-H.; Lammens, K.; Gut, F.; Bartho, J.; Alt, A.; Kessler, B.; Hopfner, K.-P. Mechanism of DNA End Sensing and Processing by the Mre11-Rad50 Complex. Mol. Cell 2019, 76, 382–394.e6. [Google Scholar] [CrossRef]
- Park, Y.B.; Chae, J.; Kim, Y.C.; Cho, Y. Crystal structure of human Mre11: Understanding tumorigenic mutations. Structure 2011, 19, 1591–1602. [Google Scholar] [CrossRef]
- Schiller, C.B.; Lammens, K.; Guerini, I.; Coordes, B.; Feldmann, H.; Schlauderer, F.; Möckel, C.; Schele, A.; Sträßer, K.; Jackson, S.P.; et al. Structure of Mre11-Nbs1 complex yields insights into ataxia-telangiectasia-like disease mutations and DNA damage signaling. Nat. Struct. Mol. Biol. 2012, 19, 693–700. [Google Scholar] [CrossRef]
- Seifert, F.U.; Lammens, K.; Hopfner, K.-P. Structure of the catalytic domain of Mre11 from Chaetomium thermophilum. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2015, 71, 752–757. [Google Scholar] [CrossRef]
- Seifert, F.U.; Lammens, K.; Stoehr, G.; Kessler, B.; Hopfner, K.-P. Structural mechanism of ATP-dependent DNA binding and DNA end bridging by eukaryotic Rad50. EMBO J. 2016, 35, 759–772. [Google Scholar] [CrossRef]
- Williams, R.S.; Dodson, G.E.; Limbo, O.; Yamada, Y.; Williams, J.S.; Guenther, G.; Classen, S.; Glover, J.N.M.; Iwasaki, H.; Russell, P.; et al. Nbs1 flexibly tethers Ctp1 and Mre11-Rad50 to coordinate DNA double-strand break processing and repair. Cell 2009, 139, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, J.; Chapman, J.R.; Clapperton, J.A.; Haire, L.F.; Hartsuiker, E.; Li, J.; Carr, A.M.; Jackson, S.P.; Smerdon, S.J. A supramodular FHA/BRCT-repeat architecture mediates Nbs1 adaptor function in response to DNA damage. Cell 2009, 139, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Matange, N.; Podobnik, M.; Visweswariah, S.S. Metallophosphoesterases: Structural fidelity with functional promiscuity. Biochem. J. 2015, 467, 201–216. [Google Scholar] [CrossRef]
- Paull, T.T. 20 Years of Mre11 Biology: No End in Sight. Mol. Cell 2018, 71, 419–427. [Google Scholar] [CrossRef]
- Furuse, M.; Nagase, Y.; Tsubouchi, H.; Murakami-Murofushi, K.; Shibata, T.; Ohta, K. Distinct roles of two separable in vitro activities of yeast Mre11 in mitotic and meiotic recombination. EMBO J. 1998, 17, 6412–6425. [Google Scholar] [CrossRef]
- Paull, T.T.; Gellert, M. The 3′ to 5′ Exonuclease Activity of Mre11 Facilitates Repair of DNA Double-Strand Breaks. Mol. Cell 1998, 1, 969–979. [Google Scholar] [CrossRef]
- Hoa, N.N.; Shimizu, T.; Zhou, Z.W.; Wang, Z.Q.; Deshpande, R.A.; Paull, T.T.; Akter, S.; Tsuda, M.; Furuta, R.; Tsusui, K.; et al. Mre11 Is Essential for the Removal of Lethal Topoisomerase 2 Covalent Cleavage Complexes. Mol. Cell 2016, 64, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, T.; Baer, R.; Gottesman, M.; Gautier, J. MRN, CtIP, and BRCA1 mediate repair of topoisomerase II-DNA adducts. J. Cell Biol. 2016, 212, 399–408. [Google Scholar] [CrossRef]
- Neale, M.J.; Pan, J.; Keeney, S. Endonucleolytic processing of covalent protein-linked DNA double-strand breaks. Nature 2005, 436, 1053–1057. [Google Scholar] [CrossRef]
