Cannabidiol Promotes Endothelial Cell Survival by Heme Oxygenase-1-Mediated Autophagy

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Cell Viability Analysis
2.4. Quantitative RT-PCR Analysis
2.5. Western Blot Analysis
2.6. siRNA Transfection
2.7. Statistics
3. Results
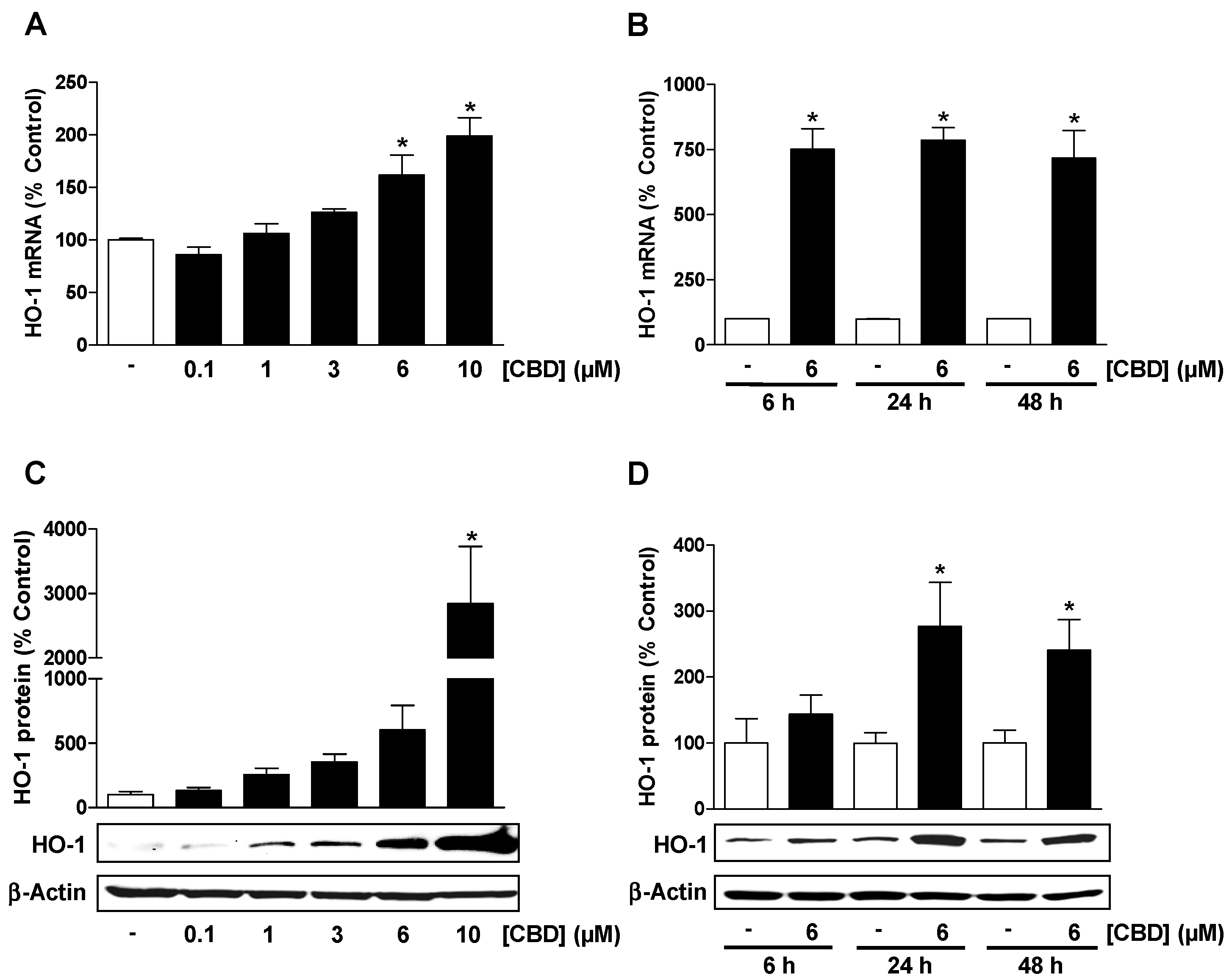
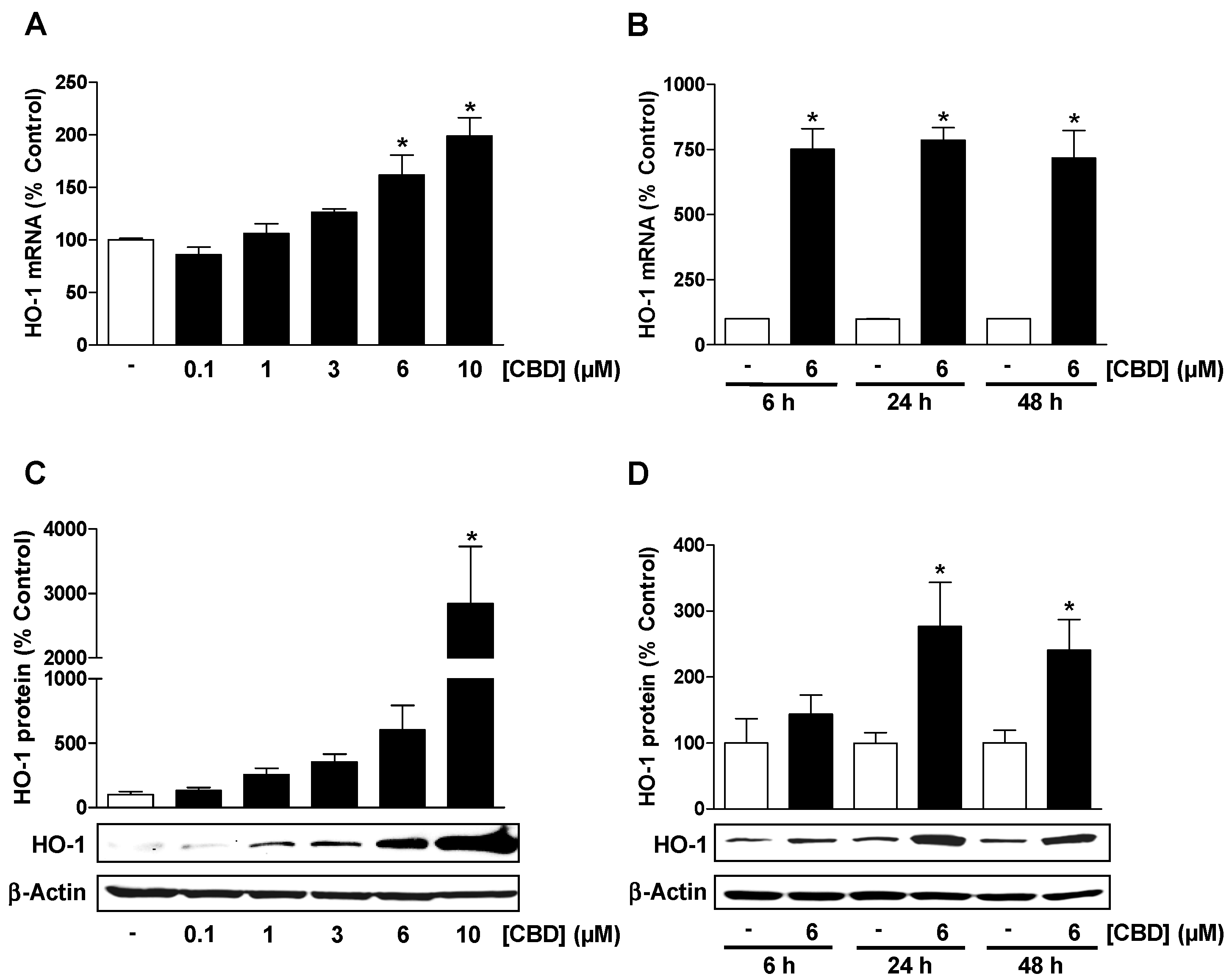
3.1. CBD Causes a Concentration- and Time-Dependent Induction of HO-1 Expression in HUVEC
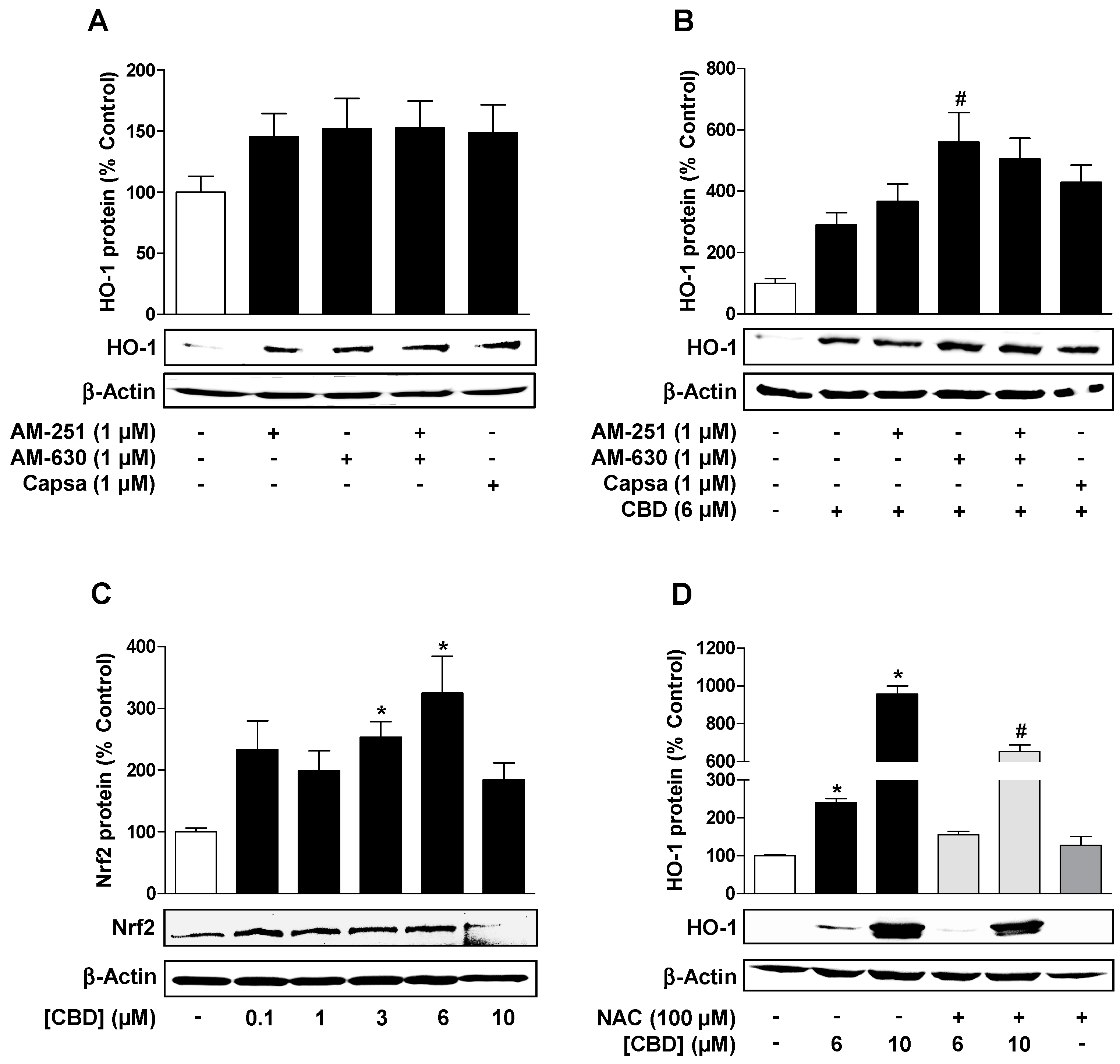
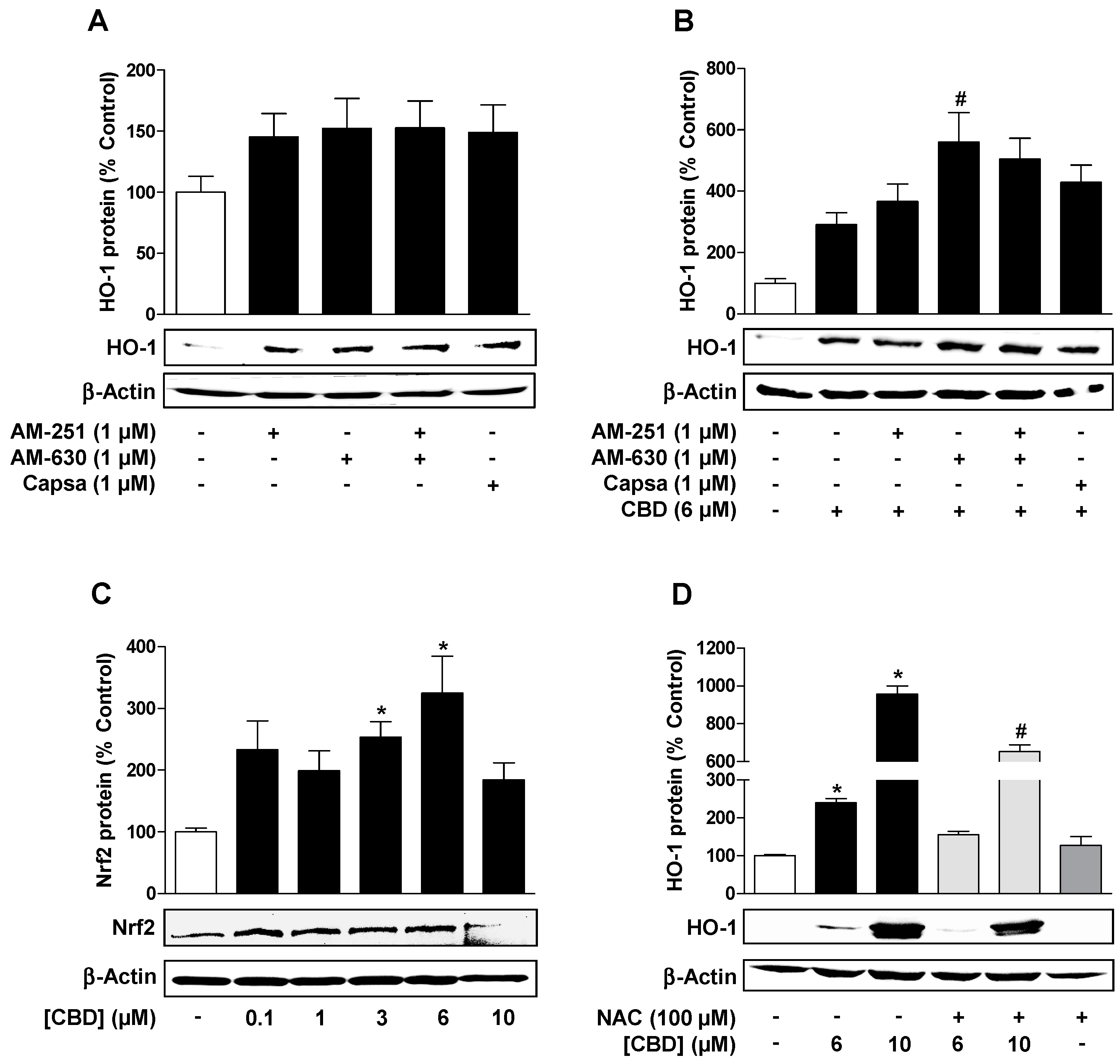
3.2. Reactive Oxygen Species but not Cannabinoid-Activated Receptors Mediate CBD-Induced HO-1 Expression in HUVEC
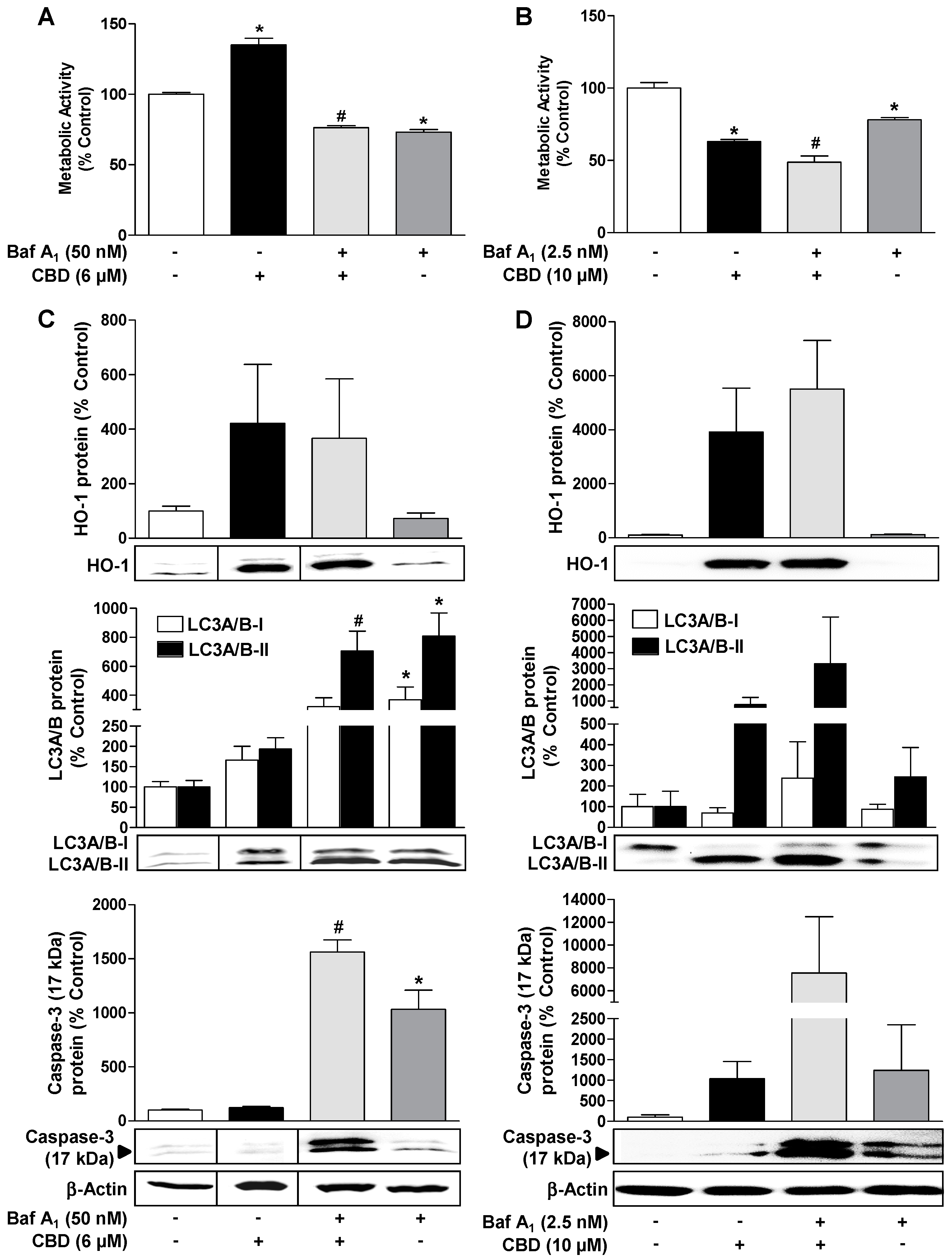
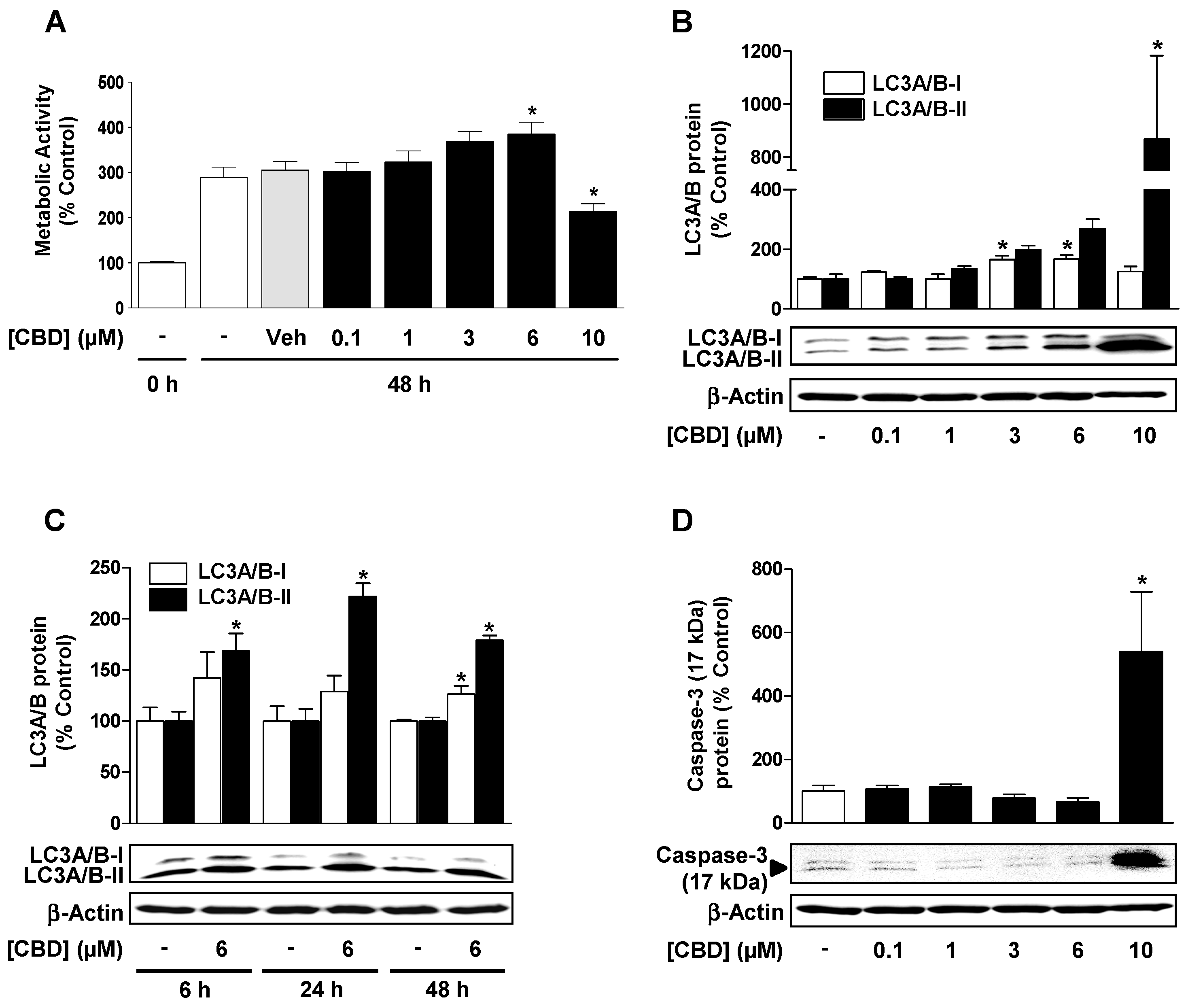
3.3. CBD Induces a Concentration-Dependent Increase in Cellular Autophagy, but Regulates Metabolic Activity and Apoptosis Differently Depending on the Concentration
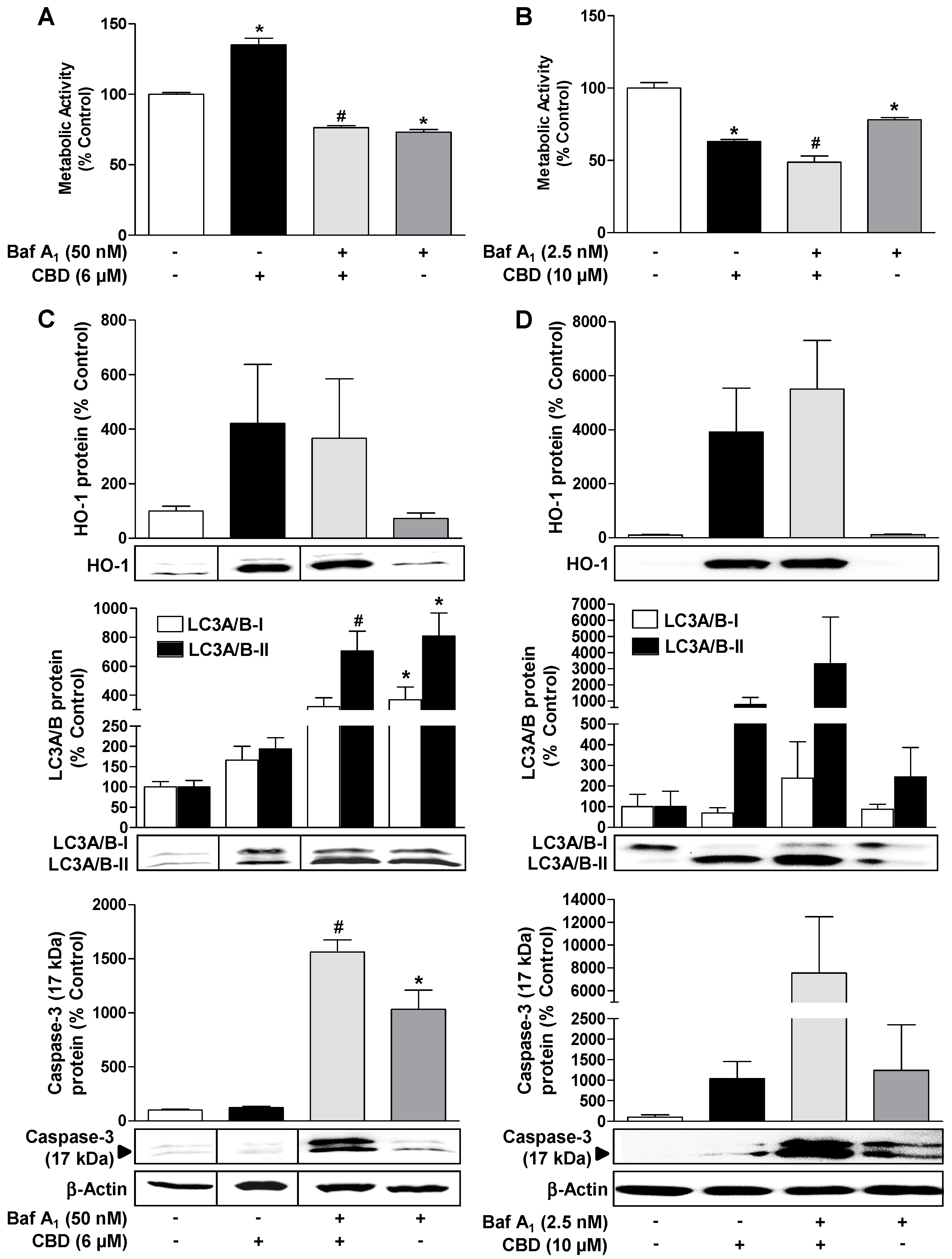
3.4. ROS Mediate the CBD (10 µM) Induced Reduction of Metabolic Activity as well as the Increase of Autophagy and Induction of Apoptosis in HUVEC
3.5. Inhibition of CBD-Induced Autophagy Leads to Increased Apoptosis and Loss of Viability of HUVEC
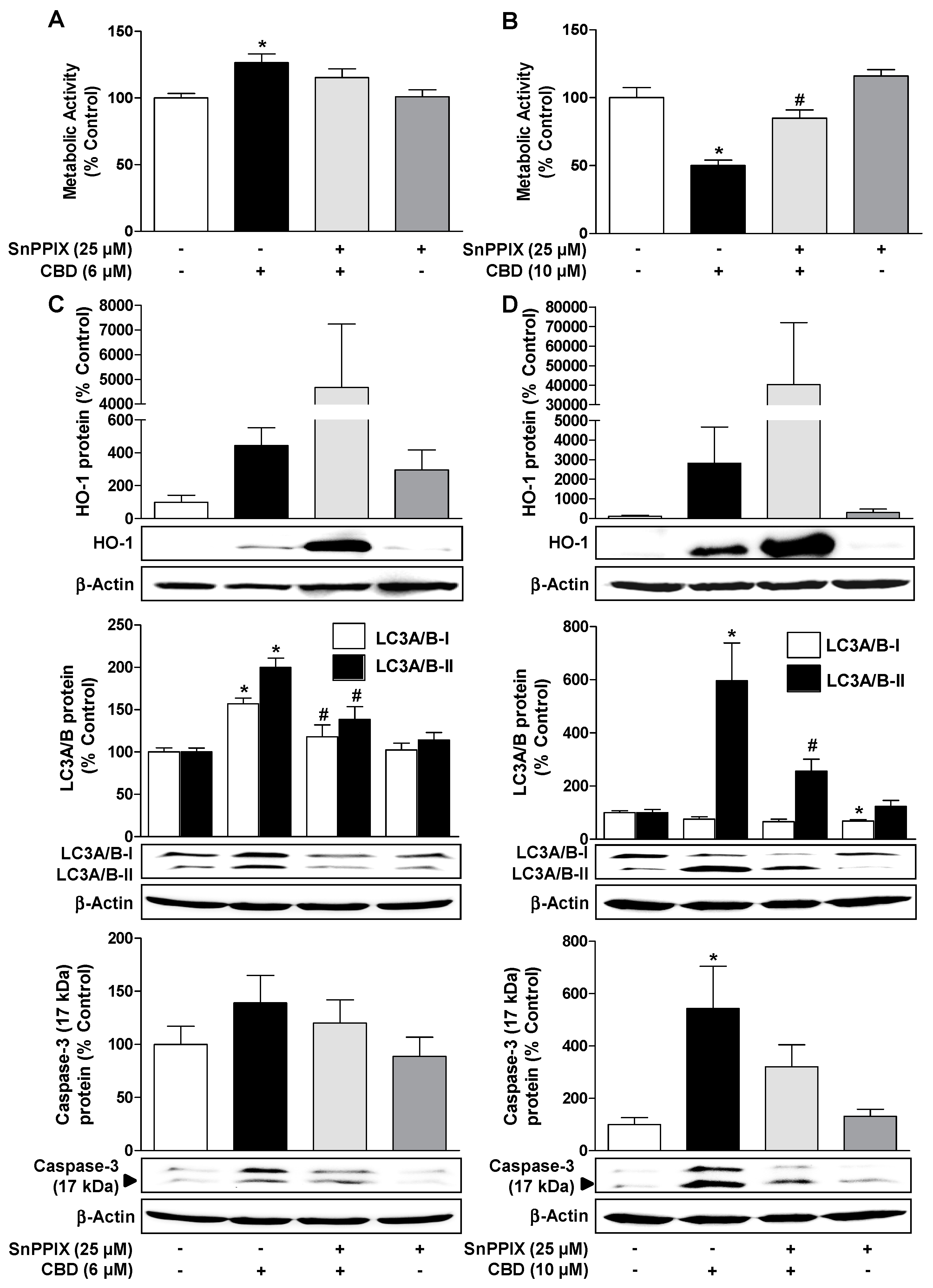
3.6. Inhibition of HO-1 Activity by SnPPIX Reduces CBD-Induced Autophagy and Attenuates the Loss of Viability Due to 10 µM CBD
3.7. Inhibition of HO-1 Expression by Nrf2 siRNA Reduces CBD-Induced Autophagy and Attenuates the Loss of Viability by 10 µM CBD
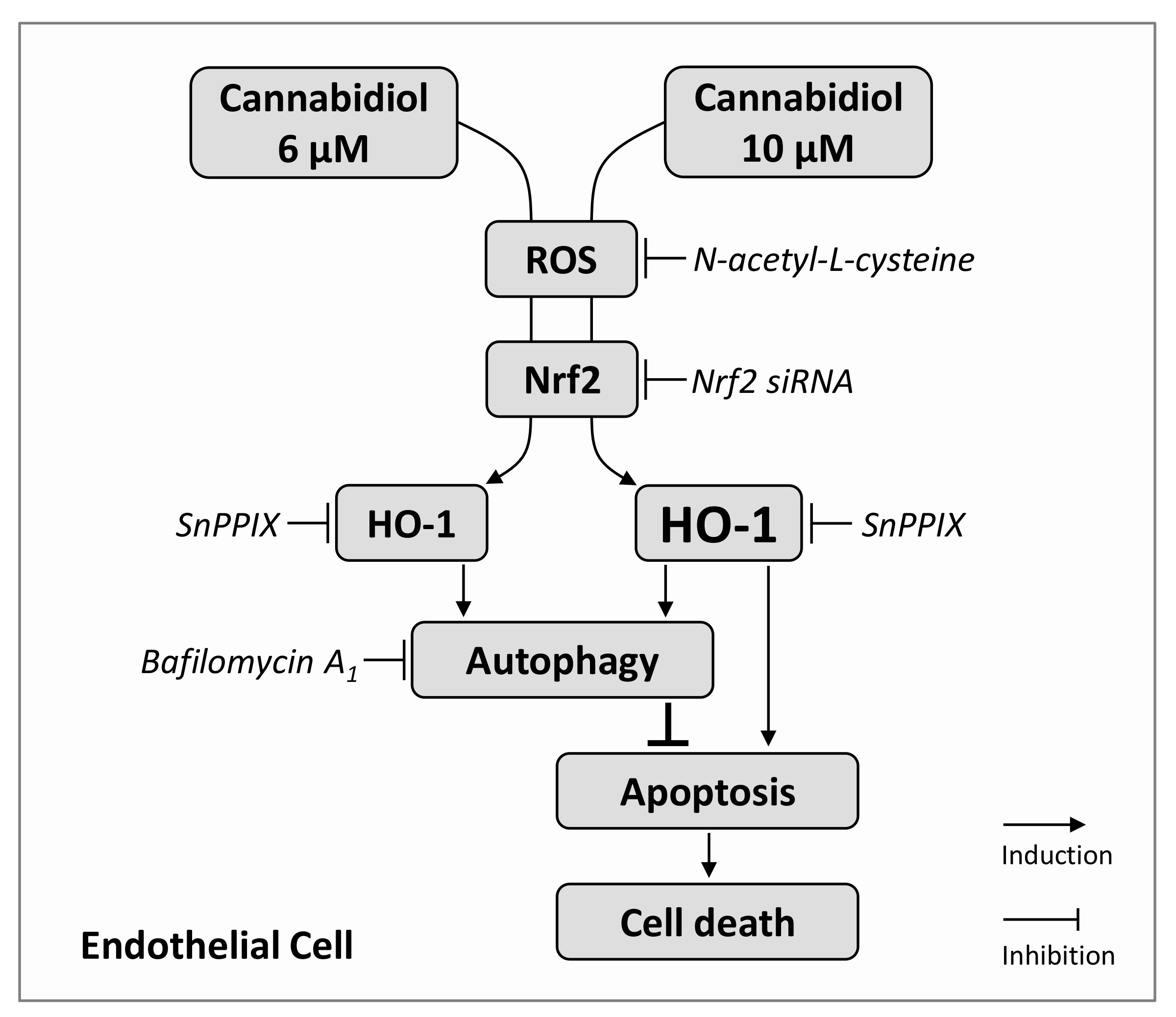
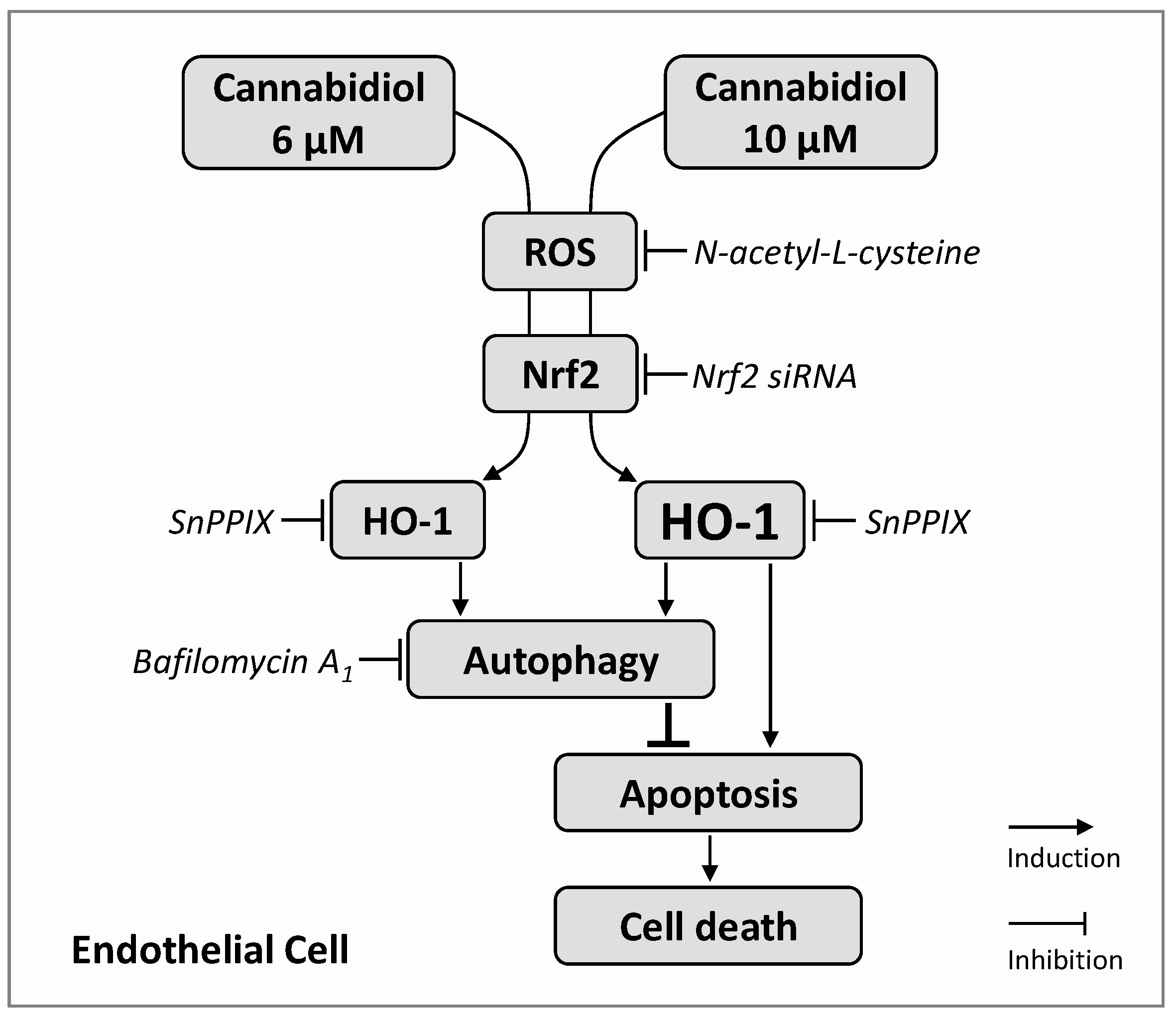
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.B.; Mengi, S.A.; Xu, Y.J.; Arneja, A.S.; Dhalla, N.S. Pathogenesis of atherosclerosis: A multifactorial process. Exp. Clin. Cardiol. 2002, 7, 40–53. [Google Scholar] [PubMed]
- Li, H.; Horke, S.; Förstermann, U. Vascular oxidative stress, nitric oxide and atherosclerosis. Atherosclerosis 2014, 237, 208–219. [Google Scholar] [CrossRef]
- Yang, X.; Li, Y.; Li, Y.; Ren, X.; Zhang, X.; Hu, D.; Gao, Y.; Xing, Y.; Shang, H. Oxidative stress-mediated atherosclerosis: Mechanisms and therapies. Front. Physiol. 2017, 8, 600. [Google Scholar] [CrossRef] [Green Version]
- Dimmeler, S.; Zeiher, A.M. Endothelial cell apoptosis in angiogenesis and vessel regression. Circ. Res. 2000, 87, 434–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rössig, L.; Dimmeler, S.; Zeiher, A.M. Apoptosis in the vascular wall and atherosclerosis. Basic Res. Cardiol. 2001, 96, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xiong, J.; Yang, B.; Zhou, Q.; Wu, Y.; Luo, H.; Zhou, H.; Liu, N.; Li, Y.; Song, Z.; et al. Endothelial cell apoptosis induces TGF-β signaling-dependent host endothelial-mesenchymal transition to promote transplant arteriosclerosis. Am. J. Transplant. 2015, 15, 3095–3111. [Google Scholar] [CrossRef]
- Paine, A.; Eiz-Vesper, B.; Blasczyk, R.; Immenschuh, S. Signaling to heme oxygenase-1 and its anti-inflammatory therapeutic potential. Biochem. Pharmacol. 2010, 80, 1895–1903. [Google Scholar] [CrossRef] [Green Version]
