Role of Mitochondria-Cytoskeleton Interactions in the Regulation of Mitochondrial Structure and Function in Cancer Stem Cells

Abstract
:1. Introduction
2. Unique Features of CSCs
2.1. Identification of CSCs
2.2. Cellular Bioenergetics in CSCs
3. Mitochondrial Structure and Function
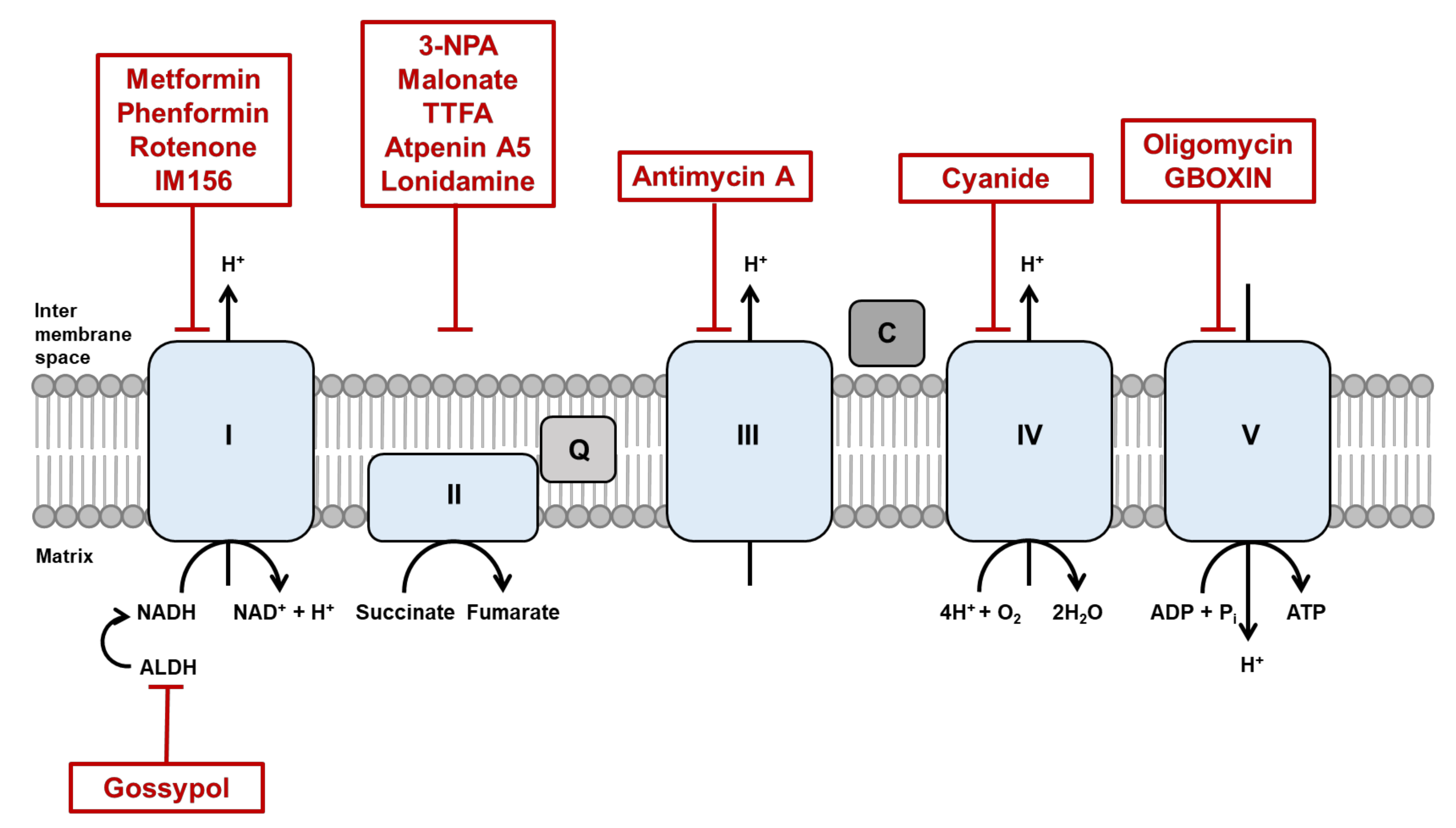
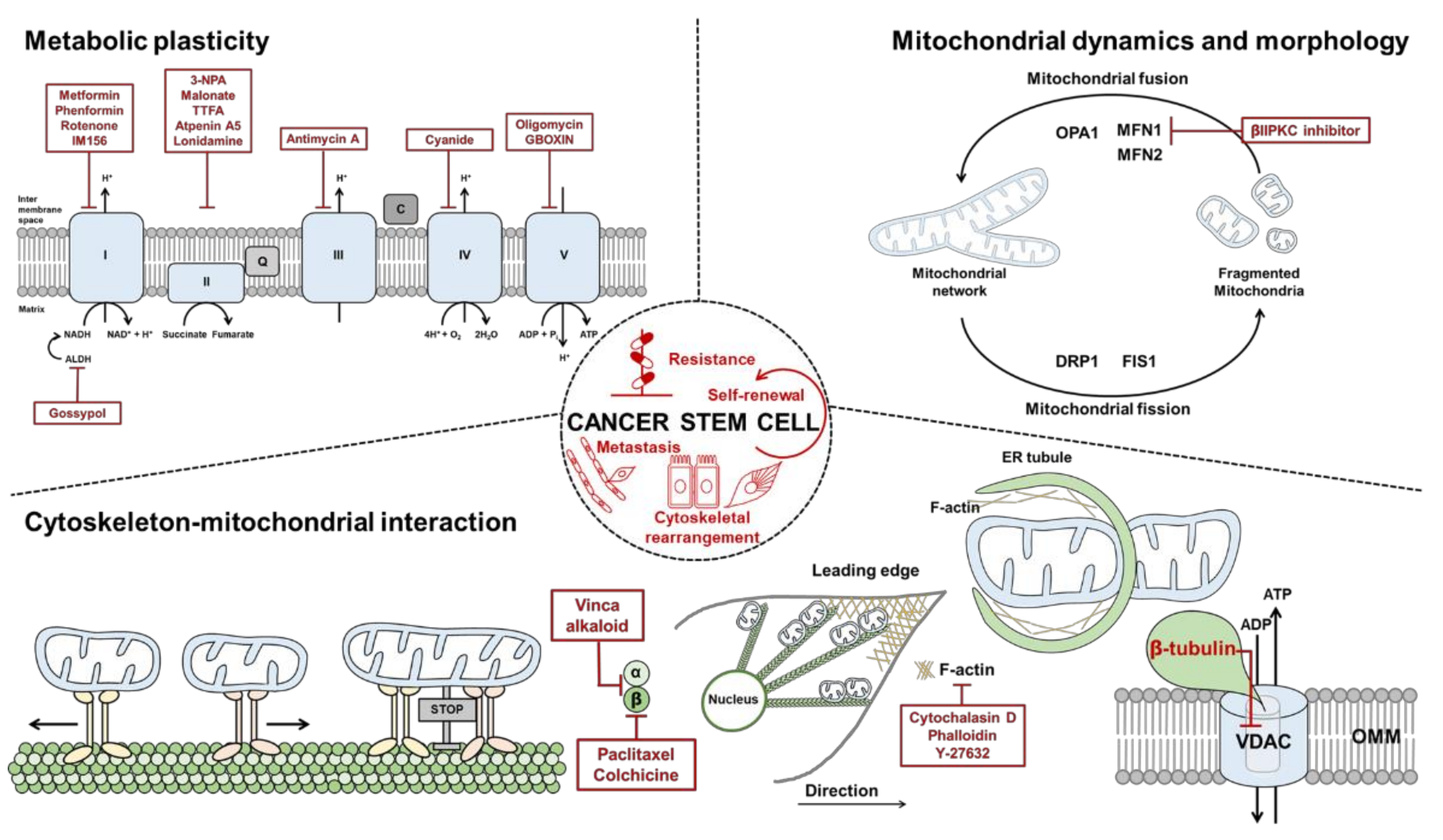
3.1. Mitochondrial Respiratory Chain
3.2. Mitochondrial Dynamics
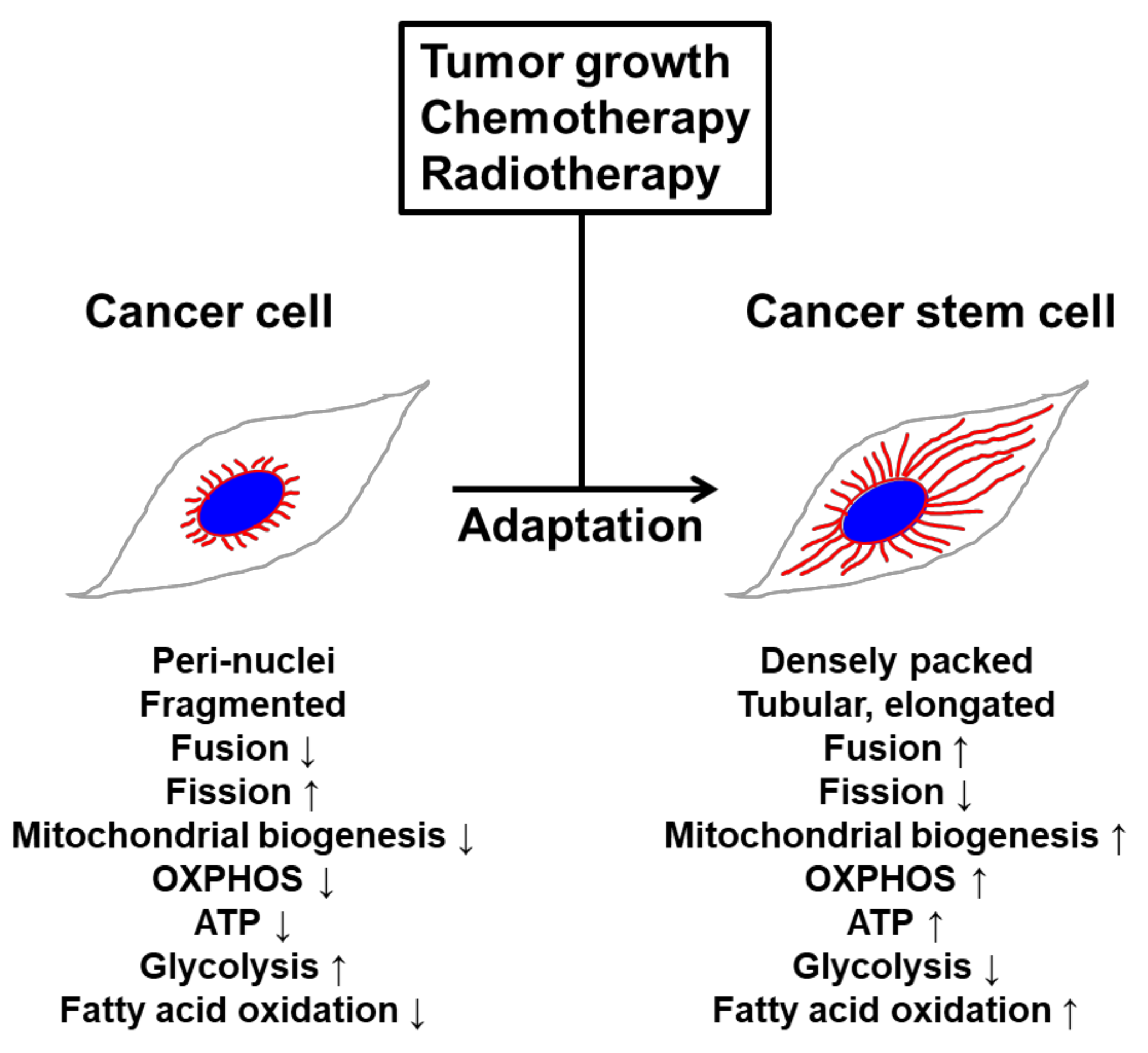
3.3. Mitochondrial Structure-Function in CSCs
4. Cytoskeleton
4.1. Role of Actin Microfilaments in the Regulation of Mitochondria
4.2. Role of Microtubules in the Regulation of Mitochondria
5. Cytoskeleton-Mitochondria Interaction
5.1. Interaction and Regulation of Mitochondrial Function by Actin
5.2. Interaction and Regulation of Mitochondrial Function by Microtubules
5.3. Cytoskeleton Rearrangement in the Regulation of Cancer Metabolism
5.4. Cytoskeleton-Mitochondria Interplay Regulates EMT
6. Therapeutic Strategies Targeting CSCs
6.1. Targeting Mitochondrial Dynamics
6.2. Targeting OXPHOS
6.3. Targeting the Cytoskeleton
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chan, D.C. Mitochondria: Dynamic organelles in disease, aging, and development. Cell 2006, 125, 1241–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suen, D.F.; Norris, K.L.; Youle, R.J. Mitochondrial dynamics and apoptosis. Genes Dev. 2008, 22, 1577–1590. [Google Scholar] [CrossRef] [Green Version]
- Trotta, A.P.; Chipuk, J.E. Mitochondrial dynamics as regulators of cancer biology. Cell Mol. Life Sci. 2017, 74, 1999–2017. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M.R.; Lewis, W.H. Mitochondria in Tissue Culture. Science 1914, 39, 330–333. [Google Scholar] [CrossRef]
- Desai, S.P.; Bhatia, S.N.; Toner, M.; Irimia, D. Mitochondrial localization and the persistent migration of epithelial cancer cells. Biophys. J. 2013, 104, 2077–2088. [Google Scholar] [CrossRef] [Green Version]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sanchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Lim, S.K.; Liang, Q.; Iyer, S.V.; Wang, H.Y.; Wang, Z.; Xie, X.; Sun, D.; Chen, Y.J.; Tabar, V.; et al. Gboxin is an oxidative phosphorylation inhibitor that targets glioblastoma. Nature 2019, 567, 341–346. [Google Scholar] [CrossRef]
- Roesch, A.; Vultur, A.; Bogeski, I.; Wang, H.; Zimmermann, K.M.; Speicher, D.; Korbel, C.; Laschke, M.W.; Gimotty, P.A.; Philipp, S.E.; et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell 2013, 23, 811–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003, 1001–1015. [Google Scholar] [CrossRef] [Green Version]
- Caino, M.C.; Seo, J.H.; Aguinaldo, A.; Wait, E.; Bryant, K.G.; Kossenkov, A.V.; Hayden, J.E.; Vaira, V.; Morotti, A.; Ferrero, S.; et al. A neuronal network of mitochondrial dynamics regulates metastasis. Nat. Commun. 2016, 7, 13730. [Google Scholar] [CrossRef]
- Maldonado, E.N.; Patnaik, J.; Mullins, M.R.; Lemasters, J.J. Free tubulin modulates mitochondrial membrane potential in cancer cells. Cancer Res. 2010, 70, 10192–10201. [Google Scholar] [CrossRef] [Green Version]
- Monge, C.; Beraud, N.; Kuznetsov, A.V.; Rostovtseva, T.; Sackett, D.; Schlattner, U.; Vendelin, M.; Saks, V.A. Regulation of respiration in brain mitochondria and synaptosomes: Restrictions of ADP diffusion in situ, roles of tubulin, and mitochondrial creatine kinase. Mol. Cell Biochem. 2008, 318, 147–165. [Google Scholar] [CrossRef]
- Noskov, S.Y.; Rostovtseva, T.K.; Bezrukov, S.M. ATP transport through VDAC and the VDAC-tubulin complex probed by equilibrium and nonequilibrium MD simulations. Biochemistry 2013, 52, 9246–9256. [Google Scholar] [CrossRef]
- Rostovtseva, T.K.; Bezrukov, S.M. VDAC regulation: Role of cytosolic proteins and mitochondrial lipids. J. Bioenerg. Biomembr. 2008, 40, 163–170. [Google Scholar] [CrossRef] [Green Version]
- Rostovtseva, T.K.; Sheldon, K.L.; Hassanzadeh, E.; Monge, C.; Saks, V.; Bezrukov, S.M.; Sackett, D.L. Tubulin binding blocks mitochondrial voltage-dependent anion channel and regulates respiration. Proc. Natl. Acad. Sci. USA 2008, 105, 18746–18751. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Schaedel, L.; Portran, D.; Aguilar, A.; Gaillard, J.; Marinkovich, M.P.; Thery, M.; Nachury, M.V. Microtubules acquire resistance from mechanical breakage through intralumenal acetylation. Science 2017, 356, 328–332. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.M.; Giltnane, J.M.; Balko, J.M.; Schwarz, L.J.; Guerrero-Zotano, A.L.; Hutchinson, K.E.; Nixon, M.J.; Estrada, M.V.; Sanchez, V.; Sanders, M.E.; et al. MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metab. 2017, 26, 633–647.e7. [Google Scholar] [CrossRef] [Green Version]
- Samardzija, C.; Greening, D.W.; Escalona, R.; Chen, M.; Bilandzic, M.; Luwor, R.; Kannourakis, G.; Findlay, J.K.; Ahmed, N. Knockdown of stem cell regulator Oct4A in ovarian cancer reveals cellular reprogramming associated with key regulators of cytoskeleton-extracellular matrix remodelling. Sci. Rep. 2017, 7, 46312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashida, T.; Jinno, H.; Kitagawa, Y.; Kitajima, M. Cooperation of cancer stem cell properties and epithelial-mesenchymal transition in the establishment of breast cancer metastasis. J. Oncol. 2011, 2011, 591427. [Google Scholar] [CrossRef] [Green Version]
