Necroptosis in Immuno-Oncology and Cancer Immunotherapy

 ,
,  ,
,  ,
,  ,
, 
Abstract
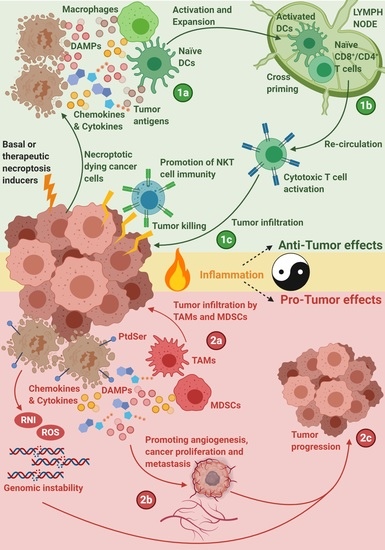
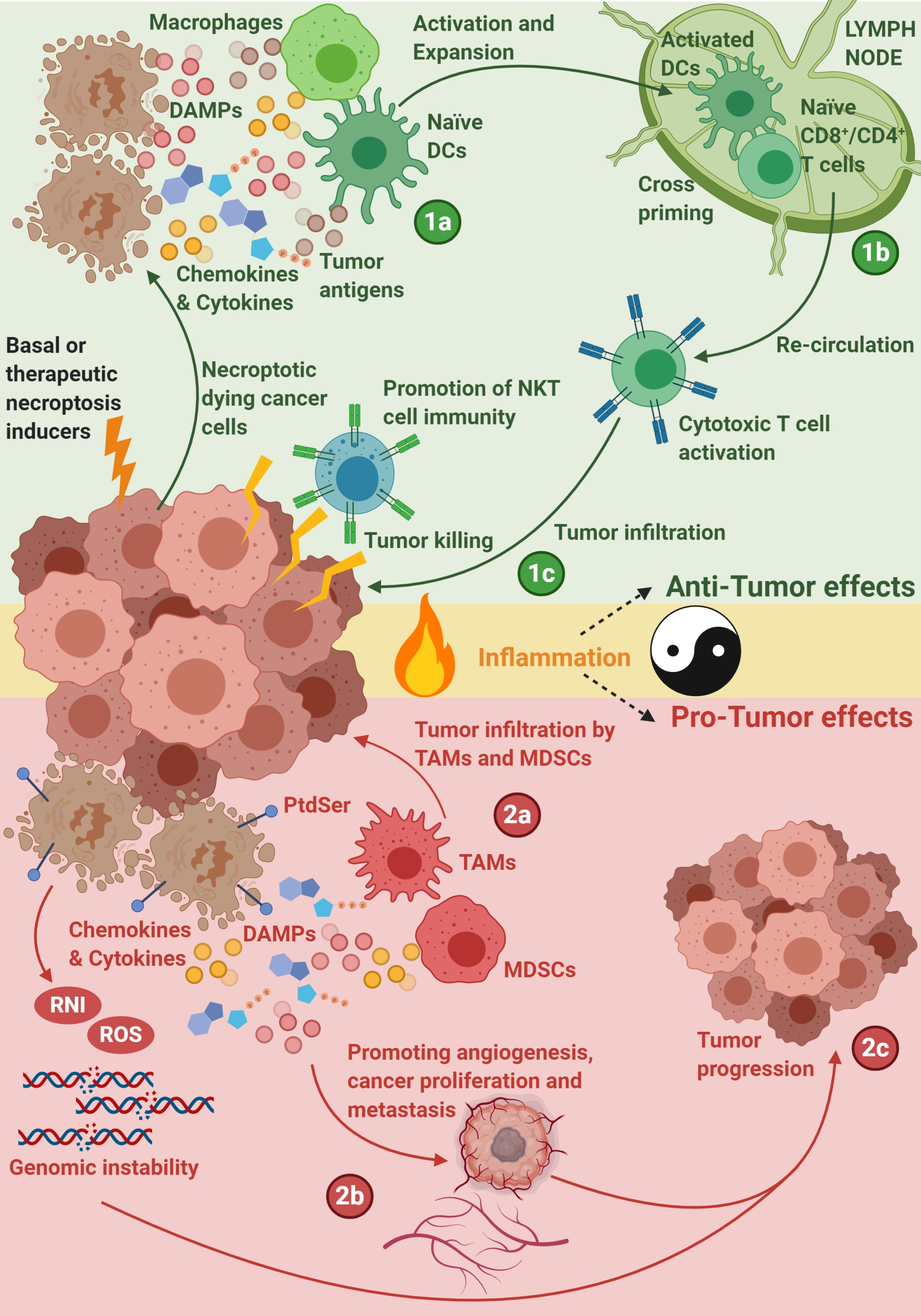
1. Introduction
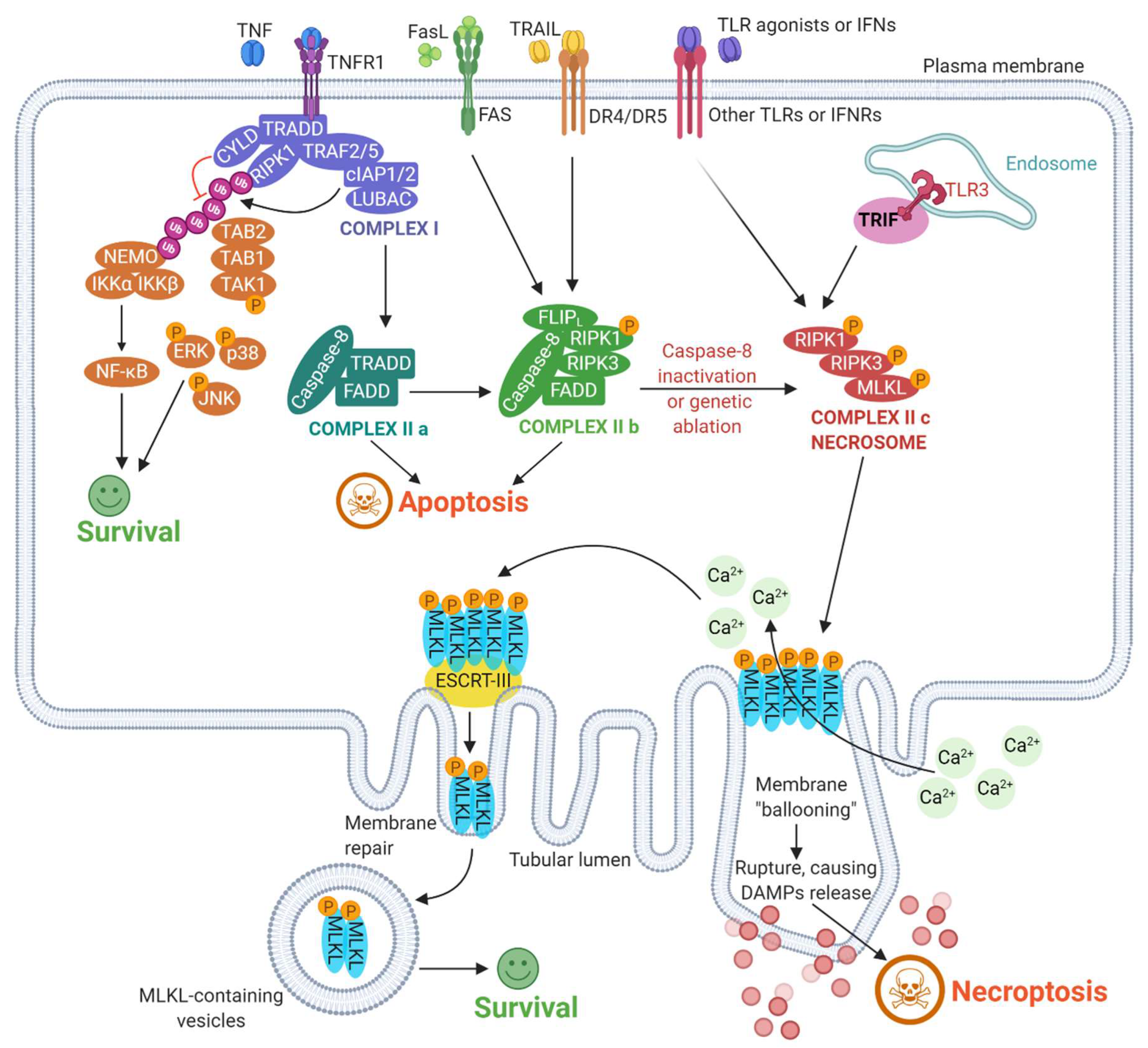
2. Mechanisms Underlying Necroptosis: A Broad Overview
3. Necroptosis and Inflammation
3.1. Necroptosis: Innate Immune Attraction and Phagocytic Clearance
3.2. Necroptosis-Driven Modulation of Immune Responses
4. Necroptosis in Oncology
5. Necroptosis in Immuno-Oncology
5.1. Necroptosis-Driven Modulation of Anticancer Immunity
5.2. Necroptosis and Combinatorial Cancer Immunotherapy
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gotwals, P.; Cameron, S.; Cipolletta, D.; Cremasco, V.; Crystal, A.; Hewes, B.; Mueller, B.; Quaratino, S.; Sabatos-Peyton, C.; Petruzzelli, L.; et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat. Rev. Cancer 2017, 17, 286–301. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Durden, D.L. Combinatorial approach to improve cancer immunotherapy: Rational drug design strategy to simultaneously hit multiple targets to kill tumor cells and to activate the immune system. J. Oncol. 2019, 2019, 5245034. [Google Scholar] [CrossRef] [PubMed]
- Rotte, A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res. 2019, 38, 255. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.K.; Arya, A.; Iams, W.; Cruz, M.R.; Chandra, S.; Choi, J.; Giles, F. Current landscape and future of dual anti-CTLA4 and PD-1/PD-L1 blockade immunotherapy in cancer; lessons learned from clinical trials with melanoma and non-small cell lung cancer (NSCLC). J. Immunother. Cancer 2018, 6, 39. [Google Scholar] [CrossRef]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155. [Google Scholar] [CrossRef]
- Łuksza, M.; Riaz, N.; Makarov, V.; Balachandran, V.P.; Hellmann, M.D.; Solovyov, A.; Rizvi, N.A.; Merghoub, T.; Levine, A.J.; Chan, T.A.; et al. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature 2017, 551, 517–520. [Google Scholar] [CrossRef]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Quaranta, V.; Schmid, M.C. Macrophage-Mediated Subversion of Anti-Tumour Immunity. Cells 2019, 8, 747. [Google Scholar] [CrossRef]
- Linnemann, C.; van Buuren, M.M.; Bies, L.; Verdegaal, E.M.E.; Schotte, R.; Calis, J.J.A.; Behjati, S.; Velds, A.; Hilkmann, H.; Atmioui, D.E.; et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat. Med. 2015, 21, 81–85. [Google Scholar] [CrossRef]
- Kotecki, N.; Awada, A. Checkpoints inhibitors in the (neo)adjuvant setting of solid tumors: Lessons learnt and perspectives. Curr. Opin. Oncol. 2019, 31, 439–444. [Google Scholar] [CrossRef]
- Garg, A.D.; Vandenberk, L.; Van Woensel, M.; Belmans, J.; Schaaf, M.; Boon, L.; De Vleeschouwer, S.; Agostinis, P. Preclinical efficacy of immune-checkpoint monotherapy does not recapitulate corresponding biomarkers-based clinical predictions in glioblastoma. Oncoimmunology 2017, 6, e1295903. [Google Scholar] [CrossRef] [PubMed]
- Dempke, W.C.M.; Fenchel, K.; Uciechowski, P.; Dale, S.P. Second- and third-generation drugs for immuno-oncology treatment-The more the better? Eur. J. Cancer 2017, 74, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.L.; Smothers, J.; Srinivasan, R.; Hoos, A. Big opportunities for small molecules in immuno-oncology. Nat. Rev. Drug Discov. 2015, 14, 603–622. [Google Scholar] [CrossRef] [PubMed]
- Paulson, K.G.; Lahman, M.C.; Chapuis, A.G.; Brownell, I. Immunotherapy for skin cancer. Int. Immunol. 2019, 31, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Crown, J. Biomarkers for Predicting Response to Immunotherapy with Immune Checkpoint Inhibitors in Cancer Patients. Clin. Chem. 2019, 65, 1228–1238. [Google Scholar] [CrossRef] [PubMed]
- Meije, C.B.; Swart, G.W.M.; Lepoole, C.; Das, P.K.; Van den Oord, J.J. Antigenic profiles of individual-matched pairs of primary and melanoma metastases. Hum. Pathol. 2009, 40, 1399–1407. [Google Scholar] [CrossRef]
- Gubin, M.M.; Artyomov, M.N.; Mardis, E.R.; Schreiber, R.D. Tumor neoantigens: Building a framework for personalized cancer immunotherapy. J. Clin. Investig. 2015, 125, 3413–3421. [Google Scholar] [CrossRef]
- Peters, H.L.; Tripathi, S.C.; Kerros, C.; Katayama, H.; Garber, H.R.; St John, L.S.; Federico, L.; Meraz, I.M.; Roth, J.A.; Sepesi, B.; et al. Serine proteases enhance immunogenic antigen presentation on lung cancer cells. Cancer Immunol. Res. 2017, 5, 319–329. [Google Scholar] [CrossRef]
- Chung, E.Y.; Liu, J.; Homma, Y.; Zhang, Y.; Brendolan, A.; Saggese, M.; Han, J.; Silverstein, R.; Selleri, L.; Ma, X. Interleukin-10 expression in macrophages during phagocytosis of apoptotic cells is mediated by homeodomain proteins Pbx1 and Prep-1. Immunity 2007, 27, 952–964. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Spranger, S.; Dai, D.; Horton, B.; Gajewski, T.F. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 2017, 31, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Okoye, I.S.; Xu, L.; Walker, J.; Elahi, S. The glucocorticoids prednisone and dexamethasone differentially modulate T cell function in response to anti-PD-1 and anti-CTLA-4 immune checkpoint blockade. Cancer Immunol. Immunother. 2020, 69, 1423–1436. [Google Scholar] [CrossRef] [PubMed]
- Chikuma, S. CTLA-4, an Essential Immune-Checkpoint for T-Cell Activation. Curr. Top. Microbiol. Immunol. 2017, 410, 99–126. [Google Scholar]
- Wei, S.C.; Levine, J.H.; Cogdill, A.P.; Zhao, Y.; Anang, N.-A.A.S.; Andrews, M.C.; Sharma, P.; Wang, J.; Wargo, J.A.; Pe’er, D.; et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell 2017, 170, 1120–1133. [Google Scholar] [CrossRef]
- Zitvogel, L.; Tesniere, A.; Kroemer, G. Cancer despite immunosurveillance: Immunoselection and immunosubversion. Nat. Rev. Immunol. 2006, 6, 715–727. [Google Scholar] [CrossRef]
- Henrickson, S.E.; Perro, M.; Loughhead, S.M.; Senman, B.; Stutte, S.; Quigley, M.; Alexe, G.; Iannacone, M.; Flynn, M.P.; Omid, S.; et al. Antigen availability determines CD8+ T cell-dendritic cell interaction kinetics and memory fate decisions. Immunity 2013, 39, 496–507. [Google Scholar] [CrossRef]
- Griffith, T.S.; Ferguson, T.A. Cell death in the maintenance and abrogation of tolerance: The five Ws of dying cells. Immunity 2011, 35, 456–466. [Google Scholar] [CrossRef]
- Kreda, S.M.; Pickles, R.J.; Lazarowski, E.R.; Boucher, R.C. G-protein-coupled receptors as targets for gene transfer vectors using natural small-molecule ligands. Nat. Biotechnol. 2000, 18, 635–640. [Google Scholar] [CrossRef]
- Komohara, Y.; Harada, M.; Ishihara, Y.; Suekane, S.; Noguchi, M.; Yamada, A.; Matsuoka, K.; Itoh, K. HLA-G as a target molecule in specific immunotherapy against renal cell carcinoma. Oncol. Rep. 2007, 18, 1463–1468. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kailayangiri, S.; Altvater, B.; Spurny, C.; Jamitzky, S.; Schelhaas, S.; Jacobs, A.H.; Wiek, C.; Roellecke, K.; Hanenberg, H.; Hartmann, W.; et al. Targeting Ewing sarcoma with activated and GD2-specific chimeric antigen receptor-engineered human NK cells induces upregulation of immune-inhibitory HLA-G. Oncoimmunology 2017, 6, e1250050. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vacchelli, E.; Bravo-San Pedro, J.-M.; Buqué, A.; Senovilla, L.; Baracco, E.E.; Bloy, N.; Castoldi, F.; Abastado, J.-P.; Agostinis, P.; et al. Classification of current anticancer immunotherapies. Oncotarget 2014, 5, 12472–12508. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Dudek, A.M.; Agostinis, P. Cancer immunogenicity, danger signals, and DAMPs: What, when, and how? Biofactors 2013, 39, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Blankenstein, T.; Coulie, P.G.; Gilboa, E.; Jaffee, E.M. The determinants of tumour immunogenicity. Nat. Rev. Cancer 2012, 12, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Palucka, K.; Banchereau, J. Cancer immunotherapy via dendritic cells. Nat. Rev. Cancer 2012, 12, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression—Implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Zitvogel, L.; Kroemer, G. The secret ally: Immunostimulation by anticancer drugs. Nat. Rev. Drug Discov. 2012, 11, 215–233. [Google Scholar] [CrossRef]