- Brouard, S.; Otterbein, L.E.; Anrather, J.; Tobiasch, E.; Bach, F.H.; Choi, A.M.; Soares, M.P. Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J. Exp. Med. 2000, 192, 1015–1026. [Google Scholar] [CrossRef]
- Kushida, T.; Li Volti, G.; Quan, S.; Goodman, A.; Abraham, N.G. Role of human heme oxygenase-1 in attenuating TNF-alpha-mediated inflammation injury in endothelial cells. J. Cell Biochem. 2002, 87, 377–385. [Google Scholar] [CrossRef]
- Asija, A.; Peterson, S.J.; Stec, D.E.; Abraham, N.G. Targeting endothelial cells with heme oxygenase-1 gene using VE-cadherin promoter attenuates hyperglycemia-mediated cell injury and apoptosis. Antioxid. Redox Signal. 2007, 9, 2065–2074. [Google Scholar] [CrossRef] [PubMed]
- Castilho, Á.; Aveleira, C.A.; Leal, E.C.; Simões, N.F.; Fernandes, C.R.; Meirinhos, R.I.; Baptista, F.I.; Ambrósio, A.F. Heme oxygenase-1 protects retinal endothelial cells against high glucose- and oxidative/nitrosative stress-induced toxicity. PLoS ONE 2012, 7, e42428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waltz, P.; Carchman, E.H.; Young, A.C.; Rao, J.; Rosengart, M.R.; Kaczorowski, D.; Zuckerbraun, B.S. Lipopolysaccaride induces autophagic signaling in macrophages via a TLR4, heme oxygenase-1 dependent pathway. Autophagy 2011, 7, 315–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, T.K.; Chen, S.D.; Chuang, Y.C.; Lin, H.Y.; Huang, C.R.; Chuang, J.H.; Wang, P.W.; Huang, S.T.; Tiao, M.M.; Chen, J.B.; et al. Resveratrol partially prevents rotenone-induced neurotoxicity in dopaminergic SH-SY5Y cells through induction of heme oxygenase-1 dependent autophagy. Int. J. Mol. Sci. 2014, 22, 1625–1646. [Google Scholar] [CrossRef]
- Surolia, R.; Karki, S.; Kim, H.; Yu, Z.; Kulkarni, T.; Mirov, S.B.; Carter, A.B.; Rowe, S.M.; Matalon, S.; Thannickal, V.J.; et al. Heme oxygenase-1-mediated autophagy protects against pulmonary endothelial cell death and development of emphysema in cadmium-treated mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L280–L292. [Google Scholar] [CrossRef] [Green Version]
- Zou, S.; Sun, H.; Candiotti, K.A.; Peng, Y.; Zhang, Q.; Xiao, W.; Zhao, S.; Wu, L.; Yang, J. Octreotide protects against hepatic ischemia/reperfusion injury via HO-1-mediated autophagy. Acta Biochim. Biophys. Sin. (Shanghai) 2018, 50, 316–318. [Google Scholar] [CrossRef] [Green Version]
- Stocker, R.; Perrella, M.A. Heme oxygenase-1: A novel drug target for atherosclerotic diseases? Circulation 2006, 114, 2178–2189. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.H.; Kuo, K.L.; Hung, S.C.; Hsu, C.C.; Chen, Y.H.; Tarng, D.C. Length polymorphism in heme oxygenase-1 and risk of CKD among patients with coronary artery disease. J. Am. Soc. Nephrol. 2014, 25, 2669–2677. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Khan, Z.A.; Barbin, Y.; Chakrabarti, S. Pro-oxidant role of heme oxygenase in mediating glucose-induced endothelial cell damage. Free Radic. Res. 2004, 38, 1301–1310. [Google Scholar] [CrossRef]