- Pasonen-Seppanen, S.; Hyttinen, J.M.; Rilla, K.; Jokela, T.; Noble, P.W.; Tammi, M.; Tammi, R. Role of CD44 in the organization of keratinocyte pericellular hyaluronan. Histochem Cell Biol. 2012, 137, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Eberlein, C.; Rooney, C.; Ross, S.J.; Farren, M.; Weir, H.M.; Barry, S.T. E-Cadherin and EpCAM expression by NSCLC tumour cells associate with normal fibroblast activation through a pathway initiated by integrin alphavbeta6 and maintained through TGFbeta signalling. Oncogene 2015, 34, 704–716. [Google Scholar] [CrossRef]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, L.; Werner, S.; Pantel, K. Biology and clinical relevance of EpCAM. Cell Stress 2019, 3, 165–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam-Cha, S.H.; Serrano-Vargas, R.; Escario, E.; Azana, J.M.; Calero-Oliver, R.; Martin, A.G.; Poblet, E. CD133 expression in normal skin and in epithelial cutaneous tumors. Biomed. Res. Int. 2013, 2013, 385604. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Shi, S.; Yen, Y.; Brown, J.; Ta, J.Q.; Le, A.D. A subpopulation of CD133(+) cancer stem-like cells characterized in human oral squamous cell carcinoma confer resistance to chemotherapy. Cancer Lett. 2010, 289, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Auvinen, P.; Tammi, R.; Tammi, M.; Johansson, R.; Kosma, V.M. Expression of CD 44 s, CD 44 v 3 and CD 44 v 6 in benign and malignant breast lesions: Correlation and colocalization with hyaluronan. Histopathology 2005, 47, 420–428. [Google Scholar] [CrossRef]
- Marcato, P.; Dean, C.A.; Giacomantonio, C.A.; Lee, P.W. Aldehyde dehydrogenase: Its role as a cancer stem cell marker comes down to the specific isoform. Cell Cycle 2011, 10, 1378–1384. [Google Scholar] [CrossRef]
- Tanei, T.; Morimoto, K.; Shimazu, K.; Kim, S.J.; Tanji, Y.; Taguchi, T.; Tamaki, Y.; Noguchi, S. Association of breast cancer stem cells identified by aldehyde dehydrogenase 1 expression with resistance to sequential Paclitaxel and epirubicin-based chemotherapy for breast cancers. Clin. Cancer Res. 2009, 15, 4234–4241. [Google Scholar] [CrossRef] [Green Version]
- Tomita, H.; Tanaka, K.; Tanaka, T.; Hara, A. Aldehyde dehydrogenase 1A1 in stem cells and cancer. Oncotarget 2016, 7, 11018–11032. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.H.; Lee, S.H.; Hong, D.; Lee, J.S.; Ahn, H.S.; Ahn, J.H.; Seong, T.W.; Lee, C.H.; Jang, H.; Hong, K.M.; et al. Aldehyde dehydrogenase is used by cancer cells for energy metabolism. Exp. Mol. Med. 2016, 48, e272. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Shim, J.K.; Kang, J.H.; Choi, J.; Chang, J.H.; Kim, S.Y.; Kang, S.G. Regulation of bioenergetics through dual inhibition of aldehyde dehydrogenase and mitochondrial complex I suppresses glioblastoma tumorspheres. Neuro. Oncol. 2018, 20, 954–965. [Google Scholar] [CrossRef] [Green Version]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 10190–10195. [Google Scholar] [CrossRef] [Green Version]
- Janiszewska, M.; Suva, M.L.; Riggi, N.; Houtkooper, R.H.; Auwerx, J.; Clement-Schatlo, V.; Radovanovic, I.; Rheinbay, E.; Provero, P.; Stamenkovic, I. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. 2012, 26, 1926–1944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasto, A.; Bellio, C.; Pilotto, G.; Ciminale, V.; Silic-Benussi, M.; Guzzo, G.; Rasola, A.; Frasson, C.; Nardo, G.; Zulato, E.; et al. Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget 2014, 5, 4305–4319. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.A.; Lin, C.H.; Chi, W.H.; Wang, C.Y.; Hsieh, Y.T.; Wei, Y.H.; Chen, Y.J. Resveratrol Impedes the Stemness, Epithelial-Mesenchymal Transition, and Metabolic Reprogramming of Cancer Stem Cells in Nasopharyngeal Carcinoma through p53 Activation. Evid. Based Complement. Alternat. Med. 2013, 2013, 590393. [Google Scholar] [CrossRef]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef] [Green Version]
- De Luca, A.; Fiorillo, M.; Peiris-Pages, M.; Ozsvari, B.; Smith, D.L.; Sanchez-Alvarez, R.; Martinez-Outschoorn, U.E.; Cappello, A.R.; Pezzi, V.; Lisanti, M.P.; et al. Mitochondrial biogenesis is required for the anchorage-independent survival and propagation of stem-like cancer cells. Oncotarget 2015, 6, 14777–14795. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.Q.; Li, Q.; Wang, G.H.; Sun, F.F.; Huang, G.J.; Bian, X.W.; Yu, S.C.; Qian, G.S. Mitochondrial and energy metabolism-related properties as novel indicators of lung cancer stem cells. Int. J. Cancer 2011, 129, 820–831. [Google Scholar] [CrossRef]
- Farnie, G.; Sotgia, F.; Lisanti, M.P. High mitochondrial mass identifies a sub-population of stem-like cancer cells that are chemo-resistant. Oncotarget 2015, 6, 30472–30486. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Yang, J.; Ku, M.; Kim, N.H.; Park, Y.; Park, C.B.; Suh, J.S.; Park, E.S.; Yook, J.I.; Mills, G.B.; et al. Metabolic stress induces a Wnt-dependent cancer stem cell-like state transition. Cell Death Dis. 2015, 6, e1805. [Google Scholar] [CrossRef] [Green Version]
- Lue, H.W.; Podolak, J.; Kolahi, K.; Cheng, L.; Rao, S.; Garg, D.; Xue, C.H.; Rantala, J.K.; Tyner, J.W.; Thornburg, K.L.; et al. Metabolic reprogramming ensures cancer cell survival despite oncogenic signaling blockade. Genes Dev. 2017, 31, 2067–2084. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Araujo, E.P.; Carvalheira, J.B.; Velloso, L.A. Disruption of metabolic pathways--perspectives for the treatment of cancer. Curr. Cancer Drug Targets 2006, 6, 77–87. [Google Scholar] [CrossRef]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s Achilles’ heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef]