- Pentimalli, F.; Grelli, S.; Di Daniele, N.; Melino, G.; Amelio, I. Cell death pathologies: Targeting death pathways and the immune system for cancer therapy. Genes Immun. 2019, 20, 539–554. [Google Scholar] [CrossRef]
- Shalapour, S.; Font-Burgada, J.; Di Caro, G.; Zhong, Z.; Sanchez-Lopez, E.; Dhar, D.; Willimsky, G.; Ammirante, M.; Strasner, A.; Hansel, D.E.; et al. Immunosuppressive plasma cells impede T-cell-dependent immunogenic chemotherapy. Nature 2015, 521, 94–98. [Google Scholar] [CrossRef]
- Smyth, M.J.; Ngiow, S.F.; Ribas, A.; Teng, M.W.L. Combination cancer immunotherapies tailored to the tumour microenvironment. Nat. Rev. Clin. Oncol. 2016, 13, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Voorwerk, L.; Slagter, M.; Horlings, H.M.; Sikorska, K.; van de Vijver, K.K.; de Maaker, M.; Nederlof, I.; Kluin, R.J.C.; Warren, S.; Ong, S.; et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: The TONIC trial. Nat. Med. 2019, 25, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Sprooten, J.; Ceusters, J.; Coosemans, A.; Agostinis, P.; De Vleeschouwer, S.; Zitvogel, L.; Kroemer, G.; Galluzzi, L.; Garg, A.D. Trial watch: Dendritic cell vaccination for cancer immunotherapy. Oncoimmunology 2019, 8, e1638212. [Google Scholar] [CrossRef] [PubMed]
- Sadeghzadeh, M.; Bornehdeli, S.; Mohahammadrezakhani, H.; Abolghasemi, M.; Poursaei, E.; Asadi, M.; Zafari, V.; Aghebati-Maleki, L.; Shanehbandi, D. Dendritic cell therapy in cancer treatment; the state-of-the-art. Life Sci. 2020, 254, 117580. [Google Scholar] [CrossRef]
- Lee, J.; Lee, Y.; Xu, L.; White, R.; Sullenger, B.A. Differential Induction of Immunogenic Cell Death and Interferon Expression in Cancer Cells by Structured ssRNAs. Mol. Ther. 2017, 25, 1295–1305. [Google Scholar] [CrossRef]
- Inoue, H.; Tani, K. Multimodal immunogenic cancer cell death as a consequence of anticancer cytotoxic treatments. Cell Death Differ. 2014, 21, 39–49. [Google Scholar] [CrossRef]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef]
- Errington, F.; Jones, J.; Merrick, A.; Bateman, A.; Harrington, K.; Gough, M.; O’Donnell, D.; Selby, P.; Vile, R.; Melcher, A. Fusogenic membrane glycoprotein-mediated tumour cell fusion activates human dendritic cells for enhanced IL-12 production and T-cell priming. Gene Ther. 2006, 13, 138–149. [Google Scholar] [CrossRef]
- Gough, M.J.; Melcher, A.A.; Crittenden, M.R.; Sanchez-Perez, L.; Voellmy, R.; Vile, R.G. Induction of cell stress through gene transfer of an engineered heat shock transcription factor enhances tumor immunogenicity. Gene Ther. 2004, 11, 1099–1104. [Google Scholar] [CrossRef][Green Version]
- Garg, A.D.; Coulie, P.G.; Van den Eynde, B.J.; Agostinis, P. Integrating Next-Generation Dendritic Cell Vaccines into the Current Cancer Immunotherapy Landscape. Trends Immunol. 2017, 38, 577–593. [Google Scholar] [CrossRef]
- Garg, A.D.; Galluzzi, L.; Apetoh, L.; Baert, T.; Birge, R.B.; Bravo-San Pedro, J.M.; Breckpot, K.; Brough, D.; Chaurio, R.; Cirone, M.; et al. Molecular and translational classifications of damps in immunogenic cell death. Front. Immunol. 2015, 6, 588. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Yang, Z.; Xu, Y.; Chen, Y.; Yu, Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol. Cancer 2015, 14, 48. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Warren, S.; Adjemian, S.; Agostinis, P.; Martinez, A.B.; Chan, T.A.; Coukos, G.; Demaria, S.; Deutsch, E.; et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J. Immunother. Cancer 2020, 8, e000337. [Google Scholar] [CrossRef] [PubMed]
- Vanmeerbeek, I.; Sprooten, J.; De Ruysscher, D.; Tejpar, S.; Vandenberghe, P.; Fucikova, J.; Spisek, R.; Zitvogel, L.; Kroemer, G.; Galluzzi, L.; et al. Trial watch: Chemotherapy-induced immunogenic cell death in immuno-oncology. Oncoimmunology 2020, 9, 1703449. [Google Scholar] [CrossRef]
- van Vloten, J.P.; Workenhe, S.T.; Wootton, S.K.; Mossman, K.L.; Bridle, B.W. Critical Interactions between Immunogenic Cancer Cell Death, Oncolytic Viruses, and the Immune System Define the Rational Design of Combination Immunotherapies. J. Immunol. 2018, 200, 450–458. [Google Scholar] [CrossRef]
- Turubanova, V.D.; Balalaeva, I.V.; Mishchenko, T.A.; Catanzaro, E.; Alzeibak, R.; Peskova, N.N.; Efimova, I.; Bachert, C.; Mitroshina, E.V.; Krysko, O.; et al. Immunogenic cell death induced by a new photodynamic therapy based on photosens and photodithazine. J. Immunother. Cancer 2019, 7, 350. [Google Scholar] [CrossRef]
- Sprooten, J.; Agostinis, P.; Garg, A.D. Type I Interferons and Dendritic Cells in Cancer Immunotherapy. Int. Rev. Cell Mol. Biol. 2019, 348, 217–262. [Google Scholar]
- Garg, A.D.; Agostinis, P. Cell death and immunity in cancer: From danger signals to mimicry of pathogen defense responses. Immunol. Rev. 2017, 280, 126–148. [Google Scholar] [CrossRef]
- Garg, A.D.; Vandenberk, L.; Fang, S.; Fasche, T.; Van Eygen, S.; Maes, J.; Van Woensel, M.; Koks, C.; Vanthillo, N.; Graf, N.; et al. Pathogen response-like recruitment and activation of neutrophils by sterile immunogenic dying cells drives neutrophil-mediated residual cell killing. Cell Death Differ. 2017, 24, 832–843. [Google Scholar] [CrossRef]
- Ravichandran, K.S. Beginnings of a good apoptotic meal: The find-me and eat-me signaling pathways. Immunity 2011, 35, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Vanpouille-Box, C.; Demaria, S.; Galluzzi, L. Immunogenic Cell Death Driven by Radiation-Impact on the Tumor Microenvironment. Cancer Treat. Res. 2020, 180, 281–296. [Google Scholar] [PubMed]
- Aaes, T.L.; Kaczmarek, A.; Delvaeye, T.; De Craene, B.; De Koker, S.; Heyndrickx, L.; Delrue, I.; Taminau, J.; Wiernicki, B.; De Groote, P.; et al. Vaccination with Necroptotic Cancer Cells Induces Efficient Anti-tumor Immunity. Cell Rep. 2016, 15, 274–287. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef]
- Kaczmarek, A.; Vandenabeele, P.; Krysko, D.V. Necroptosis: The release of damage-associated molecular patterns and its physiological relevance. Immunity 2013, 38, 209–223. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Fulda, S. Targeting apoptosis for anticancer therapy. Semin. Cancer Biol. 2015, 31, 84–88. [Google Scholar] [CrossRef]
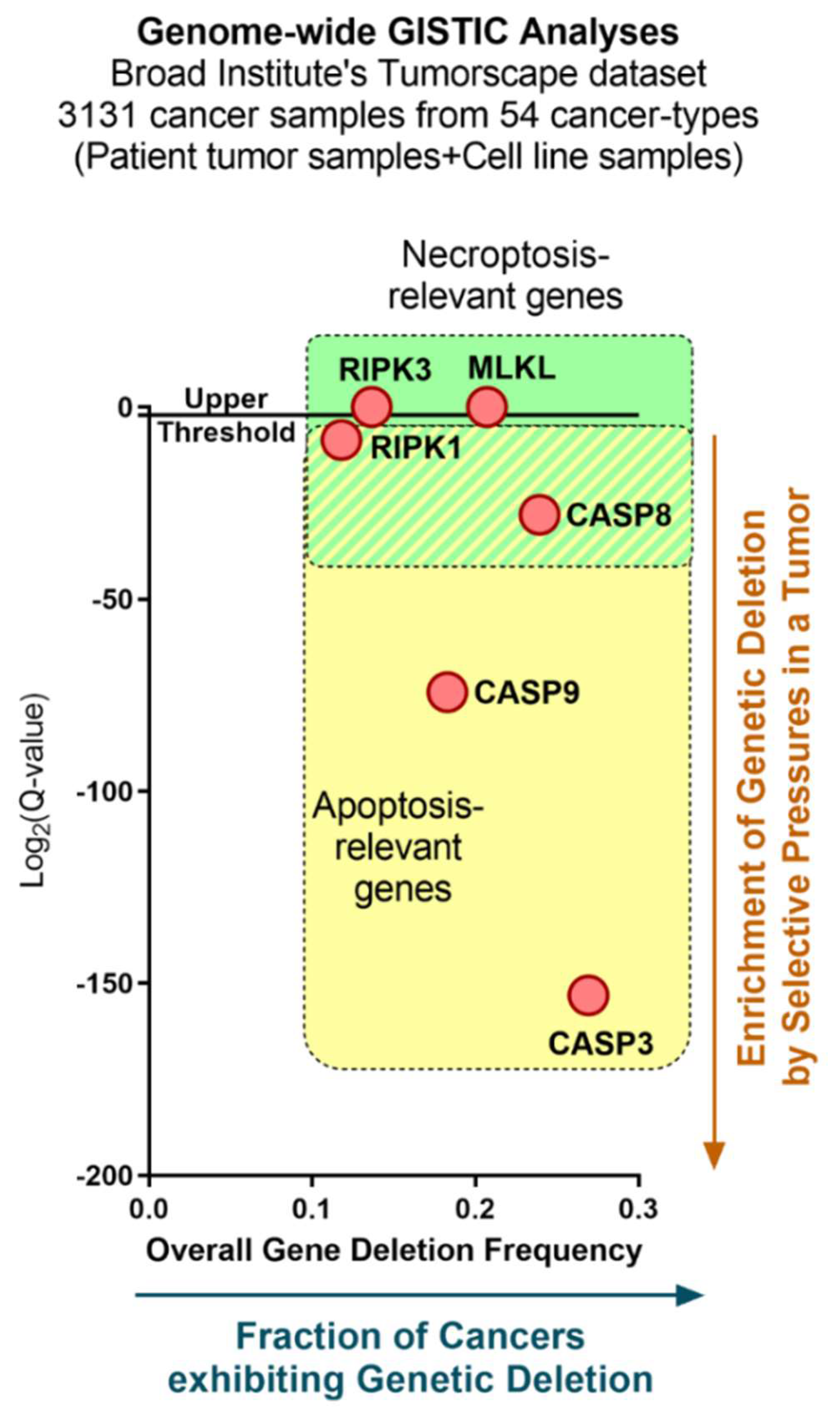
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 12, R41. [Google Scholar] [CrossRef]
- Chan, F.K.-M. Fueling the flames: Mammalian programmed necrosis in inflammatory diseases. Cold Spring Harb. Perspect. Biol. 2012, 4, a008805. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Krysko, D.V.; Conrad, M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat. Rev. Cancer 2019, 19, 405–414. [Google Scholar] [CrossRef]
- Xia, X.; Wang, X.; Cheng, Z.; Qin, W.; Lei, L.; Jiang, J.; Hu, J. The role of pyroptosis in cancer: Pro-cancer or pro-“host”? Cell Death Dis. 2019, 10, 650. [Google Scholar] [CrossRef] [PubMed]
- Vanden Berghe, T.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Minagawa, S.; Yoshida, M.; Araya, J.; Hara, H.; Imai, H.; Kuwano, K. Regulated necrosis in pulmonary disease: A focus on necroptosis and ferroptosis. Am. J. Respir. Cell Mol. Biol. 2020, 62, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Nair, P.; Lu, M.; Petersen, S.; Ashkenazi, A. Apoptosis initiation through the cell-extrinsic pathway. Meth. Enzymol. 2014, 544, 99–128. [Google Scholar]
- Fulda, S.; Debatin, K.M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef]
- Fritsch, M.; Günther, S.D.; Schwarzer, R.; Albert, M.-C.; Schorn, F.; Werthenbach, J.P.; Schiffmann, L.M.; Stair, N.; Stocks, H.; Seeger, J.M.; et al. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature 2019, 575, 683–687. [Google Scholar] [CrossRef]
- Shlomovitz, I.; Zargarian, S.; Erlich, Z.; Edry-Botzer, L.; Gerlic, M. Distinguishing Necroptosis from Apoptosis. Methods Mol. Biol. 2018, 1857, 35–51. [Google Scholar]
- Vanden Berghe, T.; Kaiser, W.J.; Bertrand, M.J.; Vandenabeele, P. Molecular crosstalk between apoptosis, necroptosis, and survival signaling. Mol. Cell. Oncol. 2015, 2, e975093. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Krautwald, S.; Kroemer, G.; Linkermann, A. Molecular mechanisms of regulated necrosis. Semin. Cell Dev. Biol. 2014, 35, 24–32. [Google Scholar] [CrossRef]
