CpG-Islands as Markers for Liquid Biopsies of Cancer Patients



Abstract
1. Introduction
2. Materials and Methods
2.1. Datasets
2.2. Data Processing
2.3. Feature Selection
2.4. Hyperparameter Tuning
3. Results
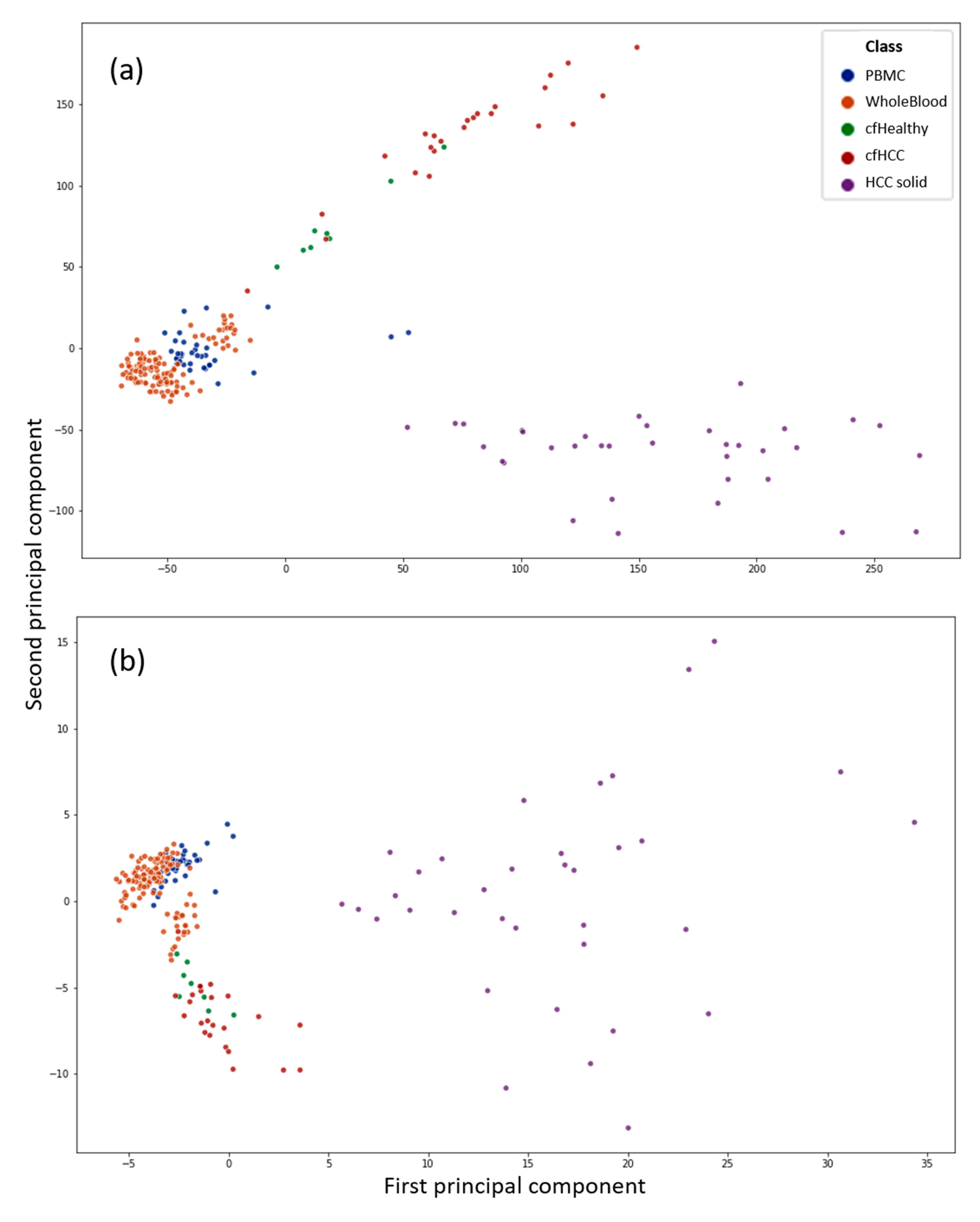
3.1. Principal Component Analysis Shows the Difference between Healthy and Cancerous cfDNA Samples
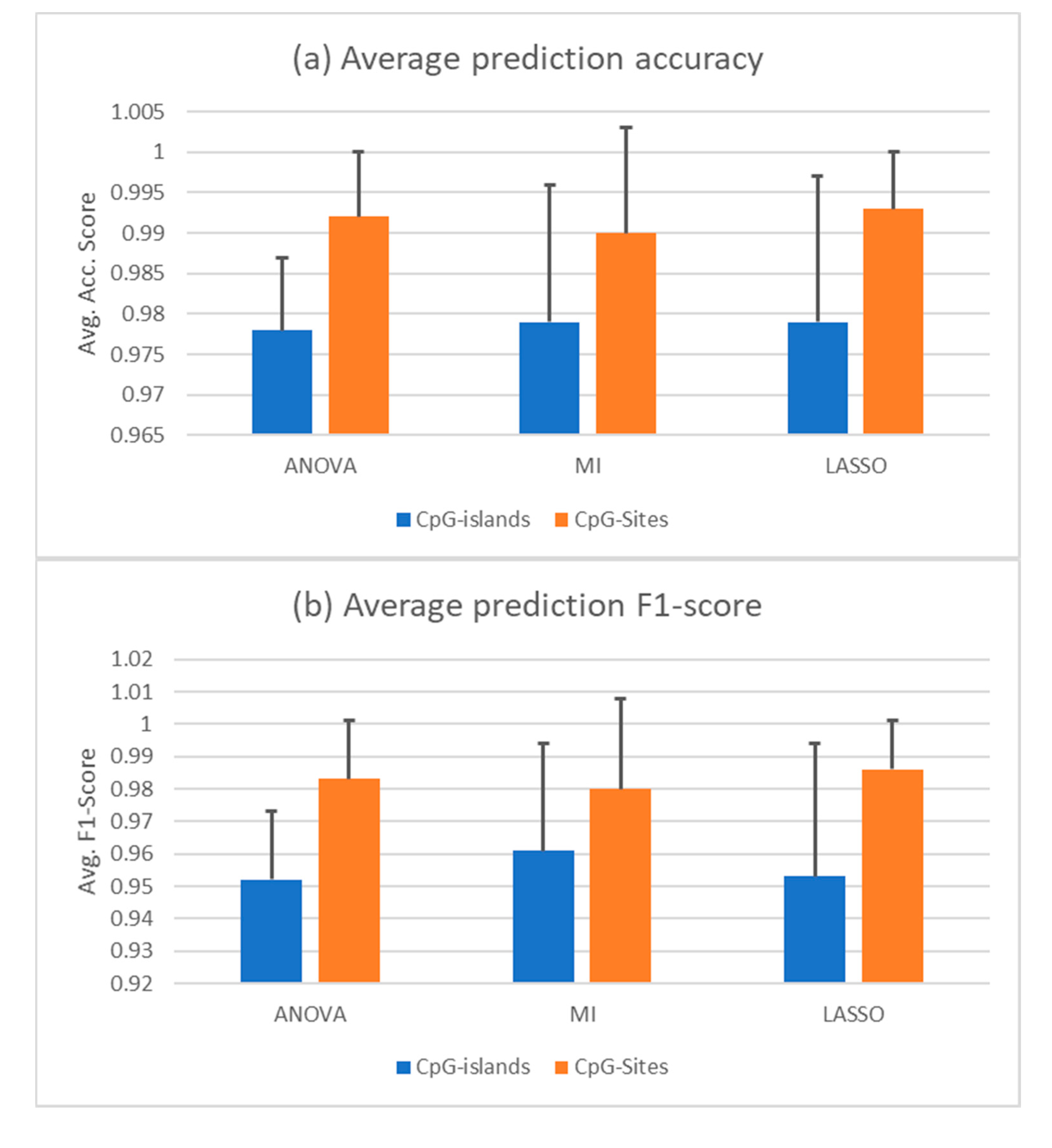
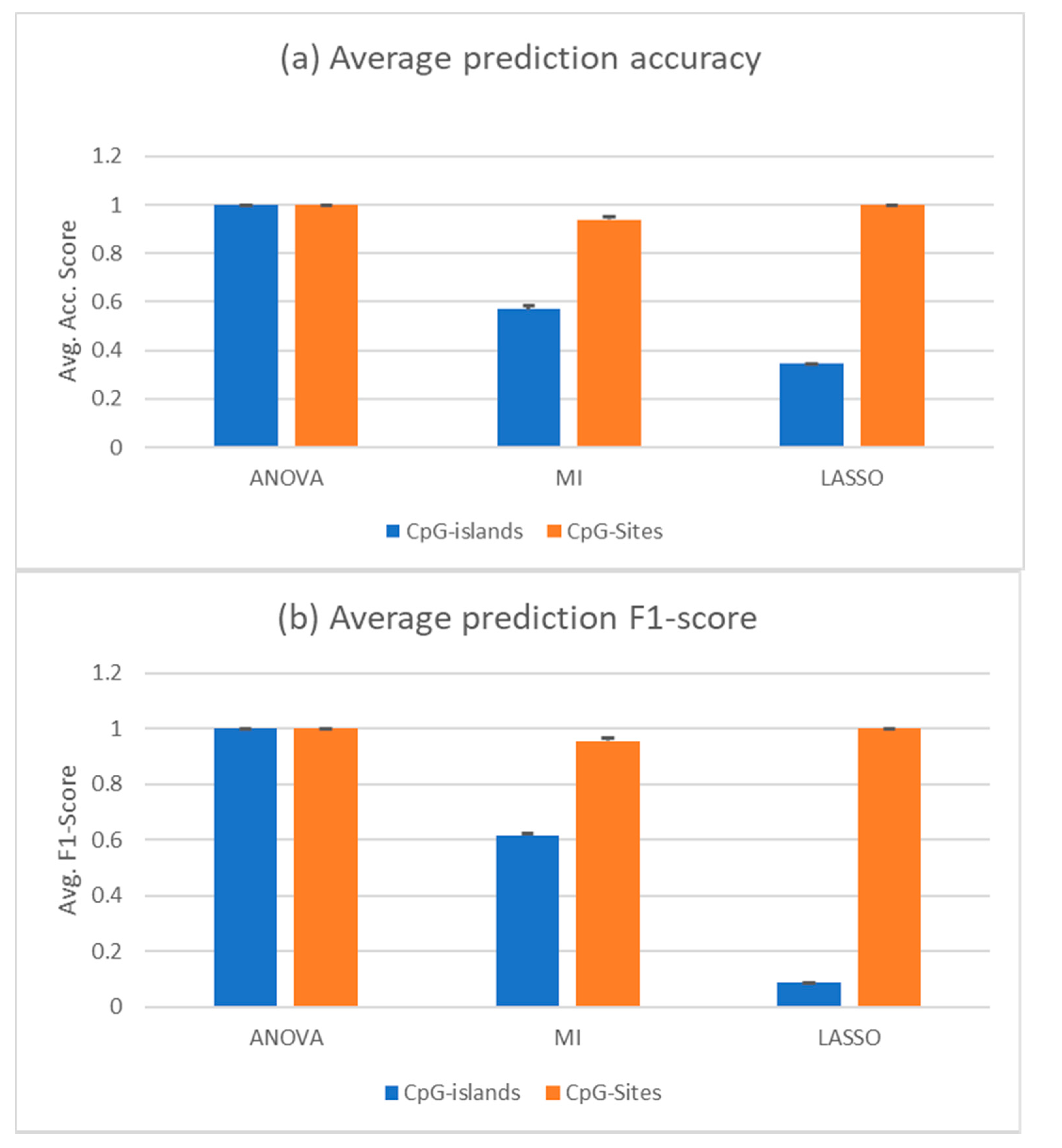
3.2. Feature Selection and Supervised Analysis Can Differentiate between Liquid Biopsies of Healthy Individuals and HCC Patients
3.3. A Random Forest Can Be Trained from PBMC Data and Solid Tumour Samples and Still Be Able to Predict cfDNA Samples Correctly
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.P.; Karajannis, M.A. DNA Methylation–Based Classifier for Accurate Molecular diagnosis of bone sarcomas. JCO Precis. Oncol. 2017, 1, 1–11. [Google Scholar]
- Kwapisz, D. The first liquid biopsy test approved. Is it a new era of mutation testing for non-small cell lung cancer? Ann. Transl. Med. 2017, 5, 46. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Li, Q.; Chen, Q.; Zhou, Y.; Park, S.; Lee, G.; Grimes, B.; Krysan, K.; Yu, M.; Wang, W.; et al. Cancer Locator: Non-invasive cancer diagnosis and tissue-of-origin prediction using methylation profiles of cell-free DNA. Genome Biol. 2017, 18, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Panagopoulou, M.; Karaglani, M.; Balgkouranidou, I.; Biziota, E.; Koukaki, T.; Karamitrousis, E.; Nena, E.; Tsamardinos, I.; Kolios, G.; Lianidou, E.; et