- Obre, E.; Rossignol, R. Emerging concepts in bioenergetics and cancer research: Metabolic flexibility, coupling, symbiosis, switch, oxidative tumors, metabolic remodeling, signaling and bioenergetic therapy. Int. J. Biochem. Cell Biol. 2015, 59, 167–181. [Google Scholar] [CrossRef]
- Kim, S.Y. Targeting cancer energy metabolism: A potential systemic cure for cancer. Arch. Pharm. Res. 2019, 42, 140–149. [Google Scholar] [CrossRef]
- Vazquez, F.; Lim, J.H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.B.; Granter, S.R.; Widlund, H.R.; Spiegelman, B.M.; et al. PGC1alpha expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 2013, 23, 287–301. [Google Scholar] [CrossRef] [Green Version]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Bou Kheir, T.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Grana, O.; et al. MYC/PGC-1alpha Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef] [Green Version]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Matsutani, T.; Masui, K.; Poulou, M.; Popescu, R.; Della Donna, L.; Evers, P.; Dekmezian, C.; et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16062–16067. [Google Scholar] [CrossRef] [Green Version]
- Morandi, A.; Indraccolo, S. Linking metabolic reprogramming to therapy resistance in cancer. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 1–6. [Google Scholar] [CrossRef]
- Zhou, Y.; Tozzi, F.; Chen, J.; Fan, F.; Xia, L.; Wang, J.; Gao, G.; Zhang, A.; Xia, X.; Brasher, H.; et al. Intracellular ATP levels are a pivotal determinant of chemoresistance in colon cancer cells. Cancer Res. 2012, 72, 304–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vellinga, T.T.; Borovski, T.; de Boer, V.C.; Fatrai, S.; van Schelven, S.; Trumpi, K.; Verheem, A.; Snoeren, N.; Emmink, B.L.; Koster, J.; et al. SIRT1/PGC1alpha-Dependent Increase in Oxidative Phosphorylation Supports Chemotherapy Resistance of Colon Cancer. Clin. Cancer Res. 2015, 21, 2870–2879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.J.; Jhe, Y.L.; Kim, J.; Lim, J.Y.; Lee, J.E.; Shin, M.K.; Cheong, J.H. FoxM1-dependent and fatty acid oxidation-mediated ROS modulation is a cell-intrinsic drug resistance mechanism in cancer stem-like cells. Redox Biol. 2020, 36, 101589. [Google Scholar] [CrossRef] [PubMed]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef]
- Reid, R.A.; Moyle, J.; Mitchell, P. Synthesis of adenosine triphosphate by a protonmotive force in rat liver mitochondria. Nature 1966, 212, 257–258. [Google Scholar] [CrossRef]
- Mitchell, P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 1961, 191, 144–148. [Google Scholar] [CrossRef]
- Greenhouse, W.V.; Lehninger, A.L. Magnitude of malate-aspartate reduced nicotinamide adenine dinucleotide shuttle activity in intact respiring tumor cells. Cancer Res. 1977, 37, 4173–4181. [Google Scholar]
- Dionisi, O.; Longhi, G.; Eboli, M.L.; Galeotti, T.; Terranova, T. Oxidation of reduced nicotinamide-adenine dinucleotide by the malate-aspartate shuttle in Ehrlich ascites tumour cells. Biochim. Biophys. Acta 1974, 333, 577–580. [Google Scholar] [CrossRef]
- Piel, S.; Ehinger, J.K.; Chamkha, I.; Frostner, E.A.; Sjovall, F.; Elmer, E.; Hansson, M.J. Bioenergetic bypass using cell-permeable succinate, but not methylene blue, attenuates metformin-induced lactate production. Intensive Care Med. Exp. 2018, 6, 22. [Google Scholar] [CrossRef]
- Frederick, R.L.; Shaw, J.M. Moving mitochondria: Establishing distribution of an essential organelle. Traffic 2007, 8, 1668–1675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, F.; Hamann, A.; Osiewacz, H.D. Mitochondrial quality control: An integrated network of pathways. Trends Biochem. Sci. 2012, 37, 284–292. [Google Scholar] [CrossRef]
- Westermann, B. Merging mitochondria matters: Cellular role and molecular machinery of mitochondrial fusion. EMBO Rep. 2002, 3, 527–531. [Google Scholar] [CrossRef] [Green Version]
- Hollenbeck, P.J.; Saxton, W.M. The axonal transport of mitochondria. J. Cell Sci. 2005, 118, 5411–5419. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, K.; Shaw, J.M. Mitochondrial morphology and dynamics in yeast and multicellular eukaryotes. Annu. Rev. Genet. 2005, 39, 503–536. [Google Scholar] [CrossRef]
- Eura, Y.; Ishihara, N.; Yokota, S.; Mihara, K. Two mitofusin proteins, mammalian homologues of FZO, with distinct functions are both required for mitochondrial fusion. J. Biochem. 2003, 134, 333–344. [Google Scholar] [CrossRef]
- Qi, Y.; Yan, L.; Yu, C.; Guo, X.; Zhou, X.; Hu, X.; Huang, X.; Rao, Z.; Lou, Z.; Hu, J. Structures of human mitofusin 1 provide insight into mitochondrial tethering. J. Cell Biol. 2016, 215, 621–629. [Google Scholar] [CrossRef] [Green Version]
- Santin, G.; Piccolini, V.M.; Barni, S.; Veneroni, P.; Giansanti, V.; Dal Bo, V.; Bernocchi, G.; Bottone, M.G. Mitochondrial fusion: A mechanism of cisplatin-induced resistance in neuroblastoma cells? Neurotoxicology 2013, 34, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Cipolat, S.; Martins de Brito, O.; Dal Zilio, B.; Scorrano, L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef] [Green Version]