- Thomas, L.R.; Johnson, R.L.; Reed, J.C.; Thorburn, A. The C-terminal tails of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and Fas receptors have opposing functions in Fas-associated death domain (FADD) recruitment and can regulate agonist-specific mechanisms of receptor activation. J. Biol. Chem. 2004, 279, 52479–52486. [Google Scholar] [CrossRef]
- Schütze, S.; Tchikov, V.; Schneider-Brachert, W. Regulation of TNFR1 and CD95 signalling by receptor compartmentalization. Nat. Rev. Mol. Cell Biol. 2008, 9, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Grootjans, S.; Vanden Berghe, T.; Vandenabeele, P. Initiation and execution mechanisms of necroptosis: An overview. Cell Death Differ. 2017, 24, 1184–1195. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Gonzalez, G.; Vandenabeele, P.; Krysko, D.V. Necroptosis: A novel cell death modality and its potential relevance for critical care medicine. Am. J. Respir. Crit. Care Med. 2016, 194, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, D.A.; Weinlich, R.; Brown, S.; Guy, C.; Fitzgerald, P.; Dillon, C.P.; Oberst, A.; Quarato, G.; Low, J.; Cripps, J.G.; et al. Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ. 2016, 23, 76–88. [Google Scholar] [CrossRef]
- Moujalled, D.M.; Cook, W.D.; Murphy, J.M.; Vaux, D.L. Necroptosis induced by RIPK3 requires MLKL but not Drp1. Cell Death Dis. 2014, 5, e1086. [Google Scholar] [CrossRef]
- Dondelinger, Y.; Darding, M.; Bertrand, M.J.M.; Walczak, H. Poly-ubiquitination in TNFR1-mediated necroptosis. Cell Mol. Life Sci. 2016, 73, 2165–2176. [Google Scholar] [CrossRef]
- Vercammen, D.; Brouckaert, G.; Denecker, G.; Van deaaa 1Craen, M.; Declercq, W.; Fiers, W.; Vandenabeele, P. Dual signaling of the Fas receptor: Initiation of both apoptotic and necrotic cell death pathways. J. Exp. Med. 1998, 188, 919–930. [Google Scholar] [CrossRef]
- Vercammen, D.; Beyaert, R.; Denecker, G.; Goossens, V.; Van Loo, G.; Declercq, W.; Grooten, J.; Fiers, W.; Vandenabeele, P. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J. Exp. Med. 1998, 187, 1477–1485. [Google Scholar] [CrossRef]
- Liu, X.; Shi, F.; Li, Y.; Yu, X.; Peng, S.; Li, W.; Luo, X.; Cao, Y. Post-translational modifications as key regulators of TNF-induced necroptosis. Cell Death Dis. 2016, 7, e2293. [Google Scholar] [CrossRef]
- Declercq, W.; Denecker, G.; Fiers, W.; Vandenabeele, P. Cooperation of both TNF receptors in inducing apoptosis: Involvement of the TNF receptor-associated factor binding domain of the TNF receptor 75. J. Immunol. 1998, 161, 390–399. [Google Scholar]
- Hsu, H.; Shu, H.B.; Pan, M.G.; Goeddel, D.V. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell 1996, 84, 299–308. [Google Scholar] [CrossRef]
- Zheng, L.; Bidere, N.; Staudt, D.; Cubre, A.; Orenstein, J.; Chan, F.K.; Lenardo, M. Competitive control of independent programs of tumor necrosis factor receptor-induced cell death by TRADD and RIP1. Mol. Cell. Biol. 2006, 26, 3505–3513. [Google Scholar] [CrossRef] [PubMed]
- Jackson-Bernitsas, D.G.; Ichikawa, H.; Takada, Y.; Myers, J.N.; Lin, X.L.; Darnay, B.G.; Chaturvedi, M.M.; Aggarwal, B.B. Evidence that TNF-TNFR1-TRADD-TRAF2-RIP-TAK1-IKK pathway mediates constitutive NF-kappaB activation and proliferation in human head and neck squamous cell carcinoma. Oncogene 2007, 26, 1385–1397. [Google Scholar] [CrossRef] [PubMed]
- Dhuriya, Y.K.; Sharma, D. Necroptosis: A regulated inflammatory mode of cell death. J. Neuroinflammation 2018, 15, 199. [Google Scholar] [CrossRef]
- Fulda, S.; Gorman, A.M.; Hori, O.; Samali, A. Cellular stress responses: Cell survival and cell death. Int. J. Cell Biol. 2010, 2010, 214074. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef]
- Huang, X.; Xiao, F.; Li, Y.; Qian, W.; Ding, W.; Ye, X. Bypassing drug resistance by triggering necroptosis: Recent advances in mechanisms and its therapeutic exploitation in leukemia. J. Exp. Clin. Cancer Res. 2018, 37, 310. [Google Scholar] [CrossRef]
- Li, H.; Kobayashi, M.; Blonska, M.; You, Y.; Lin, X. Ubiquitination of RIP is required for tumor necrosis factor alpha-induced NF-kappaB activation. J. Biol. Chem. 2006, 281, 13636–13643. [Google Scholar] [CrossRef]
- Ea, C.-K.; Deng, L.; Xia, Z.-P.; Pineda, G.; Chen, Z.J. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol. Cell 2006, 22, 245–257. [Google Scholar] [CrossRef]
- Micheau, O.; Lens, S.; Gaide, O.; Alevizopoulos, K.; Tschopp, J. NF-kappaB signals induce the expression of c-FLIP. Mol. Cell. Biol. 2001, 21, 5299–5305. [Google Scholar] [CrossRef]
- Dondelinger, Y.; Vandenabeele, P.; Bertrand, M.J. Regulation of RIPK1’s cell death function by phosphorylation. Cell Cycle 2016, 15, 5–6. [Google Scholar] [CrossRef] [PubMed]
- In, E.-J.; Lee, Y.; Koppula, S.; Kim, T.-Y.; Han, J.-H.; Lee, K.-H.; Kang, T.-B. Identification and characterization of NTB451 as a potential inhibitor of necroptosis. Molecules 2018, 23, 2884. [Google Scholar] [CrossRef] [PubMed]
- Bozec, D.; Iuga, A.C.; Roda, G.; Dahan, S.; Yeretssian, G. Critical function of the necroptosis adaptor RIPK3 in protecting from intestinal tumorigenesis. Oncotarget 2016, 7, 46384–46400. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Zhang, A.; Choksi, S.; Li, W.; Li, T.; Zhang, X.-M.; Liu, Z.-G. Activation of cell-surface proteases promotes necroptosis, inflammation and cell migration. Cell Res. 2016, 26, 886–900. [Google Scholar] [CrossRef]
- O’Donnell, M.A.; Perez-Jimenez, E.; Oberst, A.; Ng, A.; Massoumi, R.; Xavier, R.; Green, D.R.; Ting, A.T. Caspase 8 inhibits programmed necrosis by processing CYLD. Nat. Cell Biol. 2011, 13, 1437–1442. [Google Scholar] [CrossRef]
- Li, J.; McQuade, T.; Siemer, A.B.; Napetschnig, J.; Moriwaki, K.; Hsiao, Y.-S.; Damko, E.; Moquin, D.; Walz, T.; McDermott, A.; et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 2012, 150, 339–350. [Google Scholar] [CrossRef]
- Murphy, J.M.; Silke, J. Ars Moriendi; the art of dying well—New insights into the molecular pathways of necroptotic cell death. EMBO Rep. 2014, 15, 155–164. [Google Scholar] [CrossRef]
- Remijsen, Q.; Goossens, V.; Grootjans, S.; Van den Haute, C.; Vanlangenakker, N.; Dondelinger, Y.; Roelandt, R.; Bruggeman, I.; Goncalves, A.; Bertrand, M.J.M.; et al. Depletion of RIPK3 or MLKL blocks TNF-driven necroptosis and switches towards a delayed RIPK1 kinase-dependent apoptosis. Cell Death Dis. 2014, 5, e1004. [Google Scholar] [CrossRef]
- Dillon, C.P.; Weinlich, R.; Rodriguez, D.A.; Cripps, J.G.; Quarato, G.; Gurung, P.; Verbist, K.C.; Brewer, T.L.; Llambi, F.; Gong, Y.-N.; et al. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell 2014, 157, 1189–1202. [Google Scholar] [CrossRef]
- Wegner, K.W.; Saleh, D.; Degterev, A. Complex pathologic roles of RIPK1 and RIPK3: Moving beyond necroptosis. Trends Pharmacol. Sci. 2017, 38, 202–225. [Google Scholar] [CrossRef]
- Green, D.R. The coming decade of cell death research: Five riddles. Cell 2019, 177, 1094–1107. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.-N.; Crawford, J.C.; Heckmann, B.L.; Green, D.R. To the edge of cell death and back. FEBS J. 2019, 286, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Tummers, B.; Green, D.R. Caspase-8: Regulating life and death. Immunol. Rev. 2017, 277, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.M.; Czabotar, P.E.; Hildebrand, J.M.; Lucet, I.S.; Zhang, J.-G.; Alvarez-Diaz, S.; Lewis, R.; Lalaoui, N.; Metcalf, D.; Webb, A.I.; et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 2013, 39, 443–453. [Google Scholar] [CrossRef]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K.-M. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef]
- Dondelinger, Y.; Declercq, W.; Montessuit, S.; Roelandt, R.; Goncalves, A.; Bruggeman, I.; Hulpiau, P.; Weber, K.; Sehon, C.A.; Marquis, R.W.; et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014, 7, 971–981. [Google Scholar] [CrossRef]
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.-C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.-G.; Liu, Z.-G. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 2014, 16, 55–65. [Google Scholar] [CrossRef]