- Yang, C.M.; Lin, C.C.; Hsieh, H.L. High-Glucose-Derived Oxidative Stress-Dependent Heme Oxygenase-1 Expression from Astrocytes Contributes to the Neuronal Apoptosis. Mol. Neurobiol. 2017, 54, 470–483. [Google Scholar] [CrossRef]
- Suttner, D.M.; Dennery, P.A. Reversal of HO-1 related cytoprotection with increased expression is due to reactive iron. FASEB J. 1999, 13, 1800–1809. [Google Scholar] [CrossRef] [PubMed]
- Maruhashi, K.; Kasahara, Y.; Ohta, K.; Wada, T.; Ohta, K.; Nakamura, N.; Toma, T.; Koizumi, S.; Yachie, A. Paradoxical enhancement of oxidative cell injury by overexpression of heme oxygenase-1 in an anchorage-dependent cell ECV304. J. Cell Biochem. 2004, 93, 552–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amico, G.; Lam, F.; Hagen, T.; Moncada, S. Inhibition of cellular respiration by endogenously produced carbon monoxide. J. Cell Sci. 2006, 119, 2291–2298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuckerbraun, B.S.; Chin, B.Y.; Bilban, M.; d’Avila, J.C.; Rao, J.; Billiar, T.R.; Otterbein, L.E. Carbon monoxide signals via inhibition of cytochrome c oxidase and generation of mitochondrial reactive oxygen species. FASEB J. 2007, 21, 1099–1106. [Google Scholar] [CrossRef] [Green Version]
- Rajesh, M.; Mukhopadhyay, P.; Bátkai, S.; Haskó, G.; Liaudet, L.; Drel, V.R.; Obrosova, I.G.; Pacher, P. Cannabidiol attenuates high glucose-induced endothelial cell inflammatory response and barrier disruption. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H610–H619. [Google Scholar] [CrossRef] [Green Version]
- Rajesh, M.; Mukhopadhyay, P.; Bátkai, S.; Patel, V.; Saito, K.; Matsumoto, S.; Kashiwaya, Y.; Horváth, B.; Mukhopadhyay, B.; Becker, L.; et al. Cannabidiol attenuates cardiac dysfunction, oxidative stress, fibrosis, and inflammatory and cell death signaling pathways in diabetic cardiomyopathy. J. Am. Coll. Cardiol. 2010, 56, 2115–2125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mechoulam, R.; Peters, M.; Murillo-Rodriguez, E.; Hanus, L.O. Cannabidiol—Recent advances. Chem. Biodivers. 2007, 4, 1678–1692. [Google Scholar] [CrossRef]
- Jacobsson, S.O.; Wallin, T.; Fowler, C.J. Inhibition of rat C6 glioma cell proliferation by endogenous and synthetic cannabinoids. Relative involvement of cannabinoid and vanilloid receptors. J. Pharmacol. Exp. Ther. 2001, 299, 951–959. [Google Scholar]
- Mukherjee, S.; Adams, M.; Whiteaker, K.; Daza, A.; Kage, K.; Cassar, S.; Meyer, M.; Yao, B.B. Species comparison and pharmacological characterization of rat and human CB2 cannabinoid receptors. Eur. J. Pharmacol. 2004, 505, 1–9. [Google Scholar] [CrossRef]