- Otera, H.; Ishihara, N.; Mihara, K. New insights into the function and regulation of mitochondrial fission. Biochim. Biophys. Acta 2013, 1833, 1256–1268. [Google Scholar] [CrossRef] [Green Version]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archer, S.L. Mitochondrial dynamics--mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 2013, 369, 2236–2251. [Google Scholar] [CrossRef] [Green Version]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef] [Green Version]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, Y.; Krueger, E.W.; Oswald, B.J.; McNiven, M.A. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol. Cell Biol. 2003, 23, 5409–5420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, D.; Xu, L.; Yu, Y.; Zhu, W.; Andrews, D.W.; Yoon, Y.; Kuo, T.H. Regulation of Ca2+-induced permeability transition by Bcl-2 is antagonized by Drpl and hFis1. Mol. Cell Biochem. 2005, 272, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Cereghetti, G.M.; Costa, V.; Scorrano, L. Inhibition of Drp1-dependent mitochondrial fragmentation and apoptosis by a polypeptide antagonist of calcineurin. Cell Death Differ. 2010, 17, 1785–1794. [Google Scholar] [CrossRef] [Green Version]
- Olichon, A.; Baricault, L.; Gas, N.; Guillou, E.; Valette, A.; Belenguer, P.; Lenaers, G. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J. Biol. Chem. 2003, 278, 7743–7746. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Chan, D.C. Emerging functions of mammalian mitochondrial fusion and fission. Hum. Mol. Genet. 2005, 14, R283–R289. [Google Scholar] [CrossRef]
- Chen, H.; Chomyn, A.; Chan, D.C. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem. 2005, 280, 26185–26192. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Chan, D.C. Mitochondrial dynamics--fusion, fission, movement, and mitophagy--in neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef]
- Zorzano, A.; Liesa, M.; Sebastian, D.; Segales, J.; Palacin, M. Mitochondrial fusion proteins: Dual regulators of morphology and metabolism. Semin. Cell Dev. Biol. 2010, 21, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, T.P.; Bjorklund, M. Mitochondrial Function and Cell Size: An Allometric Relationship. Trends Cell Biol. 2017, 27, 393–402. [Google Scholar] [CrossRef]
- Scorrano, L.; Ashiya, M.; Buttle, K.; Weiler, S.; Oakes, S.A.; Mannella, C.A.; Korsmeyer, S.J. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev. Cell 2002, 2, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Roucou, X.; Montessuit, S.; Antonsson, B.; Martinou, J.C. Bax oligomerization in mitochondrial membranes requires tBid (caspase-8-cleaved Bid) and a mitochondrial protein. Biochem. J. 2002, 368, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Boutant, M.; Kulkarni, S.S.; Joffraud, M.; Ratajczak, J.; Valera-Alberni, M.; Combe, R.; Zorzano, A.; Canto, C. Mfn2 is critical for brown adipose tissue thermogenic function. EMBO J. 2017, 36, 1543–1558. [Google Scholar] [CrossRef] [Green Version]
- Rambold, A.S.; Cohen, S.; Lippincott-Schwartz, J. Fatty acid trafficking in starved cells: Regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev. Cell 2015, 32, 678–692. [Google Scholar] [CrossRef] [Green Version]
- Shiratori, R.; Furuichi, K.; Yamaguchi, M.; Miyazaki, N.; Aoki, H.; Chibana, H.; Ito, K.; Aoki, S. Glycolytic suppression dramatically changes the intracellular metabolic profile of multiple cancer cell lines in a mitochondrial metabolism-dependent manner. Sci. Rep. 2019, 9, 18699. [Google Scholar] [CrossRef] [Green Version]
- Buck, M.D.; O’Sullivan, D.; Klein Geltink, R.I.; Curtis, J.D.; Chang, C.H.; Sanin, D.E.; Qiu, J.; Kretz, O.; Braas, D.; van der Windt, G.J.; et al. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell 2016, 166, 63–76. [Google Scholar] [CrossRef] [Green Version]
- Serasinghe, M.N.; Wieder, S.Y.; Renault, T.T.; Elkholi, R.; Asciolla, J.J.; Yao, J.L.; Jabado, O.; Hoehn, K.; Kageyama, Y.; Sesaki, H.; et al. Mitochondrial division is requisite to RAS-induced transformation and targeted by oncogenic MAPK pathway inhibitors. Mol. Cell 2015, 57, 521–536. [Google Scholar] [CrossRef] [Green Version]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef]
- Graves, J.A.; Wang, Y.; Sims-Lucas, S.; Cherok, E.; Rothermund, K.; Branca, M.F.; Elster, J.; Beer-Stolz, D.; Van Houten, B.; Vockley, J.; et al. Mitochondrial structure, function and dynamics are temporally controlled by c-Myc. PLoS ONE 2012, 7, e37699. [Google Scholar] [CrossRef] [Green Version]
- Kashatus, J.A.; Nascimento, A.; Myers, L.J.; Sher, A.; Byrne, F.L.; Hoehn, K.L.; Counter, C.M.; Kashatus, D.F. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol. Cell 2015, 57, 537–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, J.; Wang, J.; Huang, Y.; Wu, H.; Xia, T.; Xiao, J.; Chen, X.; Li, H.; Qiu, Y.; Wang, Y.; et al. ERK/Drp1-dependent mitochondrial fission is involved in the MSC-induced drug resistance of T-cell acute lymphoblastic leukemia cells. Cell Death Dis. 2016, 7, e2459. [Google Scholar] [CrossRef] [PubMed]
- Loiseau, D.; Chevrollier, A.; Verny, C.; Guillet, V.; Gueguen, N.; Pou de Crescenzo, M.A.; Ferre, M.; Malinge, M.C.; Guichet, A.; Nicolas, G.; et al. Mitochondrial coupling defect in Charcot-Marie-Tooth type 2A disease. Ann. Neurol. 2007, 61, 315–323. [Google Scholar] [CrossRef]