- Tanzer, M.C.; Matti, I.; Hildebrand, J.M.; Young, S.N.; Wardak, A.; Tripaydonis, A.; Petrie, E.J.; Mildenhall, A.L.; Vaux, D.L.; Vince, J.E.; et al. Evolutionary divergence of the necroptosis effector MLKL. Cell Death Differ. 2016, 23, 1185–1197. [Google Scholar] [CrossRef]
- Petrie, E.J.; Birkinshaw, R.W.; Koide, A.; Denbaum, E.; Hildebrand, J.M.; Garnish, S.E.; Davies, K.A.; Sandow, J.J.; Samson, A.L.; Gavin, X.; et al. Identification of MLKL membrane translocation as a checkpoint in necroptotic cell death using Monobodies. Proc. Natl. Acad. Sci. USA 2020, 117, 8468–8475. [Google Scholar] [CrossRef]
- Schenk, B.; Fulda, S. Reactive oxygen species regulate Smac mimetic/TNFα-induced necroptotic signaling and cell death. Oncogene 2015, 34, 5796–5806. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Fang, S.; Chen, X.; Hu, H.; Chen, P.; Wang, H.; Gao, Z. MLKL forms cation channels. Cell Res. 2016, 26, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Madan, E.; Sayyid, M.; Arias-Pulido, H.; Moreno, E.; Kuppusamy, P.; Gogna, R. Oxygen regulates molecular mechanisms of cancer progression and metastasis. Cancer Metastasis Rev. 2014, 33, 183–215. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.-N.; Guy, C.; Olauson, H.; Becker, J.U.; Yang, M.; Fitzgerald, P.; Linkermann, A.; Green, D.R. ESCRT-III Acts Downstream of MLKL to Regulate Necroptotic Cell Death and Its Consequences. Cell 2017, 169, 286–300. [Google Scholar] [CrossRef]
- Svensson, A.; Sandberg, T.; Siesjö, P.; Eriksson, H. Sequestering of damage-associated molecular patterns (DAMPs): A possible mechanism affecting the immune-stimulating properties of aluminium adjuvants. Immunol. Res. 2017, 65, 1164–1175. [Google Scholar] [CrossRef]
- Vénéreau, E.; Ceriotti, C.; Bianchi, M.E. DAMPs from Cell Death to New Life. Front. Immunol. 2015, 6, 422. [Google Scholar] [CrossRef]
- Krysko, O.; Løve Aaes, T.; Bachert, C.; Vandenabeele, P.; Krysko, D.V. Many faces of DAMPs in cancer therapy. Cell Death Dis. 2013, 4, e631. [Google Scholar] [CrossRef]
- Chen, J.; Kos, R.; Garssen, J.; Redegeld, F. Molecular Insights into the Mechanism of Necroptosis: The Necrosome as a Potential Therapeutic Target. Cells 2019, 8, 1486. [Google Scholar] [CrossRef]
- Saelens, X.; Festjens, N.; Parthoens, E.; Vanoverberghe, I.; Kalai, M.; van Kuppeveld, F.; Vandenabeele, P. Protein synthesis persists during necrotic cell death. J. Cell Biol. 2005, 168, 545–551. [Google Scholar] [CrossRef]
- Vanden Berghe, T.; Kalai, M.; Denecker, G.; Meeus, A.; Saelens, X.; Vandenabeele, P. Necrosis is associated with IL-6 production but apoptosis is not. Cell Signal. 2006, 18, 328–335. [Google Scholar] [CrossRef]
- Orozco, S.L.; Daniels, B.P.; Yatim, N.; Messmer, M.N.; Quarato, G.; Chen-Harris, H.; Cullen, S.P.; Snyder, A.G.; Ralli-Jain, P.; Frase, S.; et al. RIPK3 Activation Leads to Cytokine Synthesis that Continues after Loss of Cell Membrane Integrity. Cell Rep. 2019, 28, 2275–2287. [Google Scholar] [CrossRef] [PubMed]
- Sprooten, J.; Garg, A.D. Type I interferons and endoplasmic reticulum stress in health and disease. Int. Rev. Cell Mol. Biol. 2020, 350, 63–118. [Google Scholar] [PubMed]
- Medina, C.B.; Ravichandran, K.S. Do not let death do us part: “find-me” signals in communication between dying cells and the phagocytes. Cell Death Differ. 2016, 23, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, K.S. Find-me and eat-me signals in apoptotic cell clearance: Progress and conundrums. J. Exp. Med. 2010, 207, 1807–1817. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; D’Herde, K.; Vandenabeele, P. Clearance of apoptotic and necrotic cells and its immunological consequences. Apoptosis 2006, 11, 1709–1726. [Google Scholar] [CrossRef]
- Fond, A.M.; Ravichandran, K.S. Clearance of dying cells by phagocytes: Mechanisms and implications for disease pathogenesis. Adv. Exp. Med. Biol. 2016, 930, 25–49. [Google Scholar]
- Lu, J.; Shi, W.; Liang, B.; Chen, C.; Wu, R.; Lin, H.; Zhang, Y.; Han, J. Efficient engulfment of necroptotic and pyroptotic cells by nonprofessional and professional phagocytes. Cell Discov. 2019, 5, 39. [Google Scholar] [CrossRef]
- Fang, S.; Agostinis, P.; Salven, P.; Garg, A.D. Decoding cancer cell death-driven immune cell recruitment: An in vivo method for site-of-vaccination analyses. Meth. Enzymol. 2020, 636, 185–207. [Google Scholar]
- McCracken, M.N.; Cha, A.C.; Weissman, I.L. Molecular Pathways: Activating T Cells after Cancer Cell Phagocytosis from Blockade of CD47 “Don’t Eat Me” Signals. Clin. Cancer Res. 2015, 21, 3597–3601. [Google Scholar] [CrossRef]
- Arandjelovic, S.; Ravichandran, K.S. Phagocytosis of apoptotic cells in homeostasis. Nat. Immunol. 2015, 16, 907–917. [Google Scholar] [CrossRef]
- Tyurin, V.A.; Balasubramanian, K.; Winnica, D.; Tyurina, Y.Y.; Vikulina, A.S.; He, R.R.; Kapralov, A.A.; Macphee, C.H.; Kagan, V.E. Oxidatively modified phosphatidylserines on the surface of apoptotic cells are essential phagocytic “eat-me” signals: Cleavage and inhibition of phagocytosis by Lp-PLA2. Cell Death Differ. 2014, 21, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Segawa, K.; Nagata, S. An apoptotic “eat me” signal: Phosphatidylserine exposure. Trends Cell Biol. 2015, 25, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.S.A.; Morioka, S.; Medina, C.B.; Iker Etchegaray, J.; Barron, B.; Raymond, M.H.; Lucas, C.D.; Onengut-Gumuscu, S.; Delpire, E.; Ravichandran, K.S. Interpreting an apoptotic corpse as anti-inflammatory involves a chloride sensing pathway. Nat. Cell Biol. 2019, 21, 1532–1543. [Google Scholar] [CrossRef]
- Peter, C.; Wesselborg, S.; Herrmann, M.; Lauber, K. Dangerous attraction: Phagocyte recruitment and danger signals of apoptotic and necrotic cells. Apoptosis 2010, 15, 1007–1028. [Google Scholar] [CrossRef]
- Zhu, K.; Liang, W.; Ma, Z.; Xu, D.; Cao, S.; Lu, X.; Liu, N.; Shan, B.; Qian, L.; Yuan, J. Necroptosis promotes cell-autonomous activation of proinflammatory cytokine gene expression. Cell Death Dis. 2018, 9, 500. [Google Scholar] [CrossRef]
- Kearney, C.J.; Martin, S.J. An inflammatory perspective on necroptosis. Mol. Cell 2017, 65, 965–973. [Google Scholar] [CrossRef]
- Wang, Q.; Ju, X.; Zhou, Y.; Chen, K. Necroptotic cells release find-me signal and are engulfed without proinflammatory cytokine production. In Vitro Cell. Dev. Biol. Anim. 2015, 51, 1033–1039. [Google Scholar] [CrossRef]
- Devitt, A.; Marshall, L.J. The innate immune system and the clearance of apoptotic cells. J. Leukoc. Biol. 2011, 90, 447–457. [Google Scholar] [CrossRef]
- Lolmede, K.; Campana, L.; Vezzoli, M.; Bosurgi, L.; Tonlorenzi, R.; Clementi, E.; Bianchi, M.E.; Cossu, G.; Manfredi, A.A.; Brunelli, S.; et al. Inflammatory and alternatively activated human macrophages attract vessel-associated stem cells, relying on separate HMGB1- and MMP-9-dependent pathways. J. Leukoc. Biol. 2009, 85, 779–787. [Google Scholar] [CrossRef]
- Kimura, T.; Kobayashi, S.; Hanihara-Tatsuzawa, F.; Sayama, A.; MaruYama, T.; Muta, T. Responses of macrophages to the danger signals released from necrotic cells. Int. Immunol. 2014, 26, 697–704. [Google Scholar] [CrossRef]
- Dankers, W.; Hasnat, M.A.; Swann, V.; Alharbi, A.; Lee, J.P.W.; Cristofaro, M.A.; Gantier, M.P.; Jones, S.A.; Morand, E.F.; Flynn, J.K.; et al. Necrotic cell death increases the release of Macrophage migration inhibitory factor (MIF) by monocytes/macrophages. Immunol. Cell Biol. 2020. [Google Scholar] [CrossRef]
- Krysko, D.V.; Denecker, G.; Festjens, N.; Gabriels, S.; Parthoens, E.; D’Herde, K.; Vandenabeele, P. Macrophages use different internalization mechanisms to clear apoptotic and necrotic cells. Cell Death Differ. 2006, 13, 2011–2022. [Google Scholar] [CrossRef]
- Krieser, R.J.; White, K. Engulfment mechanism of apoptotic cells. Curr. Opin. Cell Biol. 2002, 14, 734–738. [Google Scholar] [CrossRef]
- Cao, L.; Chang, H.; Shi, X.; Peng, C.; He, Y. Keratin mediates the recognition of apoptotic and necrotic cells through dendritic cell receptor DEC205/CD205. Proc. Natl. Acad. Sci. USA 2016, 113, 13438–13443. [Google Scholar] [CrossRef]