al. Circulating cell-free DNA in breast cancer: Size profiling, levels, and methylation patterns lead to prognostic and predictive classifiers. Oncogene 2019, 38, 3387–3401. [Google Scholar] [CrossRef] [PubMed]
- Saxonov, S.; Berg, P.; Brutlag, D.L. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. USA 2006, 103, 1412–1417. [Google Scholar] [CrossRef]
- Keshet, I.; Schlesinger, Y.; Farkash, S.; Rand, E.; Hecht, M.; Segal, E.; Pikarski, E.; Young, R.A.; Niveleau, A.; Cedar, H.; et al. Evidence for an instructive mechanism of de novo methylation in cancer cells. Nat. Genet. 2006, 38, 149–153. [Google Scholar] [CrossRef]
- Ferenci, P.; Fried, M.; Labrecque, D.; Bruix, J.; Sherman, M.; Omata, M.; Heathcote, J.; Piratsivuth, T.; Kew, M.; Otegbayo, J.A.; et al. Hepatocellular carcinoma (HCC): A global perspective. J. Clin. Gastroenterol. 2010, 44, 239–245. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Balogh, J.; Victor III, D.; Asham, E.H.; Burroughs, S.G.; Boktour, M.; Saharia, A.; Li, X.; Ghobrial, R.M.; Monsour Jr, H.P. Hepatocellular carcinoma: A review. J. Hepatocell. Carcinoma 2016, 3, 41–53. [Google Scholar] [CrossRef]
- Hlady, R.A.; Zhao, X.; Pan, X.; Yang, J.D.; Ahmed, F.; Antwi, S.O.; Giama, N.H.; Patel, T.; Roberts, L.R.; Liu, C.; et al. Genome-wide discovery and validation of diagnostic DNA methylation-based biomarkers for hepatocellular cancer detection in circulating cell free DNA. Theranostics 2019, 9, 7239–7250. [Google Scholar] [CrossRef] [PubMed]