- Déry, U.; Coulombe, Y.; Rodrigue, A.; Stasiak, A.; Richard, S.; Masson, J.-Y. A Glycine-Arginine Domain in Control of the Human MRE11 DNA Repair Protein. Mol. Cell. Biol. 2008, 28, 3058–3069. [Google Scholar] [CrossRef]
- Yu, Z.; Vogel, G.; Yan, C.; Dubeau, D.; Spehalski, E.; Hébert, J.; Ferguson, D.O.; Masson, J.Y.; Richard, S. The MRE11 GAR motif regulates DNA double-strand break processing and ATR activation. Cell Res. 2012, 22, 305–320. [Google Scholar] [CrossRef] [PubMed]
- Sedghi, M.; Salari, M.; Moslemi, A.R.; Kariminejad, A.; Davis, M.; Goullée, H.; Olsson, B.; Laing, N.; Tajsharghi, H. Ataxia-telangiectasia-like disorder in a family deficient for MRE11A, caused by a MRE11 variant. Neurol. Genet. 2018. [Google Scholar] [CrossRef] [PubMed]
- Uchisaka, N.; Takahashi, N.; Sato, M.; Kikuchi, A.; Mochizuki, S.; Imai, K.; Nonoyama, S.; Ohara, O.; Watanabe, F.; Mizutani, S.; et al. Two Brothers with Ataxia-Telangiectasia-like Disorder with Lung Adenocarcinoma. J. Pediatr. 2009, 155, 435–438. [Google Scholar] [CrossRef] [PubMed]
- Regal, J.A.; Festerling, T.A.; Buis, J.M.; Ferguson, D.O. Disease-associated MRE11 mutants impact ATM/ATR DNA damage signaling by distinct mechanisms. Hum. Mol. Genet. 2013, 22, 5146–5159. [Google Scholar] [CrossRef] [PubMed]
- Giannini, G.; Ristori, E.; Cerignoli, F.; Rinaldi, C.; Zani, M.; Viel, A.; Ottini, L.; Crescenzi, M.; Martinotti, S.; Bignami, M.; et al. Human MRE11 is inactivated in mismatch repair-deficient cancers. EMBO Rep. 2002, 3, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Wen, Q.; Scorah, J.; Phear, G.; Rodgers, G.; Rodgers, S.; Meuth, M. A mutant allele of MRE11 found in mismatch repair-deficient tumor cells suppresses the cellular response to DNA replication fork stress in a dominant negative manner. Mol. Biol. Cell 2008, 19, 1693–1705. [Google Scholar] [CrossRef]
- Hohl, M.; Mojumdar, A.; Hailemariam, S.; Kuryavyi, V.; Ghisays, F.; Sorenson, K.; Chang, M.; Taylor, B.S.; Patel, D.J.; Burgers, P.M.; et al. Modeling cancer genomic data in yeast reveals selection against ATM function during tumorigenesis. PLoS Genet. 2020, 16, e1008422. [Google Scholar] [CrossRef]
- Fukuda, T.; Sumiyoshi, T.; Takahashi, M.; Kataoka, T.; Asahara, T.; Inui, H.; Watatani, M.; Yasutomi, M.; Kamada, N.; Miyagawa, K. Alterations of the double-strand break repair gene MRE11 in cancer. Cancer Res. 2001, 61, 23–26. [Google Scholar]
- Matsumoto, Y.; Miyamoto, T.; Sakamoto, H.; Izumi, H.; Nakazawa, Y.; Ogi, T.; Tahara, H.; Oku, S.; Hiramoto, A.; Shiiki, T.; et al. Two unrelated patients with MRE11A mutations and Nijmegen breakage syndrome-like severe microcephaly. DNA Repair (Amst) 2011, 10, 314–321. [Google Scholar] [CrossRef]
- Lee, J.-H.; Ghirlando, R.; Bhaskara, V.; Hoffmeyer, M.R.; Gu, J.; Paull, T.T. Regulation of Mre11/Rad50 by Nbs1: Effects on nucleotide-dependent DNA binding and association with ataxiatelangiectasia-like disorder mutant complexes. J. Biol. Chem. 2003, 278, 45171–45181. [Google Scholar] [CrossRef]