- Ahrens, S.; Zelenay, S.; Sancho, D.; Hanč, P.; Kjær, S.; Feest, C.; Fletcher, G.; Durkin, C.; Postigo, A.; Skehel, M.; et al. F-actin is an evolutionarily conserved damage-associated molecular pattern recognized by DNGR-1, a receptor for dead cells. Immunity 2012, 36, 635–645. [Google Scholar] [CrossRef]
- Zargarian, S.; Shlomovitz, I.; Erlich, Z.; Hourizadeh, A.; Ofir-Birin, Y.; Croker, B.A.; Regev-Rudzki, N.; Edry-Botzer, L.; Gerlic, M. Phosphatidylserine externalization, “necroptotic bodies” release, and phagocytosis during necroptosis. PLoS Biol. 2017, 15, e2002711. [Google Scholar] [CrossRef]
- Klöditz, K.; Fadeel, B. Three cell deaths and a funeral: Macrophage clearance of cells undergoing distinct modes of cell death. Cell Death Discov. 2019, 5, 65. [Google Scholar] [CrossRef]
- Brouckaert, G.; Kalai, M.; Krysko, D.V.; Saelens, X.; Vercammen, D.; Ndlovu, M.N.; Haegeman, G.; D’Herde, K.; Vandenabeele, P. Phagocytosis of necrotic cells by macrophages is phosphatidylserine dependent and does not induce inflammatory cytokine production. Mol. Biol. Cell 2004, 15, 1089–1100. [Google Scholar] [CrossRef]
- Gerlach, B.D.; Marinello, M.; Heinz, J.; Rymut, N.; Sansbury, B.E.; Riley, C.O.; Sadhu, S.; Hosseini, Z.; Kojima, Y.; Tang, D.D.; et al. Resolvin D1 promotes the targeting and clearance of necroptotic cells. Cell Death Differ. 2020, 27, 525–539. [Google Scholar] [CrossRef]
- Rock, K.L.; Kono, H. The inflammatory response to cell death. Annu. Rev. Pathol. 2008, 3, 99–126. [Google Scholar] [CrossRef]
- Oppenheim, J.J.; Yang, D. Alarmins: Chemotactic activators of immune responses. Curr. Opin. Immunol. 2005, 17, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Chiba, S.; Baghdadi, M.; Akiba, H.; Yoshiyama, H.; Kinoshita, I.; Dosaka-Akita, H.; Fujioka, Y.; Ohba, Y.; Gorman, J.V.; Colgan, J.D.; et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat. Immunol. 2012, 13, 832–842. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.; Loh, Z.; Ullah, M.A.; Lynch, J.P.; Werder, R.B.; Collinson, N.; Zhang, V.; Dondelinger, Y.; Bertrand, M.J.M.; Everard, M.L.; et al. RSV infection promotes necroptosis and HMGB1 release by airway epithelial cells. Am. J. Respir. Crit. Care Med. 2020, 201, 1358–1371. [Google Scholar] [CrossRef]
- Xu, Z.; Jin, Y.; Yan, H.; Gao, Z.; Xu, B.; Yang, B.; He, Q.; Shi, Q.; Luo, P. High-mobility group box 1 protein-mediated necroptosis contributes to dasatinib-induced cardiotoxicity. Toxicol. Lett. 2018, 296, 39–47. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef]
- Yatim, N.; Jusforgues-Saklani, H.; Orozco, S.; Schulz, O.; Barreira da Silva, R.; Reis e Sousa, C.; Green, D.R.; Oberst, A.; Albert, M.L. RIPK1 and NF-κB signaling in dying cells determines cross-priming of CD8+ T cells. Science 2015, 350, 328–334. [Google Scholar] [CrossRef]
- Chen, G.Y.; Nuñez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef]
- Man, S.M.; Kanneganti, T.-D. Regulation of inflammasome activation. Immunol. Rev. 2015, 265, 6–21. [Google Scholar] [CrossRef]
- Knuth, A.-K.; Rösler, S.; Schenk, B.; Kowald, L.; van Wijk, S.J.L.; Fulda, S. Interferons Transcriptionally Up-Regulate MLKL Expression in Cancer Cells. Neoplasia 2019, 21, 74–81. [Google Scholar] [CrossRef]
- Land, W.G.; Agostinis, P.; Gasser, S.; Garg, A.D.; Linkermann, A. DAMP-Induced Allograft and Tumor Rejection: The Circle Is Closing. Am. J. Transplant. 2016, 16, 3322–3337. [Google Scholar] [CrossRef]
- Duprez, L.; Takahashi, N.; Van Hauwermeiren, F.; Vandendriessche, B.; Goossens, V.; Vanden Berghe, T.; Declercq, W.; Libert, C.; Cauwels, A.; Vandenabeele, P. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity 2011, 35, 908–918. [Google Scholar] [CrossRef] [PubMed]
- Wauters, E.; Thevissen, K.; Wouters, C.; Bosisio, F.M.; De Smet, F.; Gunst, J.; Humblet-Baron, S.; Lambrechts, D.; Liston, A.; Matthys, P.; et al. Establishing a Unified COVID-19 “Immunome”: Integrating Coronavirus Pathogenesis and Host Immunopathology. Front. Immunol. 2020, 11, 1642. [Google Scholar] [CrossRef] [PubMed]
- Vandenberk, L.; Garg, A.D.; Verschuere, T.; Koks, C.; Belmans, J.; Beullens, M.; Agostinis, P.; De Vleeschouwer, S.; Van Gool, S.W. Irradiation of necrotic cancer cells, employed for pulsing dendritic cells (DCs), potentiates DC vaccine-induced antitumor immunity against high-grade glioma. Oncoimmunology 2016, 5, e1083669. [Google Scholar] [CrossRef] [PubMed]
- Cohen, I.; Rider, P.; Carmi, Y.; Braiman, A.; Dotan, S.; White, M.R.; Voronov, E.; Martin, M.U.; Dinarello, C.A.; Apte, R.N. Differential release of chromatin-bound IL-1alpha discriminates between necrotic and apoptotic cell death by the ability to induce sterile inflammation. Proc. Natl. Acad. Sci. USA 2010, 107, 2574–2579. [Google Scholar] [CrossRef] [PubMed]
- Wauters, E.; Van Mol, P.; Garg, A.D.; Jansen, S.; Van Herck, Y.; Vanderbeke, L.; Bassez, A.; Boeckx, B.; Malengier-Devlies, B.; Timmerman, A.; et al. Discriminating Mild from Critical COVID-19 by Innate and Adaptive Immune Single-cell Profiling of Bronchoalveolar Lavages. BioRxiv 2020. [Google Scholar] [CrossRef]
- Richards, C.H.; Mohammed, Z.; Qayyum, T.; Horgan, P.G.; McMillan, D.C. The prognostic value of histological tumor necrosis in solid organ malignant disease: A systematic review. Future Oncol. 2011, 7, 1223–1235. [Google Scholar] [CrossRef] [PubMed]
- Foria, V.; Surendra, T.; Poller, D.N. Prognostic relevance of extensive necrosis in renal cell carcinoma. J. Clin. Pathol. 2005, 58, 39–43. [Google Scholar] [CrossRef]
- Hiraoka, N.; Ino, Y.; Sekine, S.; Tsuda, H.; Shimada, K.; Kosuge, T.; Zavada, J.; Yoshida, M.; Yamada, K.; Koyama, T.; et al. Tumour necrosis is a postoperative prognostic marker for pancreatic cancer patients with a high interobserver reproducibility in histological evaluation. Br. J. Cancer 2010, 103, 1057–1065. [Google Scholar] [CrossRef]
- Chan, F.K.M.; Moriwaki, K.; De Rosa, M.J. Detection of necrosis by release of lactate dehydrogenase activity. Methods Mol. Biol. 2013, 979, 65–70. [Google Scholar]
- Armstrong, A.J.; George, D.J.; Halabi, S. Serum lactate dehydrogenase predicts for overall survival benefit in patients with metastatic renal cell carcinoma treated with inhibition of mammalian target of rapamycin. J. Clin. Oncol. 2012, 30, 3402–3407. [Google Scholar] [CrossRef]
- Sancho, D.; Joffre, O.P.; Keller, A.M.; Rogers, N.C.; Martínez, D.; Hernanz-Falcón, P.; Rosewell, I.; Reis e Sousa, C. Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature 2009, 458, 899–903. [Google Scholar] [CrossRef] [PubMed]
- Akimoto, M.; Maruyama, R.; Kawabata, Y.; Tajima, Y.; Takenaga, K. Antidiabetic adiponectin receptor agonist AdipoRon suppresses tumour growth of pancreatic cancer by inducing RIPK1/ERK-dependent necroptosis. Cell Death Dis. 2018, 9, 804. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-L.; Tang, H.-B.; Hu, J.-T.; Zang, Z.-L.; Ding, X.; Li, S.; Yang, H. PGAM5-CypD pathway is involved in bromocriptine-induced RIP3/MLKL-dependent necroptosis of prolactinoma cells. Biomed. Pharmacother. 2019, 111, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Rohde, K.; Kleinesudeik, L.; Roesler, S.; Löwe, O.; Heidler, J.; Schröder, K.; Wittig, I.; Dröse, S.; Fulda, S. A Bak-dependent mitochondrial amplification step contributes to Smac mimetic/glucocorticoid-induced necroptosis. Cell Death Differ. 2017, 24, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, I.A.; Abhari, B.A.; Fulda, S. Identification of a synergistic combination of Smac mimetic and Bortezomib to trigger cell death in B-cell non-Hodgkin lymphoma cells. Cancer Lett. 2017, 405, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Kitatani, K.; Toyoshima, M.; Ishibashi, M.; Usui, T.; Minato, J.; Egiz, M.; Shigeta, S.; Fox, T.; Deering, T.; et al. Ceramide Nanoliposomes as a MLKL-Dependent, Necroptosis-Inducing, Chemotherapeutic Reagent in Ovarian Cancer. Mol. Cancer Ther. 2018, 17, 50–59. [Google Scholar] [CrossRef]
- Xu, Y.; Lin, Z.; Zhao, N.; Zhou, L.; Liu, F.; Cichacz, Z.; Zhang, L.; Zhan, Q.; Zhao, X. Receptor interactive protein kinase 3 promotes Cisplatin-triggered necrosis in apoptosis-resistant esophageal squamous cell carcinoma cells. PLoS ONE 2014, 9, e100127. [Google Scholar] [CrossRef][Green Version]