- Ramer, R.; Hinz, B. Inhibition of cancer cell invasion by cannabinoids via increased expression of tissue inhibitor of matrix metalloproteinases-1. J. Natl. Cancer Inst. 2008, 100, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Ramer, R.; Fischer, S.; Haustein, M.; Manda, K.; Hinz, B. Cannabinoids inhibit angiogenic capacities of endothelial cells via release of tissue inhibitor of matrix metalloproteinases-1 from lung cancer cells. Biochem. Pharmacol. 2014, 91, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Sherratt, P.J.; Huang, H.C.; Yang, C.S.; Pickett, C.B. Increased protein stability as a mechanism that enhances Nrf2-mediated transcriptional activation of the antioxidant response element. Degradation of Nrf2 by the 26 S proteasome. J. Biol. Chem. 2003, 278, 4536–4541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Na, H.K.; Surh, Y.J. Oncogenic potential of Nrf2 and its principal target protein heme oxygenase-1. Free Radic. Biol. Med. 2014, 67, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728, Erratum in: 2003, 22, 4577. [Google Scholar] [CrossRef] [PubMed]
- Barth, S.; Glick, D.; Macleod, K.F. Autophagy: Assays and artifacts. J. Pathol. 2010, 221, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Kimura, S.; Fujita, N.; Noda, T.; Yoshimori, T. Monitoring autophagy in mammalian cultured cells through the dynamics of LC3. Methods Enzymol. 2009, 452, 1–12. [Google Scholar]
- Oh, C.J.; Park, S.; Kim, J.Y.; Kim, H.J.; Jeoung, N.H.; Choi, Y.K.; Go, Y.; Park, K.G.; Lee, I.K. Dimethylfumarate attenuates restenosis after acute vascular injury by cell-specific and Nrf2-dependent mechanisms. Redox Biol. 2014, 2, 855–864. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Park, C.; Lee, J.N.; Lim, H.; Hong, G.Y.; Moon, S.K.; Lim, D.J.; Choe, S.K.; Park, R. Erdosteine protects HEI-OC1 auditory cells from cisplatin toxicity through suppression of inflammatory cytokines and induction of Nrf2 target proteins. Toxicol. Appl. Pharmacol. 2015, 288, 192–202. [Google Scholar] [CrossRef]
- Tebay, L.E.; Robertson, H.; Durant, S.T.; Vitale, S.R.; Penning, T.M.; Dinkova-Kostova, A.T.; Hayes, J.D. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic. Biol. Med. 2015, 88, 108–146. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, M.; Böckmann, S.; Hinz, B. Up-regulation of heme oxygenase-1 expression and inhibition of disease-associated features by cannabidiol in vascular smooth muscle cells. Oncotarget 2018, 9, 34595–34616. [Google Scholar] [CrossRef] [Green Version]
- Juknat, A.; Pietr, M.; Kozela, E.; Rimmerman, N.; Levy, R.; Coppola, G.; Geschwind, D.; Vogel, Z. Differential transcriptional profiles mediated by exposure to the cannabinoids cannabidiol and Δ9-tetrahydrocannabinol in BV-2 microglial cells. Br. J. Pharmacol. 2012, 165, 2512–2528. [Google Scholar] [CrossRef] [Green Version]
- Juknat, A.; Pietr, M.; Kozela, E.; Rimmerman, N.; Levy, R.; Gao, F.; Coppola, G.; Geschwind, D.; Vogel, Z. Microarray and pathway analysis reveal distinct mechanisms underlying cannabinoid-mediated modulation of LPS-induced activation of BV-2 microglial cells. PLoS ONE 2013, 8, e61462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casares, L.; García, V.; Garrido-Rodríguez, M.; Millán, E.; Collado, J.A.; García-Martín, A.; Peñarando, J.; Calzado, M.A.; de la Vega, L.; Muñoz, E. Cannabidiol induces antioxidant pathways in keratinocytes by targeting BACH1. Redox Biol. 2020, 28, 101321. [Google Scholar] [CrossRef]