- Assenov, Y.; Muller, F.; Lutsik, P.; Walter, J.; Lengauer, T.; Bock, C. Comprehensive analysis of DNA methylation data with RnBeads. Nat. Methods 2014, 11, 1138–1140. [Google Scholar] [CrossRef] [PubMed]
- Muller, F.; Scherer, M.; Assenov, Y.; Lutsik, P.; Walter, J.; Lengauer, T.; Bock, C. RnBeads 2.0: Comprehensive analysis of DNA methylation data. Genome Biol. 2019, 20, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Roth, J.A.; deVos, T.; Ramsey, S.D. Clinical and Budget Impact of Increasing Colorectal Cancer Screening by Blood- and Stool-Based Testing. Am. Health Drug Benefits 2019, 12, 256–262. [Google Scholar] [PubMed]
- Li, W.; Li, Q.; Kang, S.; Same, M.; Zhou, Y.; Sun, C.; Liu, C.C.; Matsuoka, L.; Sher, L.; Wong, W.H.; et al. CancerDetector: Ultrasensitive and non-invasive cancer detection at the resolution of individual reads using cell-free DNA methylation sequencing data. Nucleic Acids Res. 2018, 46, e89. [Google Scholar] [CrossRef]
- Mizuno, Y.; Shimada, S.; Akiyama, Y.; Watanabe, S.; Aida, T.; Ogawa, K.; Ono, H.; Mitsunori, Y.; Ban, D.; Kudo, A.; et al. DEPDC5 deficiency contributes to resistance to leucine starvation via p62 accumulation in hepatocellular carcinoma. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Meng, C.; Shen, X.; Jiang, W. Potential biomarkers of HCC based on gene expression and DNA methylation profiles. Oncol. Lett. 2018, 16, 3183–3192. [Google Scholar] [CrossRef]
- Qiu, S.; Liu, J.; Xing, F. Antizyme inhibitor 1: A potential carcinogenic molecule. Cancer Sci. 2017, 108, 163–169. [Google Scholar] [CrossRef]