- Jing, L.; Song, F.; Liu, Z.; Li, J.; Wu, B.; Fu, Z.; Jiang, J.; Chen, Z. MLKL-PITPα signaling-mediated necroptosis contributes to cisplatin-triggered cell death in lung cancer A549 cells. Cancer Lett. 2018, 414, 136–146. [Google Scholar] [CrossRef]
- Chengzhu, W.U.; Gao, M.; Shen, L.; Bohan, L.I.; Bai, X.; Gui, J.; Hongmei, L.I.; Huo, Q.; Tao, M.A. Miconazole triggers various forms of cell death in human breast cancer MDA-MB-231 cells. Pharmazie 2019, 74, 290–294. [Google Scholar]
- Yu, X.; Deng, Q.; Li, W.; Xiao, L.; Luo, X.; Liu, X.; Yang, L.; Peng, S.; Ding, Z.; Feng, T.; et al. Neoalbaconol induces cell death through necroptosis by regulating RIPK-dependent autocrine TNFα and ROS production. Oncotarget 2015, 6, 1995–2008. [Google Scholar] [CrossRef]
- Weigert, M.; Binks, A.; Dowson, S.; Leung, E.Y.L.; Athineos, D.; Yu, X.; Mullin, M.; Walton, J.B.; Orange, C.; Ennis, D.; et al. RIPK3 promotes adenovirus type 5 activity. Cell Death Dis. 2017, 8, 3206. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.Y.; Jaime-Ramirez, A.C.; Bolyard, C.; Dai, H.; Nallanagulagari, T.; Wojton, J.; Hurwitz, B.S.; Relation, T.; Lee, T.J.; Lotze, M.T.; et al. Bortezomib Treatment Sensitizes Oncolytic HSV-1-Treated Tumors to NK Cell Immunotherapy. Clin. Cancer Res. 2016, 22, 5265–5276. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Deng, B.; Liao, Y.; Shan, L.; Yin, F.; Wang, Z.; Zeng, H.; Zuo, D.; Hua, Y.; Cai, Z. The anti-tumor effect of shikonin on osteosarcoma by inducing RIP1 and RIP3 dependent necroptosis. BMC Cancer 2013, 13, 580. [Google Scholar] [CrossRef] [PubMed]
- Shahsavari, Z.; Karami-Tehrani, F.; Salami, S.; Ghasemzadeh, M. RIP1K and RIP3K provoked by shikonin induce cell cycle arrest in the triple negative breast cancer cell line, MDA-MB-468: Necroptosis as a desperate programmed suicide pathway. Tumour Biol. 2016, 37, 4479–4491. [Google Scholar] [CrossRef]
- Zhou, Z.; Lu, B.; Wang, C.; Wang, Z.; Luo, T.; Piao, M.; Meng, F.; Chi, G.; Luo, Y.; Ge, P. RIP1 and RIP3 contribute to shikonin-induced DNA double-strand breaks in glioma cells via increase of intracellular reactive oxygen species. Cancer Lett. 2017, 390, 77–90. [Google Scholar] [CrossRef]
- Zielinska, E.; Zauszkiewicz-Pawlak, A.; Wojcik, M.; Inkielewicz-Stepniak, I. Silver nanoparticles of different sizes induce a mixed type of programmed cell death in human pancreatic ductal adenocarcinoma. Oncotarget 2018, 9, 4675–4697. [Google Scholar] [CrossRef]
- Hillert, L.K.; Bettermann-Bethge, K.; Nimmagadda, S.C.; Fischer, T.; Naumann, M.; Lavrik, I.N. Targeting RIPK1 in AML cells carrying FLT3-ITD. Int. J. Cancer 2019, 145, 1558–1569. [Google Scholar] [CrossRef]
- Jin, G.; Liu, Y.; Xu, P.; Jin, G. Induction of Necroptosis in Human Breast Cancer Drug-Resistant Cells by SMAC Analog LCL161 After Caspase Inhibition Requires RIP3. Pharmazie 2019, 74, 363–368. [Google Scholar]
- He, S.; Huang, S.; Shen, Z. Biomarkers for the detection of necroptosis. Cell Mol. Life Sci. 2016, 73, 2177–2181. [Google Scholar] [CrossRef]
- Caccamo, A.; Branca, C.; Piras, I.S.; Ferreira, E.; Huentelman, M.J.; Liang, W.S.; Readhead, B.; Dudley, J.T.; Spangenberg, E.E.; Green, K.N.; et al. Necroptosis activation in Alzheimer’s disease. Nat. Neurosci. 2017, 20, 1236–1246. [Google Scholar] [CrossRef]
- Koo, G.-B.; Morgan, M.J.; Lee, D.-G.; Kim, W.-J.; Yoon, J.-H.; Koo, J.S.; Kim, S.I.; Kim, S.J.; Son, M.K.; Hong, S.S.; et al. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res. 2015, 25, 707–725. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Song, Q.; Yu, A.; Tang, H.; Peng, Z.; Wang, X. Receptor-interacting protein kinase 3 is a predictor of survival and plays a tumor suppressive role in colorectal cancer. Neoplasma 2015, 62, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Nugues, A.L.; El Bouazzati, H.; Hétuin, D.; Berthon, C.; Loyens, A.; Bertrand, E.; Jouy, N.; Idziorek, T.; Quesnel, B. RIP3 is downregulated in human myeloid leukemia cells and modulates apoptosis and caspase-mediated p65/RelA cleavage. Cell Death Dis. 2014, 5, e1384. [Google Scholar] [CrossRef] [PubMed]
- Najafov, A.; Zervantonakis, I.K.; Mookhtiar, A.K.; Greninger, P.; March, R.J.; Egan, R.K.; Luu, H.S.; Stover, D.G.; Matulonis, U.A.; Benes, C.H.; et al. BRAF and AXL oncogenes drive RIPK3 expression loss in cancer. PLoS Biol. 2018, 16, e2005756. [Google Scholar] [CrossRef]
- Shi, F.; Zhou, M.; Shang, L.; Du, Q.; Li, Y.; Xie, L.; Liu, X.; Tang, M.; Luo, X.; Fan, J.; et al. EBV(LMP1)-induced metabolic reprogramming inhibits necroptosis through the hypermethylation of the RIP3 promoter. Theranostics 2019, 9, 2424–2438. [Google Scholar] [CrossRef]
- Liu, X.; Li, Y.; Peng, S.; Yu, X.; Li, W.; Shi, F.; Luo, X.; Tang, M.; Tan, Z.; Bode, A.M.; et al. Epstein-Barr virus encoded latent membrane protein 1 suppresses necroptosis through targeting RIPK1/3 ubiquitination. Cell Death Dis. 2018, 9, 53. [Google Scholar] [CrossRef]
- Yang, C.; Li, J.; Yu, L.; Zhang, Z.; Xu, F.; Jiang, L.; Zhou, X.; He, S. Regulation of RIP3 by the transcription factor Sp1 and the epigenetic regulator UHRF1 modulates cancer cell necroptosis. Cell Death Dis. 2017, 8, e3084. [Google Scholar] [CrossRef]
- Conev, N.V.; Dimitrova, E.G.; Bogdanova, M.K.; Kashlov, Y.K.; Chaushev, B.G.; Radanova, M.A.; Petrov, D.P.; Georgiev, K.D.; Bachvarov, C.H.; Todorov, G.N.; et al. RIPK3 expression as a potential predictive and prognostic marker in metastatic colon cancer. Clin. Investig. Med. 2019, 42, E31–E38. [Google Scholar] [CrossRef]
- McCormick, K.D.; Ghosh, A.; Trivedi, S.; Wang, L.; Coyne, C.B.; Ferris, R.L.; Sarkar, S.N. Innate immune signaling through differential RIPK1 expression promote tumor progression in head and neck squamous cell carcinoma. Carcinogenesis 2016, 37, 522–529. [Google Scholar] [CrossRef]
- Schneider, A.T.; Gautheron, J.; Feoktistova, M.; Roderburg, C.; Loosen, S.H.; Roy, S.; Benz, F.; Schemmer, P.; Büchler, M.W.; Nachbur, U.; et al. RIPK1 Suppresses a TRAF2-Dependent Pathway to Liver Cancer. Cancer Cell 2017, 31, 94–109. [Google Scholar] [CrossRef]
- Liu, X.Y.; Lai, F.; Yan, X.G.; Jiang, C.C.; Guo, S.T.; Wang, C.Y.; Croft, A.; Tseng, H.-Y.; Wilmott, J.S.; Scolyer, R.A.; et al. RIP1 kinase is an oncogenic driver in melanoma. Cancer Res. 2015, 75, 1736–1748. [Google Scholar] [CrossRef] [PubMed]
- Krysko, O.; Aaes, T.L.; Kagan, V.E.; D’Herde, K.; Bachert, C.; Leybaert, L.; Vandenabeele, P.; Krysko, D.V. Necroptotic cell death in anti-cancer therapy. Immunol. Rev. 2017, 280, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. The mechanism of necroptosis in normal and cancer cells. Cancer Biol. Ther. 2013, 14, 999–1004. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kepp, O.; Chan, F.K.-M.; Kroemer, G. Necroptosis: Mechanisms and relevance to disease. Annu. Rev. Pathol. 2017, 12, 103–130. [Google Scholar] [CrossRef]
- Najafov, A.; Chen, H.; Yuan, J. Necroptosis and Cancer. Trends Cancer 2017, 3, 294–301. [Google Scholar] [CrossRef]
- Lalaoui, N.; Brumatti, G. Relevance of necroptosis in cancer. Immunol. Cell Biol. 2017, 95, 137–145. [Google Scholar] [CrossRef]
- Schumacher, D.; Strilic, B.; Sivaraj, K.K.; Wettschureck, N.; Offermanns, S. Platelet-derived nucleotides promote tumor-cell transendothelial migration and metastasis via P2Y2 receptor. Cancer Cell 2013, 24, 130–137. [Google Scholar] [CrossRef]
- Hänggi, K.; Vasilikos, L.; Valls, A.F.; Yerbes, R.; Knop, J.; Spilgies, L.M.; Rieck, K.; Misra, T.; Bertin, J.; Gough, P.J.; et al. RIPK1/RIPK3 promotes vascular permeability to allow tumor cell extravasation independent of its necroptotic function. Cell Death Dis. 2017, 8, e2588. [Google Scholar] [CrossRef]
- Ruan, J.; Mei, L.; Zhu, Q.; Shi, G.; Wang, H. Mixed lineage kinase domain-like protein is a prognostic biomarker for cervical squamous cell cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 15035–15038. [Google Scholar]
- Strilic, B.; Yang, L.; Albarrán-Juárez, J.; Wachsmuth, L.; Han, K.; Müller, U.C.; Pasparakis, M.; Offermanns, S. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature 2016, 536, 215–218. [Google Scholar] [CrossRef]
- Ando, Y.; Ohuchida, K.; Otsubo, Y.; Kibe, S.; Takesue, S.; Abe, T.; Iwamoto, C.; Shindo, K.; Moriyama, T.; Nakata, K.; et al. Necroptosis in pancreatic cancer promotes cancer cell migration and invasion by release of CXCL5. PLoS ONE 2020, 15, e0228015. [Google Scholar] [CrossRef] [PubMed]