- Louvet, A.; Teixeira-Clerc, F.; Chobert, M.N.; Deveaux, V.; Pavoine, C.; Zimmer, A.; Pecker, F.; Mallat, A.; Lotersztajn, S. Cannabinoid CB2 receptors protect against alcoholic liver disease by regulating Kupffer cell polarization in mice. Hepatology 2011, 54, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Steib, C.J.; Gmelin, L.; Pfeiler, S.; Schewe, J.; Brand, S.; Göke, B.; Gerbes, A.L. Functional relevance of the cannabinoid receptor 2—heme oxygenase pathway: A novel target for the attenuation of portal hypertension. Life Sci. 2013, 93, 543–551. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, S.; Wang, Q.; Hu, W.; Wang, D.; Li, X.; Su, T.; Qin, X.; Zhang, X.; Ma, K.; et al. Effects of cannabinoid receptor type 2 on endogenous myocardial regeneration by activating cardiac progenitor cells in mouse infarcted heart. Sci. China Life Sci. 2014, 57, 201–208. [Google Scholar] [CrossRef] [Green Version]
- Blázquez, C.; González-Feria, L.; Alvarez, L.; Haro, A.; Casanova, M.L.; Guzmán, M. Cannabinoids inhibit the vascular endothelial growth factor pathway in gliomas. Cancer Res. 2004, 64, 5617–5623. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Wood, J.T.; Whitten, K.M.; Vadivel, S.K.; Seng, S.; Makriyannis, A.; Avraham, H.K. Inhibition of fatty acid amide hydrolase activates Nrf2 signalling and induces heme oxygenase 1 transcription in breast cancer cells. Br. J. Pharmacol. 2013, 170, 489–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Liang, B.; Shirwany, N.A.; Zou, M.H. 2-Deoxy-D-glucose treatment of endothelial cells induces autophagy by reactive oxygen species-mediated activation of the AMP-activated protein kinase. PLoS ONE 2011, 6, e17234. [Google Scholar] [CrossRef] [Green Version]
- Teng, R.J.; Du, J.; Welak, S.; Guan, T.; Eis, A.; Shi, Y.; Konduri, G.G. Cross talk between NADPH oxidase and autophagy in pulmonary artery endothelial cells with intrauterine persistent pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 302, L651–L663. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, F. Autophagy in vascular endothelial cells. Clin. Exp. Pharmacol. Physiol. 2016, 43, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Sardana, M.K.; Kappas, A. Dual control mechanism for heme oxygenase: Tin(IV)- protoporphyrin potently inhibits enzyme activity while markedly increasing content of enzyme protein in liver. Proc. Natl. Acad. Sci. USA 1987, 84, 2464–2468. [Google Scholar] [CrossRef] [Green Version]