- Limbo, O.; Moiani, D.; Kertokalio, A.; Wyman, C.; Tainer, J.A.; Russell, P. Mre11 ATLD17/18 mutation retains Tel1/ATM activity but blocks DNA double-strand break repair. Nucleic Acids Res. 2012, 40, 11435–11449. [Google Scholar] [CrossRef]
- Bartkova, J.; Tommiska, J.; Oplustilova, L.; Aaltonen, K.; Tamminen, A.; Heikkinen, T.; Mistrik, M.; Aittomäki, K.; Blomqvist, C.; Heikkilä, P.; et al. Aberrations of the MRE11-RAD50-NBS1 DNA damage sensor complex in human breast cancer: MRE11 as a candidate familial cancer-predisposing gene. Mol. Oncol. 2008, 2, 296–316. [Google Scholar] [CrossRef]
- Fernet, M.; Gribaa, M.; Salih, M.A.M.; Seidahmed, M.Z.; Hall, J.; Koenig, M. Identification and functional consequences of a novel MRE11 mutation affecting 10 Saudi Arabian patients with the ataxia telangiectasia-like disorder. Hum. Mol. Genet. 2005, 14, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Cummins, J.M.; Shen, D.; Cahill, D.P.; Jallepalli, P.V.; Wang, T.-L.; Parsons, D.W.; Traverso, G.; Awad, M.; Silliman, N.; et al. Three classes of genes mutated in colorectal cancers with chromosomal instability. Cancer Res. 2004, 64, 2998–3001. [Google Scholar] [CrossRef]
- Sjöblom, T.; Jones, S.; Wood, L.D.; Parsons, D.W.; Lin, J.; Barber, T.D.; Mandelker, D.; Leary, R.J.; Ptak, J.; Silliman, N.; et al. The Consensus Coding Sequences of Human Breast and Colorectal Cancers. Science 2006, 314, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Heikkinen, K.; Karppinen, S.-M.; Soini, Y.; Mäkinen, M.; Winqvist, R. Mutation screening of Mre11 complex genes: Indication of RAD50 involvement in breast and ovarian cancer susceptibility. J. Med. Genet. 2003, 40, e131. [Google Scholar] [CrossRef] [PubMed]
- Delia, D.; Paine, M.; Buscemi, G.; Savio, C.; Palmeri, S.; Lulli, P.; Carlessi, L.; Fontanella, E.; Chessa, L. MRE11 mutations and impaired ATM-dependent responses in an Italian family with ataxia-telangiectasia-like disorder. Hum. Mol. Genet. 2004, 13, 2155–2163. [Google Scholar] [CrossRef]
- Hopfner, K.-P.; Tainer, J.A. Rad50/SMC proteins and ABC transporters: Unifying concepts from high-resolution structures. Curr. Opin. Struct. Biol. 2003, 13, 249–255. [Google Scholar] [CrossRef]
- Hohl, M.; Kochańczyk, T.; Tous, C.; Aguilera, A.; Krężel, A.; Petrini, J.H.J. Interdependence of the rad50 hook and globular domain functions. Mol. Cell 2015, 57, 479–491. [Google Scholar] [CrossRef]
- Hohl, M.; Kwon, Y.; Galván, S.M.; Xue, X.; Tous, C.; Aguilera, A.; Sung, P.; Petrini, J.H.J. The Rad50 coiled-coil domain is indispensable for Mre11 complex functions. Nat. Struct. Mol. Biol. 2011, 18, 1124–1131. [Google Scholar] [CrossRef]
- Boswell, Z.K.; Canny, M.D.; Buschmann, T.A.; Sang, J.; Latham, M.P. Adjacent mutations in the archaeal Rad50 ABC ATPase D-loop disrupt allosteric regulation of ATP hydrolysis through different mechanisms. Nucleic Acids Res. 2020, 48, 2457–2472. [Google Scholar] [CrossRef] [PubMed]