- Conos, S.A.; Chen, K.W.; De Nardo, D.; Hara, H.; Whitehead, L.; Núñez, G.; Masters, S.L.; Murphy, J.M.; Schroder, K.; Vaux, D.L.; et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc. Natl. Acad. Sci. USA 2017, 114, E961–E969. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Kuo, W.T.; Huang, Y.C.; Lee, T.C.; Yu, L.C.H. Resistance to hypoxia-induced necroptosis is conferred by glycolytic pyruvate scavenging of mitochondrial superoxide in colorectal cancer cells. Cell Death Dis. 2013, 4, e622. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, C. Death by inflammation: Drug makers chase the master controller. Nat. Biotechnol. 2019, 37, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-H.; Wu, Z.-Q.; Qian, D.; Zaorsky, N.G.; Qiu, M.-H.; Cheng, J.-J.; Jiang, C.; Wang, J.; Zeng, X.-L.; Liu, C.-L.; et al. Ablative Hypofractionated Radiation Therapy Enhances Non-Small Cell Lung Cancer Cell Killing via Preferential Stimulation of Necroptosis In Vitro and In Vivo. Int. J. Radiat. Oncol. Biol. Phys. 2018, 101, 49–62. [Google Scholar] [CrossRef]
- Bist, P.; Leow, S.C.; Phua, Q.H.; Shu, S.; Zhuang, Q.; Loh, W.T.; Nguyen, T.H.; Zhou, J.B.; Hooi, S.C.; Lim, L.H.K. Annexin-1 interacts with NEMO and RIP1 to constitutively activate IKK complex and NF-κB: Implication in breast cancer metastasis. Oncogene 2011, 30, 3174–3185. [Google Scholar] [CrossRef]
- Park, S.; Hatanpaa, K.J.; Xie, Y.; Mickey, B.E.; Madden, C.J.; Raisanen, J.M.; Ramnarain, D.B.; Xiao, G.; Saha, D.; Boothman, D.A.; et al. The receptor interacting protein 1 inhibits p53 induction through NF-kappaB activation and confers a worse prognosis in glioblastoma. Cancer Res. 2009, 69, 2809–2816. [Google Scholar] [CrossRef]
- Li, X.; Guo, J.; Ding, A.-P.; Qi, W.-W.; Zhang, P.-H.; Lv, J.; Qiu, W.-S.; Sun, Z.-Q. Association of Mixed Lineage Kinase Domain-Like Protein Expression with Prognosis in Patients with Colon Cancer. Technol. Cancer Res. Treat. 2017, 16, 428–434. [Google Scholar] [CrossRef]
- Li, L.; Yu, S.; Zang, C. Low Necroptosis Process Predicts Poor Treatment Outcome of Human Papillomavirus Positive Cervical Cancers by Decreasing Tumor-Associated Macrophages M1 Polarization. Gynecol. Obstet. Investig. 2018, 83, 259–267. [Google Scholar] [CrossRef]
- He, L.; Peng, K.; Liu, Y.; Xiong, J.; Zhu, F.-F. Low expression of mixed lineage kinase domain-like protein is associated with poor prognosis in ovarian cancer patients. Onco Targets Ther. 2013, 6, 1539–1543. [Google Scholar]
- Colbert, L.E.; Fisher, S.B.; Hardy, C.W.; Hall, W.A.; Saka, B.; Shelton, J.W.; Petrova, A.V.; Warren, M.D.; Pantazides, B.G.; Gandhi, K.; et al. Pronecrotic mixed lineage kinase domain-like protein expression is a prognostic biomarker in patients with early-stage resected pancreatic adenocarcinoma. Cancer 2013, 119, 3148–3155. [Google Scholar] [CrossRef]
- Jiao, D.; Cai, Z.; Choksi, S.; Ma, D.; Choe, M.; Kwon, H.-J.; Baik, J.Y.; Rowan, B.G.; Liu, C.; Liu, Z.-G. Necroptosis of tumor cells leads to tumor necrosis and promotes tumor metastasis. Cell Res. 2018, 28, 868–870. [Google Scholar] [CrossRef] [PubMed]
- Ertao, Z.; Jianhui, C.; Kang, W.; Zhijun, Y.; Hui, W.; Chuangqi, C.; Changjiang, Q.; Sile, C.; Yulong, H.; Shirong, C. Prognostic value of mixed lineage kinase domain-like protein expression in the survival of patients with gastric caner. Tumour Biol. 2016, 37, 13679–13685. [Google Scholar] [CrossRef] [PubMed]
- Wellenstein, M.D.; de Visser, K.E. Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape. Immunity 2018, 48, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The immune landscape of cancer. Immunity 2018, 48, 812–830. [Google Scholar] [CrossRef]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef]
- Garg, A.D.; Romano, E.; Rufo, N.; Agostinis, P. Immunogenic versus tolerogenic phagocytosis during anticancer therapy: Mechanisms and clinical translation. Cell Death Differ. 2016, 23, 938–951. [Google Scholar] [CrossRef]
- Yang, H.; Ma, Y.; Chen, G.; Zhou, H.; Yamazaki, T.; Klein, C.; Pietrocola, F.; Vacchelli, E.; Souquere, S.; Sauvat, A.; et al. Contribution of RIP3 and MLKL to immunogenic cell death signaling in cancer chemotherapy. Oncoimmunology 2016, 5, e1149673. [Google Scholar] [CrossRef]
- Smola, S. RIPK3-a predictive marker for personalized immunotherapy? Oncoimmunology 2016, 5, e1075695. [Google Scholar] [CrossRef]
- Schmidt, S.V.; Seibert, S.; Walch-Rückheim, B.; Vicinus, B.; Kamionka, E.-M.; Pahne-Zeppenfeld, J.; Solomayer, E.-F.; Kim, Y.-J.; Bohle, R.M.; Smola, S. RIPK3 expression in cervical cancer cells is required for PolyIC-induced necroptosis, IL-1α release, and efficient paracrine dendritic cell activation. Oncotarget 2015, 6, 8635–8647. [Google Scholar] [CrossRef] [PubMed]
- Koks, C.A.; Garg, A.D.; Ehrhardt, M.; Riva, M.; Vandenberk, L.; Boon, L.; De Vleeschouwer, S.; Agostinis, P.; Graf, N.; Van Gool, S.W. Newcastle disease virotherapy induces long-term survival and tumor-specific immune memory in orthotopic glioma through the induction of immunogenic cell death. Int. J. Cancer 2015, 136, E313–E325. [Google Scholar] [CrossRef] [PubMed]
- Dudek-Perić, A.M.; Ferreira, G.B.; Muchowicz, A.; Wouters, J.; Prada, N.; Martin, S.; Kiviluoto, S.; Winiarska, M.; Boon, L.; Mathieu, C.; et al. Antitumor immunity triggered by melphalan is potentiated by melanoma cell surface-associated calreticulin. Cancer Res. 2015, 75, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Vandenberk, L.; Koks, C.; Verschuere, T.; Boon, L.; Van Gool, S.W.; Agostinis, P. Dendritic cell vaccines based on immunogenic cell death elicit danger signals and T cell-driven rejection of high-grade glioma. Sci. Transl. Med. 2016, 8, 328ra27. [Google Scholar] [CrossRef]
- Aaes, T.L.; Verschuere, H.; Kaczmarek, A.; Heyndrickx, L.; Wiernicki, B.; Delrue, I.; De Craene, B.; Taminau, J.; Delvaeye, T.; Bertrand, M.J.M.; et al. Immunodominant AH1 Antigen-Deficient Necroptotic, but Not Apoptotic, Murine Cancer Cells Induce Antitumor Protection. J. Immunol. 2020, 204, 775–787. [Google Scholar] [CrossRef]
- Krysko, D.V.; Brouckaert, G.; Kalai, M.; Vandenabeele, P.; D’Herde, K. Mechanisms of internalization of apoptotic and necrotic L929 cells by a macrophage cell line studied by electron microscopy. J. Morphol. 2003, 258, 336–345. [Google Scholar] [CrossRef]
- Gamrekelashvili, J.; Ormandy, L.A.; Heimesaat, M.M.; Kirschning, C.J.; Manns, M.P.; Korangy, F.; Greten, T.F. Primary sterile necrotic cells fail to cross-prime CD8(+) T cells. Oncoimmunology 2012, 1, 1017–1026. [Google Scholar] [CrossRef]
- Kearney, C.J.; Cullen, S.P.; Tynan, G.A.; Henry, C.M.; Clancy, D.; Lavelle, E.C.; Martin, S.J. Necroptosis suppresses inflammation via termination of TNF- or LPS-induced cytokine and chemokine production. Cell Death Differ. 2015, 22, 1313–1327. [Google Scholar] [CrossRef]
- Liu, Z.-Y.; Wu, B.; Guo, Y.-S.; Zhou, Y.-H.; Fu, Z.-G.; Xu, B.-Q.; Li, J.-H.; Jing, L.; Jiang, J.-L.; Tang, J.; et al. Necrostatin-1 reduces intestinal inflammation and colitis-associated tumorigenesis in mice. Am. J. Cancer Res. 2015, 5, 3174–3185. [Google Scholar]
- Krysko, O.; De Ridder, L.; Cornelissen, M. Phosphatidylserine exposure during early primary necrosis (oncosis) in JB6 cells as evidenced by immunogold labeling technique. Apoptosis 2004, 9, 495–500. [Google Scholar] [CrossRef]
- Yang, L.; Joseph, S.; Sun, T.; Hoffmann, J.; Thevissen, S.; Offermanns, S.; Strilic, B. TAK1 regulates endothelial cell necroptosis and tumor metastasis. Cell Death Differ. 2019, 26, 1987–1997. [Google Scholar] [CrossRef] [PubMed]
- Seifert, L.; Werba, G.; Tiwari, S.; Giao Ly, N.N.; Alothman, S.; Alqunaibit, D.; Avanzi, A.; Barilla, R.; Daley, D.; Greco, S.H.; et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature 2016, 532, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Jafari, S.; Molavi, O.; Kahroba, H.; Hejazi, M.S.; Maleki-Dizaji, N.; Barghi, S.; Kiaie, S.H.; Jadidi-Niaragh, F. Clinical application of immune checkpoints in targeted immunotherapy of prostate cancer. Cell. Mol. Life Sci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Jiang, W.; Kim, B.Y.S.; Zhang, C.C.; Fu, Y.-X.; Weissman, I.L. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat. Rev. Cancer 2019, 19, 568–586. [Google Scholar] [CrossRef] [PubMed]