- Chang, T.; Wu, L.; Wang, R. Inhibition of vascular smooth muscle cell proliferation by chronic hemin treatment. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H999–H1007. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.J.; Ryter, S.W.; Xu, J.F.; Nakahira, K.; Kim, H.P.; Choi, A.M.; Kim, Y.S. Carbon monoxide activates autophagy via mitochondrial reactive oxygen species formation. Am. J. Respir. Cell Mol. Biol. 2011, 45, 867–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Constantin, M.; Choi, A.J.; Cloonan, S.M.; Ryter, S.W. Therapeutic potential of heme oxygenase-1/carbon monoxide in lung disease. Int. J. Hypertens. 2012, 2012, 859235–859254. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.K.; Por, E.D.; Kwon, Y.G.; Kim, Y.M. Regulation of ROS production and vascular function by carbon monoxide. Oxid. Med. Cell Longev. 2012, 2012, 794237–794254. [Google Scholar] [CrossRef] [Green Version]
- Kaczara, P.; Motterlini, R.; Kus, K.; Zakrzewska, A.; Abramov, A.Y.; Chlopicki, S. Carbon monoxide shifts energetic metabolism from glycolysis to oxidative phosphorylation in endothelial cells. FEBS Lett. 2016, 590, 3469–3480. [Google Scholar] [CrossRef]
- Berridge, M.V.; Herst, P.M.; Tan, A.S. Tetrazolium dyes as tools in cell biology: New insights into their cellular reduction. Biotechnol. Annu. Rev. 2005, 11, 127–152. [Google Scholar]
- Kimura, T.; Takahashi, A.; Takabatake, Y.; Namba, T.; Yamamoto, T.; Kaimori, J.Y.; Matsui, I.; Kitamura, H.; Niimura, F.; Matsusaka, T.; et al. Autophagy protects kidney proximal tubule epithelial cells from mitochondrial metabolic stress. Autophagy 2013, 9, 1876–1886. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.Y.; Teng, X.; Laddha, S.V.; Ma, S.; Van Nostrand, S.C.; Yang, Y.; Khor, S.; Chan, C.S.; Rabinowitz, J.D.; White, E. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. 2016, 30, 1704–1717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, A.; Weintraub, N.L.; Tang, Y. Enhancing stem cell survival in an ischemic heart by CRISPR-dCas9-based gene regulation. Med. Hypotheses 2014, 83, 702–705. [Google Scholar] [CrossRef] [Green Version]
- Uberti, F.; Lattuada, D.; Morsanuto, V.; Nava, U.; Bolis, G.; Vacca, G.; Squarzanti, D.F.; Cisari, C.; Molinari, C. Vitamin D protects human endothelial cells from oxidative stress through the autophagic and survival pathways. J. Clin. Endocrinol. Metab. 2014, 99, 1367–1374. [Google Scholar] [CrossRef] [Green Version]
- Dong, G.; Yang, S.; Cao, X.; Yu, N.; Yu, J.; Qu, X. Low shear stress-induced autophagy alleviates cell apoptosis in HUVECs. Mol. Med. Rep. 2017, 15, 3076–3082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bharath, L.P.; Cho, J.M.; Park, S.K.; Ruan, T.; Li, Y.; Mueller, R.; Bean, T.; Reese, V.; Richardson, R.S.; Cai, J.; et al. Endothelial Cell Autophagy Maintains Shear Stress-Induced Nitric Oxide Generation via Glycolysis-Dependent Purinergic Signaling to Endothelial Nitric Oxide Synthase. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1646–1656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yau, J.W.; Singh, K.K.; Hou, Y.; Lei, X.; Ramadan, A.; Quan, A.; Teoh, H.; Kuebler, W.M.; Al-Omran, M.; Yanagawa, B.; et al. Endothelial-specific deletion of autophagy-related 7 (ATG7) attenuates arterial thrombosis in mice. J. Thorac. Cardiovasc. Surg. 2017, 154, 978–988.e1. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.; Tian, C.; Chen, H. Oxidative stress mediates chemerin-induced autophagy in endothelial cells. Free Radic. Biol. Med. 2013, 55, 73–82. [Google Scholar] [CrossRef]