- Picard, M.; Shirihai, O.S.; Gentil, B.J.; Burelle, Y. Mitochondrial morphology transitions and functions: Implications for retrograde signaling? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, R393–R406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaseva, A.V.; Blake, D.R.; Gilbert, T.S.K.; Ng, S.; Hostetter, G.; Azam, S.H.; Ozkan-Dagliyan, I.; Gautam, P.; Bryant, K.L.; Pearce, K.H.; et al. KRAS Suppression-Induced Degradation of MYC Is Antagonized by a MEK5-ERK5 Compensatory Mechanism. Cancer Cell 2018, 34, 807–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, M.K.; Cheong, J.H. Mitochondria-centric bioenergetic characteristics in cancer stem-like cells. Arch. Pharm. Res. 2019, 42, 113–127. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Jiang, Y.; Meisenhelder, J.; Yang, W.; Hawke, D.H.; Zheng, Y.; Xia, Y.; Aldape, K.; He, J.; Hunter, T.; et al. Mitochondria-Translocated PGK1 Functions as a Protein Kinase to Coordinate Glycolysis and the TCA Cycle in Tumorigenesis. Mol. Cell 2016, 61, 705–719. [Google Scholar] [CrossRef] [Green Version]
- Murphy, T.A.; Dang, C.V.; Young, J.D. Isotopically nonstationary 13C flux analysis of Myc-induced metabolic reprogramming in B-cells. Metab. Eng. 2013, 15, 206–217. [Google Scholar] [CrossRef] [Green Version]
- Morrish, F.; Noonan, J.; Perez-Olsen, C.; Gafken, P.R.; Fitzgibbon, M.; Kelleher, J.; VanGilst, M.; Hockenbery, D. Myc-dependent mitochondrial generation of acetyl-CoA contributes to fatty acid biosynthesis and histone acetylation during cell cycle entry. J. Biol. Chem. 2010, 285, 36267–36274. [Google Scholar] [CrossRef] [Green Version]
- Camarda, R.; Williams, J.; Goga, A. In vivo Reprogramming of Cancer Metabolism by MYC. Front Cell Dev. Biol. 2017, 5, 35. [Google Scholar] [CrossRef] [Green Version]
- Greer, Y.E.; Porat-Shliom, N.; Nagashima, K.; Stuelten, C.; Crooks, D.; Koparde, V.N.; Gilbert, S.F.; Islam, C.; Ubaldini, A.; Ji, Y.; et al. ONC201 kills breast cancer cells in vitro by targeting mitochondria. Oncotarget 2018, 9, 18454–18479. [Google Scholar] [CrossRef] [Green Version]
- Graves, P.R.; Aponte-Collazo, L.J.; Fennell, E.M.J.; Graves, A.C.; Hale, A.E.; Dicheva, N.; Herring, L.E.; Gilbert, T.S.K.; East, M.P.; McDonald, I.M.; et al. Mitochondrial Protease ClpP is a Target for the Anticancer Compounds ONC201 and Related Analogues. ACS Chem. Biol. 2019, 14, 1020–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, A.; Wang, Z.; Coyaud, E.; Voisin, V.; Gronda, M.; Jitkova, Y.; Mattson, R.; Hurren, R.; Babovic, S.; Maclean, N.; et al. Inhibition of the Mitochondrial Protease ClpP as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell 2015, 27, 864–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogales, E. Structural insight into microtubule function. Annu. Rev. Biophys. Biomol. Struct. 2001, 30, 397–420. [Google Scholar] [CrossRef]
- Heald, R.; Nogales, E. Microtubule dynamics. J. Cell Sci. 2002, 115, 3–4. [Google Scholar]
- Fife, C.M.; McCarroll, J.A.; Kavallaris, M. Movers and shakers: Cell cytoskeleton in cancer metastasis. Br. J. Pharm. 2014, 171, 5507–5523. [Google Scholar] [CrossRef] [Green Version]
- Appaix, F.; Kuznetsov, A.V.; Usson, Y.; Kay, L.; Andrienko, T.; Olivares, J.; Kaambre, T.; Sikk, P.; Margreiter, R.; Saks, V. Possible role of cytoskeleton in intracellular arrangement and regulation of mitochondria. Exp. Physiol. 2003, 88, 175–190. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsov, A.V.; Tiivel, T.; Sikk, P.; Kaambre, T.; Kay, L.; Daneshrad, Z.; Rossi, A.; Kadaja, L.; Peet, N.; Seppet, E.; et al. Striking differences between the kinetics of regulation of respiration by ADP in slow-twitch and fast-twitch muscles in vivo. Eur. J. Biochem. 1996, 241, 909–915. [Google Scholar] [CrossRef]
- Rao, J.; Li, N. Microfilament actin remodeling as a potential target for cancer drug development. Curr. Cancer Drug Targets 2004, 4, 345–354. [Google Scholar] [CrossRef]
- Pollard, T.D.; Blanchoin, L.; Mullins, R.D. Actin dynamics. J. Cell Sci. 2001, 114, 3–4. [Google Scholar] [PubMed]
- Pollard, T.D.; Blanchoin, L.; Mullins, R.D. Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 545–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korobova, F.; Ramabhadran, V.; Higgs, H.N. An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science 2013, 339, 464–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korobova, F.; Gauvin, T.J.; Higgs, H.N. A role for myosin II in mammalian mitochondrial fission. Curr. Biol. 2014, 24, 409–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manor, U.; Bartholomew, S.; Golani, G.; Christenson, E.; Kozlov, M.; Higgs, H.; Spudich, J.; Lippincott-Schwartz, J. A mitochondria-anchored isoform of the actin-nucleating spire protein regulates mitochondrial division. Elife 2015, 4. [Google Scholar] [CrossRef]
- Langford, G.M. Actin- and microtubule-dependent organelle motors: Interrelationships between the two motility systems. Curr. Opin. Cell Biol. 1995, 7, 82–88. [Google Scholar] [CrossRef]
- Morris, R.L.; Hollenbeck, P.J. Axonal transport of mitochondria along microtubules and F-actin in living vertebrate neurons. J. Cell Biol. 1995, 131, 1315–1326. [Google Scholar] [CrossRef] [Green Version]
- Sheetz, M.P.; Steuer, E.R.; Schroer, T.A. The mechanism and regulation of fast axonal transport. Trends Neurosci. 1989, 12, 474–478. [Google Scholar] [CrossRef]
- Moore, A.S.; Wong, Y.C.; Simpson, C.L.; Holzbaur, E.L. Dynamic actin cycling through mitochondrial subpopulations locally regulates the fission-fusion balance within mitochondrial networks. Nat. Commun. 2016, 7, 12886. [Google Scholar] [CrossRef]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [Green Version]
- Ji, W.K.; Hatch, A.L.; Merrill, R.A.; Strack, S.; Higgs, H.N. Actin filaments target the oligomeric maturation of the dynamin GTPase Drp1 to mitochondrial fission sites. Elife 2015, 4, e11553. [Google Scholar] [CrossRef]