- Heikkinen, K.; Rapakko, K.; Karppinen, S.M.; Erkko, H.; Knuutila, S.; Lundán, T.; Mannermaa, A.; Børresen-Dale, A.L.; Borg, Å.; Barkardottir, R.B.; et al. RAD50 and NBS1 are breast cancer susceptibility genes associated with genomic instability. Carcinogenesis 2006, 27, 1593–1599. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Zhang, J.; Ouyang, T.; Li, J.; Wang, T.; Fan, Z.; Fan, T.; Lin, B.; Xie, Y. RAD50 germline mutations are associated with poor survival in BRCA1/2–negative breast cancer patients. Int. J. Cancer 2018, 143, 1935–1942. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.R.; Rowley, S.M.; Li, N.; McInerny, S.; Devereux, L.; Wong-Brown, M.W.; Trainer, A.H.; Mitchell, G.; Scott, R.J.; James, P.A.; et al. Panel testing for familial breast cancer: Calibrating the tension between research and clinical care. J. Clin. Oncol. 2016, 34, 1455–1459. [Google Scholar] [CrossRef] [PubMed]
- García-Sanz, P.; Triviño, J.C.; Mota, A.; Pérez López, M.; Colás, E.; Rojo-Sebastián, A.; García, Á.; Gatius, S.; Ruiz, M.; Prat, J.; et al. Chromatin remodelling and DNA repair genes are frequently mutated in endometrioid endometrial carcinoma. Int. J. Cancer 2017, 140, 1551–1563. [Google Scholar] [CrossRef]
- Maresca, L.; Lodovichi, S.; Lorenzoni, A.; Cervelli, T.; Monaco, R.; Spugnesi, L.; Tancredi, M.; Falaschi, E.; Zavaglia, K.; Landucci, E.; et al. Functional Interaction Between BRCA1 and DNA Repair in Yeast May Uncover a Role of RAD50, RAD51, MRE11A, and MSH6 Somatic Variants in Cancer Development. Front. Genet. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Young, E.L.; Thompson, B.A.; Neklason, D.W.; Firpo, M.A.; Werner, T.; Bell, R.; Berger, J.; Fraser, A.; Gammon, A.; Koptiuch, C.; et al. Pancreatic cancer as a sentinel for hereditary cancer predisposition. BMC Cancer 2018, 18, 697. [Google Scholar] [CrossRef]
- Deshpande, R.A.; Williams, G.J.; Limbo, O.; Williams, R.S.; Kuhnlein, J.; Lee, J.-H.; Classen, S.; Guenther, G.; Russell, P.; Tainer, J.A.; et al. ATP-driven Rad50 conformations regulate DNA tethering, end resection, and ATM checkpoint signaling. EMBO J. 2014, 33, 482–500. [Google Scholar] [CrossRef]
- Boswell, Z.K.; Rahman, S.; Canny, M.D.; Latham, M.P. A dynamic allosteric pathway underlies Rad50 ABC ATPase function in DNA repair. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef]
- Kaymaz, Y.; Oduor, C.I.; Yu, H.; Otieno, J.A.; Ong’echa, J.M.; Moormann, A.M.; Bailey, J.A. Comprehensive transcriptome and mutational profiling of Endemic Burkitt lymphoma reveals EBV type-specific differences. Mol. Cancer Res. 2017, 15, 563–576. [Google Scholar] [CrossRef]
- Al-Ahmadie, H.; Iyer, G.; Hohl, M.; Asthana, S.; Inagaki, A.; Schultz, N.; Hanrahan, A.J.; Scott, S.N.; Brannon, A.R.; McDermott, G.C.; et al. Synthetic Lethality in ATM-Deficient RAD50-Mutant Tumors Underlies Outlier Response to Cancer Therapy. Cancer Discov. 2014, 4, 1014–1021. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, J.; Tauchi, H.; Sakamoto, S.; Nakamura, A.; Morishima, K.; Matsuura, S.; Kobayashi, T.; Tamai, K.; Tanimoto, K.; Komatsu, K. NBS1 localizes to γ-H2AX foci through interaction with the FHA/BRCT domain. Curr. Biol. 2002, 12, 1846–1851. [Google Scholar] [CrossRef]