- Hayashi, S.; Yamamoto, A.; You, F. The stent-eluting drugs sirolimus and paclitaxel suppress healing of the endothelium by induction of autophagy. Am. J. Pathol. 2009, 175, 2226–2234. [Google Scholar] [CrossRef] [Green Version]
- Borrelli, F.; Aviello, G.; Romano, B.; Orlando, P.; Capasso, R.; Maiello, F.; Guadagno, F.; Petrosino, S.; Capasso, F.; Di Marzo, V.; et al. Cannabidiol, a safe and non-psychotropic ingredient of the marijuana plant Cannabis sativa, is protective in a murine model of colitis. J. Mol. Med. (Berl.) 2009, 87, 1111–1121. [Google Scholar] [CrossRef]
- Pan, H.; Mukhopadhyay, P.; Rajesh, M.; Patel, V.; Mukhopadhyay, B.; Gao, B.; Haskó, G.; Pacher, P. Cannabidiol attenuates cisplatin-induced nephrotoxicity by decreasing oxidative/nitrosative stress, inflammation, and cell death. J. Pharmacol. Exp. Ther. 2009, 328, 708–714. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Ruiz, J.; Sagredo, O.; Pazos, M.R.; García, C.; Pertwee, R.; Mechoulam, R.; Martínez-Orgado, J. Cannabidiol for neurodegenerative disorders: Important new clinical applications for this phytocannabinoid? Br. J. Clin. Pharmacol. 2013, 75, 323–333. [Google Scholar] [CrossRef] [PubMed]
- McKallip, R.J.; Jia, W.; Schlomer, J.; Warren, J.W.; Nagarkatti, P.S.; Nagarkatti, M. Cannabidiol-induced apoptosis in human leukemia cells: A novel role of Cannabidiol in the regulation of p22phox and NOX4 expression. Mol. Pharmacol. 2006, 70, 897–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.Y.; Wey, S.P.; Liao, M.H.; Hsu, W.L.; Wu, H.Y.; Jan, T.R. A comparative study on cannabidiol-induced apoptosis in murine thymocytes and EL-4 thymoma cells. Int. Immunopharmacol. 2008, 8, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.Y.; Chu, R.M.; Wang, C.C.; Lee, C.Y.; Lin, S.H.; Jan, T.R. Cannabidiol-induced apoptosis in primary lymphocytes is associated with oxidative stress-dependent activation of caspase-8. Toxicol. Appl. Pharmacol. 2008, 226, 260–270. [Google Scholar] [CrossRef]
- Wu, H.Y.; Jan, T.R. Cannabidiol hydroxyquinone-induced apoptosis of splenocytes is mediated predominantly by thiol depletion. Toxicol. Lett. 2010, 195, 68–74. [Google Scholar] [CrossRef]
- Wu, H.Y.; Huang, C.H.; Lin, Y.H.; Wang, C.C.; Jan, T.R. Cannabidiol induced apoptosis in human monocytes through mitochondrial permeability transition pore-mediated ROS production. Free Radic. Biol. Med. 2018, 124, 311–318. [Google Scholar] [CrossRef]
- Mecha, M.; Torrao, A.S.; Mestre, L.; Carrillo-Salinas, F.J.; Mechoulam, R.; Guaza, C. Cannabidiol protects oligodendrocyte progenitor cells from inflammation-induced apoptosis by attenuating endoplasmic reticulum stress. Cell Death Dis. 2012, 3, e331. [Google Scholar] [CrossRef] [Green Version]