- Kruppa, A.J.; Kishi-Itakura, C.; Masters, T.A.; Rorbach, J.E.; Grice, G.L.; Kendrick-Jones, J.; Nathan, J.A.; Minczuk, M.; Buss, F. Myosin VI-Dependent Actin Cages Encapsulate Parkin-Positive Damaged Mitochondria. Dev. Cell 2018, 44, 484–499.e6. [Google Scholar] [CrossRef]
- Desai, A.; Mitchison, T.J. Microtubule polymerization dynamics. Annu. Rev. Cell Dev. Biol. 1997, 13, 83–117. [Google Scholar] [CrossRef] [Green Version]
- Downing, K.H. Structural basis for the interaction of tubulin with proteins and drugs that affect microtubule dynamics. Annu. Rev. Cell Dev. Biol. 2000, 16, 89–111. [Google Scholar] [CrossRef] [PubMed]
- Straight, A.F.; Field, C.M. Microtubules, membranes and cytokinesis. Curr. Biol. 2000, 10, R760–R770. [Google Scholar] [CrossRef] [Green Version]
- Dompierre, J.P.; Godin, J.D.; Charrin, B.C.; Cordelieres, F.P.; King, S.J.; Humbert, S.; Saudou, F. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington’s disease by increasing tubulin acetylation. J. Neurosci. 2007, 27, 3571–3583. [Google Scholar] [CrossRef] [PubMed]
- Reed, N.A.; Cai, D.; Blasius, T.L.; Jih, G.T.; Meyhofer, E.; Gaertig, J.; Verhey, K.J. Microtubule acetylation promotes kinesin-1 binding and transport. Curr. Biol. 2006, 16, 2166–2172. [Google Scholar] [CrossRef] [Green Version]
- Bulinski, J.C. Microtubule modification: Acetylation speeds anterograde traffic flow. Curr. Biol. 2007, 17, R18–R20. [Google Scholar] [CrossRef] [Green Version]
- Cagalinec, M.; Safiulina, D.; Liiv, M.; Liiv, J.; Choubey, V.; Wareski, P.; Veksler, V.; Kaasik, A. Principles of the mitochondrial fusion and fission cycle in neurons. J. Cell Sci. 2013, 126, 2187–2197. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Weaver, D.; Shirihai, O.; Hajnoczky, G. Mitochondrial ‘kiss-and-run’: Interplay between mitochondrial motility and fusion-fission dynamics. EMBO J. 2009, 28, 3074–3089. [Google Scholar] [CrossRef] [Green Version]
- Martz, D.; Lasek, R.J.; Brady, S.T.; Allen, R.D. Mitochondrial motility in axons: Membranous organelles may interact with the force generating system through multiple surface binding sites. Cell Motil. 1984, 4, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Vallee, R.B.; Bloom, G.S. Mechanisms of fast and slow axonal transport. Annu. Rev. Neurosci. 1991, 14, 59–92. [Google Scholar] [CrossRef] [PubMed]
- Ligon, L.A.; Steward, O. Role of microtubules and actin filaments in the movement of mitochondria in the axons and dendrites of cultured hippocampal neurons. J. Comp. Neurol. 2000, 427, 351–361. [Google Scholar] [CrossRef]
- Saxton, W.M.; Hollenbeck, P.J. The axonal transport of mitochondria. J. Cell Sci. 2012, 125, 2095–2104. [Google Scholar] [CrossRef] [Green Version]
- Schwarzer, C.; Barnikol-Watanabe, S.; Thinnes, F.P.; Hilschmann, N. Voltage-dependent anion-selective channel (VDAC) interacts with the dynein light chain Tctex1 and the heat-shock protein PBP74. Int. J. Biochem. Cell Biol. 2002, 34, 1059–1070. [Google Scholar] [CrossRef]
- Chang, D.T.; Honick, A.S.; Reynolds, I.J. Mitochondrial trafficking to synapses in cultured primary cortical neurons. J. Neurosci. 2006, 26, 7035–7045. [Google Scholar] [CrossRef]
- Rintoul, G.L.; Filiano, A.J.; Brocard, J.B.; Kress, G.J.; Reynolds, I.J. Glutamate decreases mitochondrial size and movement in primary forebrain neurons. J. Neurosci. 2003, 23, 7881–7888. [Google Scholar] [CrossRef] [Green Version]
- Brady, S.T.; Lasek, R.J.; Allen, R.D.; Yin, H.L.; Stossel, T.P. Gelsolin inhibition of fast axonal transport indicates a requirement for actin microfilaments. Nature 1984, 310, 56–58. [Google Scholar] [CrossRef] [PubMed]
- Infante, A.S.; Stein, M.S.; Zhai, Y.; Borisy, G.G.; Gundersen, G.G. Detyrosinated (Glu) microtubules are stabilized by an ATP-sensitive plus-end cap. J. Cell Sci. 2000, 113 (Pt 22), 3907–3919. [Google Scholar]
- Park, K.C.; Kim, S.W.; Jeon, J.Y.; Jo, A.R.; Choi, H.J.; Kim, J.; Lee, H.G.; Kim, Y.; Mills, G.B.; Noh, S.H.; et al. Survival of Cancer Stem-Like Cells Under Metabolic Stress via CaMK2alpha-mediated Upregulation of Sarco/Endoplasmic Reticulum Calcium ATPase Expression. Clin. Cancer Res. 2018, 24, 1677–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roman, I.; Figys, J.; Steurs, G.; Zizi, M. Direct measurement of VDAC-actin interaction by surface plasmon resonance. Biochim. Biophys. Acta 2006, 1758, 479–486. [Google Scholar] [CrossRef] [Green Version]
- DeLuca, M.; Hall, N.; Rice, R.; Kaplan, N.O. Creatine kinase isozymes in human tumors. Biochem. Biophys. Res. Commun. 1981, 99, 189–195. [Google Scholar] [CrossRef]
- Dang, C.V.; Semenza, G.L. Oncogenic alterations of metabolism. Trends Biochem. Sci. 1999, 24, 68–72. [Google Scholar] [CrossRef]
- Guzun, R.; Timohhina, N.; Tepp, K.; Monge, C.; Kaambre, T.; Sikk, P.; Kuznetsov, A.V.; Pison, C.; Saks, V. Regulation of respiration controlled by mitochondrial creatine kinase in permeabilized cardiac cells in situ. Importance of system level properties. Biochim. Biophys. Acta 2009, 1787, 1089–1105. [Google Scholar] [CrossRef]
- Heggeness, M.H.; Simon, M.; Singer, S.J. Association of mitochondria with microtubules in cultured cells. Proc. Natl. Acad. Sci. USA 1978, 75, 3863–3866. [Google Scholar] [CrossRef] [Green Version]
- Saetersdal, T.; Greve, G.; Dalen, H. Associations between beta-tubulin and mitochondria in adult isolated heart myocytes as shown by immunofluorescence and immunoelectron microscopy. Histochemistry 1990, 95, 1–10. [Google Scholar] [CrossRef]