- Lee, J.H.; Lim, D.S. Dual role of Nbs1 in the ataxia telangiectasia mutated-dependent DNA damage response. FEBS J. 2006, 273, 1630–1636. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, K.; Bucher, P. The FHA domain: A putative nuclear signalling domain found in protein kinases and transcription factors. Trends Biochem. Sci. 1995, 20, 347–349. [Google Scholar] [CrossRef]
- Durocher, D.; Henckel, J.; Fersht, A.R.; Jackson, S.P. The FHA domain is a modular phosphopeptide recognition motif. Mol. Cell 1999. [Google Scholar] [CrossRef]
- Koonin, E.V.; Altschul, S.F.; Bork, P. BRCA1 Protein Products ... Functional Motifs. Nat. Genet. 1996, 13, 266–268. [Google Scholar] [CrossRef]
- Becker, E.; Meyer, V.; Madaoui, H.; Guerois, R. Detection of a tandem BRCT in Nbs1 and Xrs2 with functional implications in the DNA damage response. Bioinformatics 2006, 22, 1289–1292. [Google Scholar] [CrossRef]
- Seemanová, E.; Sperling, K.; Neitzel, H.; Varon, R.; Hadac, J.; Butova, O.; Schröck, E.; Seeman, P.; Digweed, M. Nijmegen breakage syndrome (NBS) with neurological abnormalities and without chromosomal instability. J. Med. Genet. 2006, 43, 218–224. [Google Scholar] [CrossRef]
- Varon, R.; Seemanova, E.; Chrzanowska, K.; Hnateyko, O.; Piekutowska-Abramczuk, D.; Krajewska-Walasek, M.; Sykut-Cegielska, J.; Sperling, K.; Reis, A. Clinical ascertainment of Nijmegen breakage syndrome (NBS) and prevalence of the major mutation, 657del5, in three Slav populations. Eur. J. Hum. Genet. 2000, 8, 900–902. [Google Scholar] [CrossRef]
- Maser, R.S.; Zinkel, R.; Petrini, J.H.J. An alternative mode of translation permits production of a variant NBS1 protein from the common Nijmegen breakage syndrome allele. Nat. Genet. 2001, 27, 417–421. [Google Scholar] [CrossRef]
- Wu, X.; Ranganathant, V.; Weisman, D.S.; Helne, W.F.; Ciccone, D.N.; O’Neill, T.B.; Crick, K.E.; Pierce, K.A.; Lane, W.S.; Rathbun, G.; et al. ATM phosphorylation of Nijmegen breakage syndrome protein is required in a DNA damage response. Nature 2000, 405, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Grosbart, M.; Anand, R.; Wyman, C.; Cejka, P.; Petrini, J.H.J. The Mre11-Nbs1 Interface Is Essential for Viability and Tumor Suppression. Cell Rep. 2017, 18, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Ebi, H.; Matsuo, K.; Sugito, N.; Suzuki, M.; Osada, H.; Tajima, K.; Ueda, R.; Takahashi, T. Novel NBS1 heterozygous germ line mutation causing MRE11-binding domain loss predisposes to common types of cancer. Cancer Res. 2007, 67, 11158–11165. [Google Scholar] [CrossRef]
- Varon, R.; Vissinga, C.; Platzer, M.; Cerosaletti, K.M.; Chrzanowska, K.H.; Saar, K.; Beckmann, G.; Seemanová, E.; Cooper, P.R.; Nowak, N.J.; et al. Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell 1998, 93, 467–476. [Google Scholar] [CrossRef]
- Watanabe, T.; Nobusawa, S.; Lu, S.; Huang, J.; Mittelbronn, M.; Ohgaki, H. Mutational inactivation of the nijmegen breakage syndrome gene (NBS1) in glioblastomas is associated with multiple TP53 mutations. J. Neuropathol. Exp. Neurol. 2009, 68, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Grotzer, M.A.; Watanabe, T.; Hewer, E.; Pietsch, T.; Rutkowski, S.; Ohgaki, H. Mutations in the nijmegen breakage syndrome gene in medulloblastomas. Clin. Cancer Res. 2008, 14, 4053–4058. [Google Scholar] [CrossRef]