- Chen, Z.; Gulzar, Z.G.; Hill, C.A.S.; Walcheck, B.; Brooks, J.D. Increased expression of GCNT1 is associated with altered O-glycosylation of PSA, PAP, and MUC1 in human prostate cancers. Prostate 2014, 74. [Google Scholar] [CrossRef]
- Jiang, R.; Gao, Q.; Chen, M.; Yu, T. Elk-1 transcriptionally regulates ZC3H4 expression to promote silica-induced epithelial-mesenchymal transition. Lab. Invest. 2020, 100, 959–973. [Google Scholar] [CrossRef] [PubMed]
- Bai, D.S.; Zhang, C.; Chen, P.; Jin, S.J.; Jiang, G.Q. The prognostic correlation of AFP level at diagnosis with pathological grade, progression, and survival of patients with hepatocellular carcinoma. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Sample | GEO Identifier | Exemplary Identifier in This Work | n = |
|---|---|---|---|
| Healthy blood: cfDNA | GSE110185 | cf_Moss_x | 8 |
| Healthy blood: cfDNA | GSE122126 | cfDNA_NCF_pool_x | 2 |
| Healthy blood: PBMCs | GSE130748 | PBMC_x | 37 |
| Healthy blood: whole blood | GSE77056 | blood_x | 24 |
| Hepatocellular carcinoma: solid tumour | GSE77269 | HCC_2_x | 20 |
| Hepatocellular carcinoma: solid tumour | GSE99036 | Hepatocellular carcinoma YSHxxx | 15 |
| Hepatocellular carcinoma: cfDNA | GSE129374 | cfDNA_603xxxx_Cirrhosis_with_HCC | 22 |
| Healthy blood: whole blood | GSE40279 | GEO Accession (GSM989xxx) | 101 |
| CpG Islands (Selected Features) | Genes | Region | Status in PBMC | Status in HCC |
|---|---|---|---|---|