- Bernier-Valentin, F.; Aunis, D.; Rousset, B. Evidence for tubulin-binding sites on cellular membranes: Plasma membranes, mitochondrial membranes, and secretory granule membranes. J. Cell Biol. 1983, 97, 209–216. [Google Scholar] [CrossRef]
- Carre, M.; Andre, N.; Carles, G.; Borghi, H.; Brichese, L.; Briand, C.; Braguer, D. Tubulin is an inherent component of mitochondrial membranes that interacts with the voltage-dependent anion channel. J. Biol. Chem. 2002, 277, 33664–33669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cicchillitti, L.; Penci, R.; Di Michele, M.; Filippetti, F.; Rotilio, D.; Donati, M.B.; Scambia, G.; Ferlini, C. Proteomic characterization of cytoskeletal and mitochondrial class III beta-tubulin. Mol. Cancer 2008, 7, 2070–2079. [Google Scholar] [CrossRef] [Green Version]
- Magri, A.; Reina, S.; De Pinto, V. VDAC1 as Pharmacological Target in Cancer and Neurodegeneration: Focus on Its Role in Apoptosis. Front. Chem. 2018, 6, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chae, Y.C.; Angelin, A.; Lisanti, S.; Kossenkov, A.V.; Speicher, K.D.; Wang, H.; Powers, J.F.; Tischler, A.S.; Pacak, K.; Fliedner, S.; et al. Landscape of the mitochondrial Hsp90 metabolome in tumours. Nat. Commun. 2013, 4, 2139. [Google Scholar] [CrossRef] [Green Version]
- Maldonado, E.N.; Sheldon, K.L.; DeHart, D.N.; Patnaik, J.; Manevich, Y.; Townsend, D.M.; Bezrukov, S.M.; Rostovtseva, T.K.; Lemasters, J.J. Voltage-dependent anion channels modulate mitochondrial metabolism in cancer cells: Regulation by free tubulin and erastin. J. Biol. Chem. 2013, 288, 11920–11929. [Google Scholar] [CrossRef] [Green Version]
- Gourlay, C.W.; Carpp, L.N.; Timpson, P.; Winder, S.J.; Ayscough, K.R. A role for the actin cytoskeleton in cell death and aging in yeast. J. Cell Biol. 2004, 164, 803–809. [Google Scholar] [CrossRef] [Green Version]
- Drubin, D.G.; Jones, H.D.; Wertman, K.F. Actin structure and function: Roles in mitochondrial organization and morphogenesis in budding yeast and identification of the phalloidin-binding site. Mol. Biol. Cell 1993, 4, 1277–1294. [Google Scholar] [CrossRef]
- Li, J.; Li, Q.; Xie, C.; Zhou, H.; Wang, Y.; Zhang, N.; Shao, H.; Chan, S.C.; Peng, X.; Lin, S.C.; et al. Beta-actin is required for mitochondria clustering and ROS generation in TNF-induced, caspase-independent cell death. J. Cell Sci. 2004, 117, 4673–4680. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Juvekar, A.; Lyssiotis, C.A.; Lien, E.C.; Albeck, J.G.; Oh, D.; Varma, G.; Hung, Y.P.; Ullas, S.; Lauring, J.; et al. Phosphoinositide 3-Kinase Regulates Glycolysis through Mobilization of Aldolase from the Actin Cytoskeleton. Cell 2016, 164, 433–446. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.S.; Tian, J.H.; Pan, P.Y.; Zald, P.; Li, C.; Deng, C.; Sheng, Z.H. Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell 2008, 132, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Caino, M.C.; Seo, J.H.; Wang, Y.; Rivadeneira, D.B.; Gabrilovich, D.I.; Kim, E.T.; Weeraratna, A.T.; Languino, L.R.; Altieri, D.C. Syntaphilin controls a mitochondrial rheostat for proliferation-motility decisions in cancer. J. Clin. Invest. 2017, 127, 3755–3769. [Google Scholar] [CrossRef] [Green Version]
- Pongrakhananon, V.; Wattanathamsan, O.; Takeichi, M.; Chetprayoon, P.; Chanvorachote, P. Loss of CAMSAP3 promotes EMT via the modification of microtubule-Akt machinery. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [Green Version]
- Ketschek, A.; Gallo, G. Nerve growth factor induces axonal filopodia through localized microdomains of phosphoinositide 3-kinase activity that drive the formation of cytoskeletal precursors to filopodia. J. Neurosci. 2010, 30, 12185–12197. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, J.; Yu, M.; Xie, Y.; Huang, Y.; Wolff, D.W.; Abel, P.W.; Tu, Y. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 2013, 32, 4814–4824. [Google Scholar] [CrossRef] [PubMed]
- Shankar, J.; Nabi, I.R. Actin cytoskeleton regulation of epithelial mesenchymal transition in metastatic cancer cells. PLoS ONE 2015, 10, e0119954. [Google Scholar] [CrossRef] [Green Version]
- Tojkander, S.; Gateva, G.; Lappalainen, P. Actin stress fibers--assembly, dynamics and biological roles. J. Cell Sci. 2012, 125, 1855–1864. [Google Scholar] [CrossRef] [Green Version]
- Taulet, N.; Delorme-Walker, V.D.; DerMardirossian, C. Reactive oxygen species regulate protrusion efficiency by controlling actin dynamics. PLoS ONE 2012, 7, e41342. [Google Scholar] [CrossRef]
- Schuler, M.H.; Lewandowska, A.; Caprio, G.D.; Skillern, W.; Upadhyayula, S.; Kirchhausen, T.; Shaw, J.M.; Cunniff, B. Miro1-mediated mitochondrial positioning shapes intracellular energy gradients required for cell migration. Mol. Biol. Cell 2017, 28, 2159–2169. [Google Scholar] [CrossRef]
- Caino, M.C.; Ghosh, J.C.; Chae, Y.C.; Vaira, V.; Rivadeneira, D.B.; Faversani, A.; Rampini, P.; Kossenkov, A.V.; Aird, K.M.; Zhang, R.; et al. PI3K therapy reprograms mitochondrial trafficking to fuel tumor cell invasion. Proc. Natl. Acad. Sci. USA 2015, 112, 8638–8643. [Google Scholar] [CrossRef] [Green Version]
- Caino, M.C.; Chae, Y.C.; Vaira, V.; Ferrero, S.; Nosotti, M.; Martin, N.M.; Weeraratna, A.; O’Connell, M.; Jernigan, D.; Fatatis, A.; et al. Metabolic stress regulates cytoskeletal dynamics and metastasis of cancer cells. J. Clin. Invest. 2013, 123, 2907–2920. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Lu, S. Transforming growth factor-beta1-induced epithelial to mesenchymal transition increases mitochondrial content in the A549 non-small cell lung cancer cell line. Mol. Med. Rep. 2015, 11, 417–421. [Google Scholar] [CrossRef]