- Chinai, J.M.; Janakiram, M.; Chen, F.; Chen, W.; Kaplan, M.; Zang, X. New immunotherapies targeting the PD-1 pathway. Trends Pharmacol. Sci. 2015, 36, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Paluch, C.; Santos, A.M.; Anzilotti, C.; Cornall, R.J.; Davis, S.J. Immune checkpoints as therapeutic targets in autoimmunity. Front. Immunol. 2018, 9, 2306. [Google Scholar] [CrossRef]
- Lucca, L.E.; Hafler, D.A. Co-inhibitory blockade while preserving tolerance: Checkpoint inhibitors for glioblastoma. Immunol. Rev. 2017, 276, 9–25. [Google Scholar] [CrossRef]
- Barrueto, L.; Caminero, F.; Cash, L.; Makris, C.; Lamichhane, P.; Deshmukh, R.R. Resistance to checkpoint inhibition in cancer immunotherapy. Transl. Oncol. 2020, 13, 100738. [Google Scholar] [CrossRef]
- Nakamura, K.; Smyth, M.J. Myeloid immunosuppression and immune checkpoints in the tumor microenvironment. Cell. Mol. Immunol. 2020, 17, 1–12. [Google Scholar] [CrossRef]
- Snyder, A.G.; Hubbard, N.W.; Messmer, M.N.; Kofman, S.B.; Hagan, C.E.; Orozco, S.L.; Chiang, K.; Daniels, B.P.; Baker, D.; Oberst, A. Intratumoral activation of the necroptotic pathway components RIPK1 and RIPK3 potentiates antitumor immunity. Sci. Immunol. 2019, 4, eaaw2004. [Google Scholar] [CrossRef]
- Kang, T.; Huang, Y.; Zhu, Q.; Cheng, H.; Pei, Y.; Feng, J.; Xu, M.; Jiang, G.; Song, Q.; Jiang, T.; et al. Necroptotic cancer cells-mimicry nanovaccine boosts anti-tumor immunity with tailored immune-stimulatory modality. Biomaterials 2018, 164, 80–97. [Google Scholar] [CrossRef] [PubMed]
- Van Hoecke, L.; Van Lint, S.; Roose, K.; Van Parys, A.; Vandenabeele, P.; Grooten, J.; Tavernier, J.; De Koker, S.; Saelens, X. Treatment with mRNA coding for the necroptosis mediator MLKL induces antitumor immunity directed against neo-epitopes. Nat. Commun. 2018, 9, 3417. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Li, H.; Wu, J.; Cai, Z.-Y.; Li, B.; Ni, H.; Qiu, X.; Chen, H.; Liu, W.; Yang, Z.-H.; et al. Gut stem cell necroptosis by genome instability triggers bowel inflammation. Nature 2020, 580, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhao, Y.; Shi, L.; Li, W.; Chen, K.; Li, M.; Chen, X.; Zhang, H.; Li, T.; Matsuzawa-Ishimoto, Y.; et al. Gut epithelial TSC1/mTOR controls RIPK3-dependent necroptosis in intestinal inflammation and cancer. J. Clin. Investig. 2020, 130, 2111–2128. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
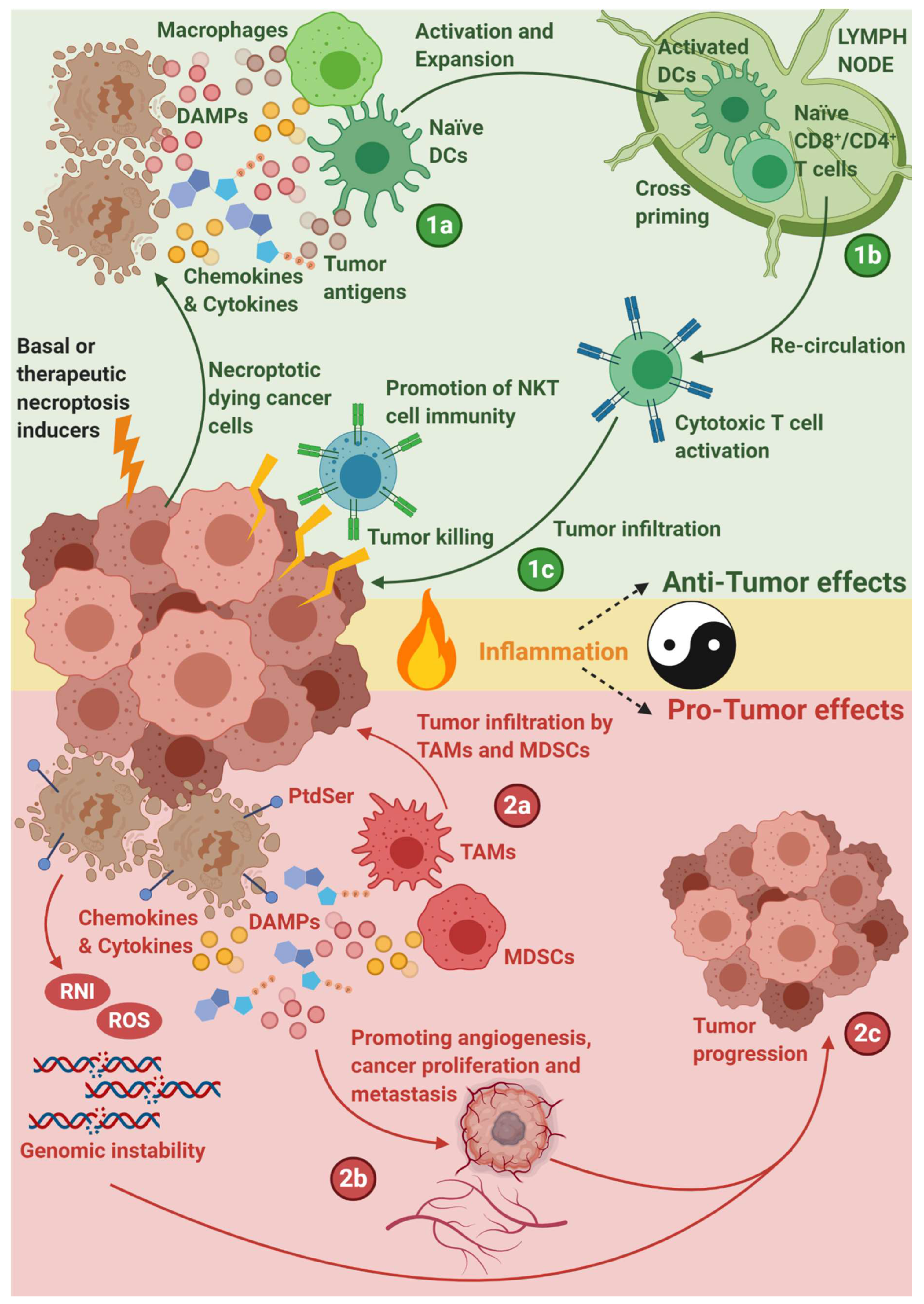{kind=link}
| Therapeutic Agent | Cancer Type | Pro-Necroptosis Roles | Refs. |
|---|---|---|---|
| AdipoRon | Human pancreatic cancer cells | Induces necroptosis through p38, MAPK and RIPK1 activation. | [182] |
| Bromocriptine | Prolactinoma | Cell death induced by bromocriptine, which is a dopamine antagonist, relies on necroptosis | [183] |
| BV6 + dexa co-treatment | ALL (acute lymphoid leukaemia) | Cell death depends on RIPK3 and MLKL. | [184] |
| BV6 and Bortezomib | B-cell non-Hodgkin Lymphoma | Induction of necroptosis, even if apoptosis is blocked. | [185] |
| Ceramide nanoliposomes | Ovarian cancer cell xenograft model | Suppressed metastatic growth through inducing necroptosis | [186] |
| Cisplatin | Oesophageal cancer | RIPK3 regulates cisplatin sensitivity and could predict chemosensitivity. | [187] |
| Cisplatin | Lung cancer | Cisplatin induces both apoptosis and necroptotic-like cell death in lung cancer cells. | [188] |
| Miconazole | Breast cancer cells | Inhibits the proliferation and induces apoptosis and necroptosis. | [189] |
| Neoalbaconolol | Human nasopharyngeal carcinoma cells | Induces necroptosis by remodeling cellular energy metabolism. | [190] |
| Oncolytic viruses | Various cancer-types | Mechanism unknown. | [191] |
| Proteasome inhibitors | Glioblastoma | Proteasome inhibitors and oncolytic HSV induce necroptosis, increase the production of mitochondrial ROS and JNK phosphorylation and significantly enhance NK cell activation. | [192] |
| Shikonin | Lung cancer, triple negative breast cancer and glioma | Induces necroptosis in cancer cells. | [193,194,195] |
| Silver nanoparticles | Pancreatic ductal adenocarcinoma | Silver nanoparticles have the potential to overcome barriers involved in chemotherapy failure. | [196] |
| SMAC mimetic (BV6) | AML (acute myeloid leukaemia) | Sensitizes cell to apoptosis and necroptosis. RIPK1 seems to play the major role in AML. | [197] |
| SMAC mimetic (LCL161) | Drug resistant breast cancer | Activation of the RIPK1-RIPK3-MLKL necroptosis. | [198] |
| Silencing Method | Mechanism | Refs. |
|---|---|---|
| Epstein Barr Virus (EBV) | EBV infection suppresses RIPK3 expression via hypermethylation of the RIPK3 promotor. | [205,206] |
| Methylation | RIPK3 can be silenced in cancer cells due to genomic methylation close to its transcriptional start site, thereby inhibiting RIPK3-dependent necroptosis by chemotherapeutics. | [201,207] |
| Sp1 | Zinc-finger transcription factor, named Sp1, regulates the expression of RIPK3 in a direct way. The knockdown of this transcription factor decreases the transcription of RIPK3 and vice versa. | [207] |
| Expression | Cancer | Prognosis | Refs. |
|---|---|---|---|
| RIPK3 expression | |||
| High expression | Non-small cell lung cancer | Improved local control and progression-free survival in treatment with hypofractionated radiation therapy (HFRT) | [225] |
| Primary CRC (colon rectal cancer) | Longer mean overall survival after treatment with 5-fluorouracil (5-FU) | [208] | |
| Low expression | Breast cancer | Worse prognosis | [201] |
| Colon rectal cancer (CRC) | Worse overall survival and disease-free interval. | [202] | |
| RIPK1 expression | |||
| Low expression | Head and neck cancer (HCC) | Worse prognosis | [209] |
| Head and neck cancer (HCC) | Worse prognosis | [210] | |
| High expression | Breast cancer | Promotes metastasis | [226] |
| Glioblastoma | Worse prognosis | [227] | |
| MLKL expression | |||
| Low expression | Colorectal cancer | Decreased overall survival in treatment with adjuvant chemotherapy. | [228] |
| HR-HPV cervical cancer (high risk- human papillomavirus) | Decreased overall survival and disease-free survival | [229] | |
| Ovarian cancer | Decreased overall survival. | [230] | |
| Pancreatic adenocarcinoma | Decreased overall survival in patients with resected tumor and decreased RFS and OS in the subset of patients with resected tumors who receive adjuvant chemotherapy. | [231] | |
| High expression | Breast cancer | Worse prognosis. | [232] |
| Cervical SCC (squamous cell carcinoma) | Dual: Higher MLKL expression in cervical SCC than in normal tissue. Though low expression in cervical SCC indicated poor prognosis. | [219] | |
| Gastric cancer | Tumor suppressing and a potential prognostic biomarker. | [233] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sprooten, J.; De Wijngaert, P.; Vanmeerbeek, I.; Martin, S.; Vangheluwe, P.; Schlenner, S.; Krysko, D.V.; Parys, J.B.; Bultynck, G.; Vandenabeele, P.; et al. Necroptosis in Immuno-Oncology and Cancer Immunotherapy. Cells 2020, 9, 1823. https://doi.org/10.3390/cells9081823
Sprooten J, De Wijngaert P, Vanmeerbeek I, Martin S, Vangheluwe P, Schlenner S, Krysko DV, Parys JB, Bultynck G, Vandenabeele P, et al. Necroptosis in Immuno-Oncology and Cancer Immunotherapy. Cells. 2020; 9(8):1823. https://doi.org/10.3390/cells9081823
Chicago/Turabian StyleSprooten, Jenny, Pieter De Wijngaert, Isaure Vanmeerbeek, Shaun Martin, Peter Vangheluwe, Susan Schlenner, Dmitri V. Krysko, Jan B. Parys, Geert Bultynck, Peter Vandenabeele, and et al. 2020. "Necroptosis in Immuno-Oncology and Cancer Immunotherapy" Cells 9, no. 8: 1823. https://doi.org/10.3390/cells9081823
APA StyleSprooten, J., De Wijngaert, P., Vanmeerbeek, I., Martin, S., Vangheluwe, P., Schlenner, S., Krysko, D. V., Parys, J. B., Bultynck, G., Vandenabeele, P., & Garg, A. D. (2020). Necroptosis in Immuno-Oncology and Cancer Immunotherapy. Cells, 9(8), 1823. https://doi.org/10.3390/cells9081823