| chr19:47614409-47614661 | Zinc Finger CCCH-Type Containing 4, ZC3H4 | Intron 2 | Non-methylated | Semi-methylated |
| chr9:79073908-79074561 | Beta-1,3-galactosyl-O-glycosylglycoprotein beta-1,6-Nacetylglucosaminyltransferase, GCNT1 | Promoter Region, Exon 1, Intron 1 | Non-methylated | Varying |
| chr1:2979276-2980758 | PRDM16 Divergent Transcript | Hypermethylated | Semi-methylated |
| CpG-Islands (Selected Features) | Genes | Region | Status in PBMC | Status in HCC |
|---|---|---|---|---|
| chr12:50361368-50361652 | Aquaporin 6, AQP6 | Intron 1 | Non methylated | hypomethylated |
| chr8:103875223-103877084 | Antizyme inhibitor 1, AZIN1 | Promoter, exon 1 | hypomethylated | Non methylated |
| chr22:32149763-32150064 | DEP domain-containing 5, DEPDC5 | Promoter, exon 1 | Non methylated | hypomethylated |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sprang, M.; Paret, C.; Faber, J. CpG-Islands as Markers for Liquid Biopsies of Cancer Patients. Cells 2020, 9, 1820. https://doi.org/10.3390/cells9081820
Sprang M, Paret C, Faber J. CpG-Islands as Markers for Liquid Biopsies of Cancer Patients. Cells. 2020; 9(8):1820. https://doi.org/10.3390/cells9081820
Chicago/Turabian StyleSprang, Maximilian, Claudia Paret, and Joerg Faber. 2020. "CpG-Islands as Markers for Liquid Biopsies of Cancer Patients" Cells 9, no. 8: 1820. https://doi.org/10.3390/cells9081820
APA StyleSprang, M., Paret, C., & Faber, J. (2020). CpG-Islands as Markers for Liquid Biopsies of Cancer Patients. Cells, 9(8), 1820. https://doi.org/10.3390/cells9081820