- Wang, Y.; Hong, Y.; Li, M.; Long, J.; Zhao, Y.P.; Zhang, J.X.; Li, Q.; You, H.; Tong, W.M.; Jia, J.D.; et al. Mutation inactivation of Nijmegen breakage syndrome gene (NBS1) in hepatocellular carcinoma and intrahepatic cholangiocarcinoma. PLoS ONE 2013, 8, e82426. [Google Scholar] [CrossRef]
- Saito, Y.; Komatsu, K. Functional role of NBS1 in radiation damage response and translesion DNA synthesis. Biomolecules 2015, 5, 1990–2002. [Google Scholar] [CrossRef]
- Varon, R.; Reis, A.; Henze, G.; Einsiedel, H.G.V.; Sperling, K.; Seeger, K. Mutations in the Nijmegen Breakage Syndrome gene (NBS1) in childhood acute lymphoblastic leukemia (ALL). Cancer Res. 2001, 61, 3570–3572. [Google Scholar]
- Desjardins, S.; Beauparlant, J.C.; Labrie, Y.; Ouellette, G.; Durocher, F. Variations in the NBN/NBS1 gene and the risk of breast cancer in non-BRCA1/2French Canadian families with high risk of breast cancer. BMC Cancer 2009, 9, 181. [Google Scholar] [CrossRef]
- Ziólkowska, I.; Mosor, M.; Wierzbicka, M.; Rydzanicz, M.; Pernak-Schwarz, M.; Nowak, J. Increased risk of larynx cancer in heterozygous carriers of the I171V mutation of the NBS1 gene. Cancer Sci. 2007, 98, 1701–1705. [Google Scholar] [CrossRef] [PubMed]
- Hebbring, S.J.; Fredriksson, H.; White, K.A.; Maier, C.; Ewing, C.; McDonnell, S.K.; Jacobsen, S.J.; Cerhan, J.; Schaid, D.J.; Ikonen, T.; et al. Role of the Nijmegen breakage syndrome 1 gene in familial and sporadic prostate cancer. Cancer Epidemiol. Biomarkers Prev. 2006, 15, 935–938. [Google Scholar] [CrossRef] [PubMed]
- Shimada, H.; Shimizu, K.; Mimaki, S.; Sakiyama, T.; Mori, T.; Shimasaki, N.; Yokota, J.; Nakachi, K.; Ohta, T.; Ohki, M. First case of aplastic anemia in a Japanese child with a homozygous missense mutation in the NBS1 gene (I171V) associated with genomic instability. Hum. Genet. 2004, 115, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Masi, A.; Antoccia, A. NBS1 Heterozygosity and Cancer Risk. Curr. Genom. 2008, 9, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Ma, N.; Li, M.; Tian, Q.B.; Liu, D.W. Functional variants in NBS1 and cancer risk: Evidence from a meta-analysis of 60 publications with 111 individual studies. Mutagenesis 2013, 28, 683–697. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Miyamoto, M.; Tatsuda, D.; Kubo, M.; Nakagama, H.; Nakamura, Y.; Satoh, H.; Matsuda, K.; Watanabe, T.; Ohta, T. A rare polymorphic variant of NBS1 reduces DNA repair activity and elevates chromosomal instability. Cancer Res. 2014, 74, 3707–3715. [Google Scholar] [CrossRef]
- Fang, W.; Qiu, F.; Zhang, L.; Deng, J.; Zhang, H.; Yang, L.; Zhou, Y.; Lu, J. The functional polymorphism of NBS1 p.Glu185Gln is associated with an increased risk of lung cancer in Chinese populations: Case-control and a meta-analysis. Mutat. Res. 2014, 770, 61–68. [Google Scholar] [CrossRef]
- He, Y.Z.; Chi, X.S.; Zhang, Y.C.; Deng, X.B.; Wang, J.R.; Lv, W.Y.; Zhou, Y.H.; Wang, Z.Q. NBS1 Glu185Gln polymorphism and cancer risk: Update on current evidence. Tumor Biol. 2014, 35, 675–687. [Google Scholar] [CrossRef]