- Kim, S.Y. Cancer Energy Metabolism: Shutting Power off Cancer Factory. Biomol. Ther. 2018, 26, 39–44. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y. Cancer Metabolism: A Hope for Curing Cancer. Biomol. Ther. 2018, 26, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Ralff, M.D.; Lulla, A.R.; Wagner, J.; El-Deiry, W.S. ONC201: A new treatment option being tested clinically for recurrent glioblastoma. Transl Cancer Res. 2017, 6, S1239–S1243. [Google Scholar] [CrossRef] [PubMed]
- Pich, S.; Bach, D.; Briones, P.; Liesa, M.; Camps, M.; Testar, X.; Palacin, M.; Zorzano, A. The Charcot-Marie-Tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Hum. Mol. Genet. 2005, 14, 1405–1415. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.C.B.; Campos, J.C.; Qvit, N.; Qi, X.; Bozi, L.H.M.; Bechara, L.R.G.; Lima, V.M.; Queliconi, B.B.; Disatnik, M.H.; Dourado, P.M.M.; et al. A selective inhibitor of mitofusin 1-betaIIPKC association improves heart failure outcome in rats. Nat. Commun. 2019, 10, 329. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, E.; Cioffi, M.; Sancho, P.; Sanchez-Ripoll, Y.; Trabulo, S.M.; Dorado, J.; Balic, A.; Hidalgo, M.; Heeschen, C. Metformin targets the metabolic achilles heel of human pancreatic cancer stem cells. PLoS ONE 2013, 8, e76518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife 2014, 3, e02242. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Lee, J.H.; Koh, I.; Shim, J.K.; Park, J.; Jeon, J.Y.; Yun, M.; Kim, S.H.; Yook, J.I.; Kim, E.H.; et al. Inhibiting stemness and invasive properties of glioblastoma tumorsphere by combined treatment with temozolomide and a newly designed biguanide (HL156A). Oncotarget 2016, 7, 65643–65659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, T.G.; Jeong, Y.S.; Kim, S.A.; Ahn, S.G. New metformin derivative HL156A prevents oral cancer progression by inhibiting the insulin-like growth factor/AKT/mammalian target of rapamycin pathways. Cancer Sci. 2018, 109, 699–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koyama, T.; Oike, M.; Ito, Y. Involvement of Rho-kinase and tyrosine kinase in hypotonic stress-induced ATP release in bovine aortic endothelial cells. J. Physiol. 2001, 532, 759–769. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Categories | Compound | Target | Conditions | Clinical Trials | Clinical Trials Identifier/Ref. |
|---|---|---|---|---|---|
| Mitochondrial dynamics and function | βIIPKC inhibitor | MFN1 | Heart failure | Experimental | [156,157] |
| ONC201 | Mitochondrial respiration | Multiple cancers | Several trials in process | [171] | |
| NADH production | Gossypol | Aldehyde dehydrogenase | Multiple cancers | Several trials in process | [158] |
| Microtubule | Paclitaxel | β-Tubulin subunit | Multiple cancers | Several trials in process | [159,160] |
| Colchicine | β-Tubulin subunit | Multiple cancers | Several trials in process | [161,162] | |
| Vinca alkaloid | Tubulin dimer | Multiple cancers | Several trials in process | [163,164] | |
| Actin | Cytochalasin D | Actin monomer | Infertility | Preclinical | NCT03677492 |
| Phalloidin | F-actin | Depression | Preclinical recruiting | NCT04137458 | |
| Y-27632 | Rho-associated kinase | Hippocampal neurons | Preclinical | [165] | |
| ETC | Metformin | Complex I | Multiple cancers | Several trials in progress | NCT03477162 |
| Phenformin | Complex I | Diabetes mellitus, type 2 | Preclinical completed | NCT02475499 | |
| Rotenone | Complex I | Parkinson’s disease | Preclinical enrolling | NCT04287543 | |
| IM156 | Complex I | Advanced solid tumor and lymphoma | Phase I active | NCT03272256 | |
| 3-NPA | Complex II | Parkinson’s disease | Preclinical | [166] | |
| Malonate | Complex II | Parkinson’s disease | Phase I terminated | NCT01476085 | |
| TTFA | Complex II | Cancer | Experimental | [167] | |
| Atpenin A5 | Complex II | Cardiac ischemia-reperfusion injury | Preclinical | [168] | |
| Lonidamine | Complex II | Symptomatic benign prostatic hyperplasia, enlarged prostate | Phase 2, 3 terminated | NCT00237536, NCT00435448 | |
| Antimycin A | Complex III | Lung cancer | Experimental | [169] | |
| Cyanide | Complex IV | Malignant glioma | Phase 2 recruiting | NCT00075387 | |
| Oligomycin | Complex V | Cancer cachexia, transthyretin amyloidosis | Preclinical recruiting | NCT03144128, NCT03328338 | |
| GBOXIN | Complex V | Glioblastoma | Preclinical | [170] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.; Cheong, J.-H. Role of Mitochondria-Cytoskeleton Interactions in the Regulation of Mitochondrial Structure and Function in Cancer Stem Cells. Cells 2020, 9, 1691. https://doi.org/10.3390/cells9071691
Kim J, Cheong J-H. Role of Mitochondria-Cytoskeleton Interactions in the Regulation of Mitochondrial Structure and Function in Cancer Stem Cells. Cells. 2020; 9(7):1691. https://doi.org/10.3390/cells9071691
Chicago/Turabian StyleKim, Jungmin, and Jae-Ho Cheong. 2020. "Role of Mitochondria-Cytoskeleton Interactions in the Regulation of Mitochondrial Structure and Function in Cancer Stem Cells" Cells 9, no. 7: 1691. https://doi.org/10.3390/cells9071691
APA StyleKim, J., & Cheong, J.-H. (2020). Role of Mitochondria-Cytoskeleton Interactions in the Regulation of Mitochondrial Structure and Function in Cancer Stem Cells. Cells, 9(7), 1691. https://doi.org/10.3390/cells9071691