- Schönhofen, P.; de Medeiros, L.M.; Bristot, I.J.; Lopes, F.M.; De Bastiani, M.A.; Kapczinski, F.; Crippa, J.A.; Castro, M.A.; Parsons, R.B.; Klamt, F. Cannabidiol exposure during neuronal differentiation sensitizes cells against redox-active neurotoxins. Mol. Neurobiol. 2015, 52, 26–37. [Google Scholar] [CrossRef]
- Usami, N.; Yamamoto, I.; Watanabe, K. Generation of reactive oxygen species during mouse hepatic microsomal metabolism of cannabidiol and cannabidiol hydroxy-quinone. Life Sci. 2008, 83, 717–724. [Google Scholar] [CrossRef]
- Méndez-García, L.A.; Martínez-Castillo, M.; Villegas-Sepúlveda, N.; Orozco, L.; Córdova, E.J. Curcumin induces p53-independent inactivation of Nrf2 during oxidative stress-induced apoptosis. Hum. Exp. Toxicol. 2019, 38, 951–961. [Google Scholar] [CrossRef]
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Redox-regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J. Biol. Chem. 2004, 279, 31556–31567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solinas, M.; Massi, P.; Cantelmo, A.R.; Cattaneo, M.G.; Cammarota, R.; Bartolini, D.; Cinquina, V.; Valenti, M.; Vicentini, L.M.; Noonan, D.M.; et al. Cannabidiol inhibits angiogenesis by multiple mechanisms. Br. J. Pharmacol. 2012, 167, 1218–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aird, W.C. Endothelial cell heterogeneity. Cold Spring Harb. Perspect. Med. 2012, 2, a006429. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment Group | Metabolic Activity (%) | HO-1 Expression (%) | LC3A/B-I Expression (%) | LC3A/B-II Expression (%) | Caspase-3 (17 kDa) Expression (%) |
|---|---|---|---|---|---|
| Vehicle | 100.0 ± 6.7 | 100.0 ± 8.3 | 100.0 ± 20.7 | 100.0 ± 19.2 | 100.0 ± 39.2 |
| 6 µM CBD | 120.6 ± 7.3 | 282.3 ± 28.8* | 143.1 ± 9.9 | 191.7 ± 11.9 | 83.5 ± 13.7 |
| CuPPIX + CBD | 132.0 ± 7.5 | 334.2 ± 37.2 | 316.0 ± 45.1# | 386.8 ± 53.1# | 238.8 ± 46.0 |
| CuPPIX | 111.9 ± 5.8 | 168.9 ± 17.0 | 190.4 ± 24.8 | 198.1 ± 31.3 | 160.0 ± 50.5 |
| Vehicle | 100.0 ± 4.5 | 100.0 ± 17.4 | 100.0 ± 9.0 | 100.0 ± 9.4 | 100.0 ± 5.1 |
| 10 µM CBD | 51.6 ± 5.3* | 3683.2 ± 978.0 | 106.5 ± 21.3 | 424.7 ± 85.7* | 256.9 ± 17.5* |
| CuPPIX + CBD | 69.3 ± 5.1 | 5755.6 ± 1525.5 | 157.6 ± 8.3 | 615.3 ± 88.0# | 333.4 ± 54.7 |
| CuPPIX | 108.7 ± 5.9 | 300.0 ± 69.5 | 188.6 ± 38.7 | 204.7 ± 14.3 | 142.1 ± 2.6 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Böckmann, S.; Hinz, B. Cannabidiol Promotes Endothelial Cell Survival by Heme Oxygenase-1-Mediated Autophagy. Cells 2020, 9, 1703. https://doi.org/10.3390/cells9071703
Böckmann S, Hinz B. Cannabidiol Promotes Endothelial Cell Survival by Heme Oxygenase-1-Mediated Autophagy. Cells. 2020; 9(7):1703. https://doi.org/10.3390/cells9071703
Chicago/Turabian StyleBöckmann, Sabine, and Burkhard Hinz. 2020. "Cannabidiol Promotes Endothelial Cell Survival by Heme Oxygenase-1-Mediated Autophagy" Cells 9, no. 7: 1703. https://doi.org/10.3390/cells9071703
APA StyleBöckmann, S., & Hinz, B. (2020). Cannabidiol Promotes Endothelial Cell Survival by Heme Oxygenase-1-Mediated Autophagy. Cells, 9(7), 1703. https://doi.org/10.3390/cells9071703