- Zheng, J.; Zhang, C.; Jiang, L.; You, Y.; Liu, Y.; Lu, J.; Zhou, Y. Functional NBS1 polymorphism is associated with occurrence and advanced disease status of nasopharyngeal carcinoma. Mol. Carcinog. 2011, 50, 689–696. [Google Scholar] [CrossRef]
- Mosor, M.; Ziółkowska, I.; Pernak-Schwarz, M.; Januszkiewicz-Lewandowska, D.; Nowak, J. Association of the heterozygous germline I171V mutation of the NBS1 gene with childhood acute lymphoblastic leukemia. Leukemia 2006, 20, 1454–1456. [Google Scholar] [CrossRef]
- di Masi, A.; Viganotti, M.; Polticelli, F.; Ascenzi, P.; Tanzarella, C.; Antoccia, A. The R215W mutation in NBS1 impairs γ-H2AX binding and affects DNA repair: Molecular bases for the severe phenotype of 657del5/R215W Nijmegen breakage syndrome patients. Biochem. Biophys. Res. Commun. 2008, 369, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. The alphabet of intrinsic disorder. Intrinsically Disord. Proteins 2013, 1, e24684. [Google Scholar] [CrossRef] [PubMed]
- Tseng, S.F.; Chang, C.Y.; Wu, K.J.; Teng, S.C. Importin KPNA2 is required for proper nuclear localization and multiple functions of NBS1. J. Biol. Chem. 2005, 280, 39594–39600. [Google Scholar] [CrossRef] [PubMed]
- Yanagihara, H.; Kobayashi, J.; Tateishi, S.; Kato, A.; Matsuura, S.; Tauchi, H.; Yamada, K.; Takezawa, J.; Sugasawa, K.; Masutani, C.; et al. NBS1 Recruits RAD18 via a RAD6-like Domain and Regulates Pol η-Dependent Translesion DNA Synthesis. Mol. Cell 2011, 43, 788–797. [Google Scholar] [CrossRef]
- Varela, I.; Tarpey, P.; Raine, K.; Huang, D.; Ong, C.K.; Stephens, P.; Davies, H.; Jones, D.; Lin, M.L.; Teague, J.; et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature 2011, 469, 539–542. [Google Scholar] [CrossRef]
- Vilar, E.; Bartnik, C.M.; Stenzel, S.L.; Raskin, L.; Ahn, J.; Moreno, V.; Mukherjee, B.; Iniesta, M.D.; Morgan, E.A.; Rennert, G.; et al. MRE11 deficiency increases sensitivity to poly(ADP-ribose) polymerase inhibition in microsatellite unstable colorectal cancers. Cancer Res. 2011, 71, 2632–2642. [Google Scholar] [CrossRef]
- Bian, L.; Meng, Y.; Zhang, M.; Li, D. MRE11-RAD50-NBS1 complex alterations and DNA damage response: Implications for cancer treatment. Mol. Cancer 2019, 18, 1–14. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, S.; Canny, M.D.; Buschmann, T.A.; Latham, M.P. A Survey of Reported Disease-Related Mutations in the MRE11-RAD50-NBS1 Complex. Cells 2020, 9, 1678. https://doi.org/10.3390/cells9071678
Rahman S, Canny MD, Buschmann TA, Latham MP. A Survey of Reported Disease-Related Mutations in the MRE11-RAD50-NBS1 Complex. Cells. 2020; 9(7):1678. https://doi.org/10.3390/cells9071678
Chicago/Turabian StyleRahman, Samiur, Marella D. Canny, Tanner A. Buschmann, and Michael P. Latham. 2020. "A Survey of Reported Disease-Related Mutations in the MRE11-RAD50-NBS1 Complex" Cells 9, no. 7: 1678. https://doi.org/10.3390/cells9071678
APA StyleRahman, S., Canny, M. D., Buschmann, T. A., & Latham, M. P. (2020). A Survey of Reported Disease-Related Mutations in the MRE11-RAD50-NBS1 Complex. Cells, 9(7), 1678. https://doi.org/10.3390/cells9071678