Brain-Specific Serine-47 Modification of Cytochrome c Regulates Cytochrome c Oxidase Activity Attenuating ROS Production and Cell Death: Implications for Ischemia/Reperfusion Injury and Akt Signaling



Abstract
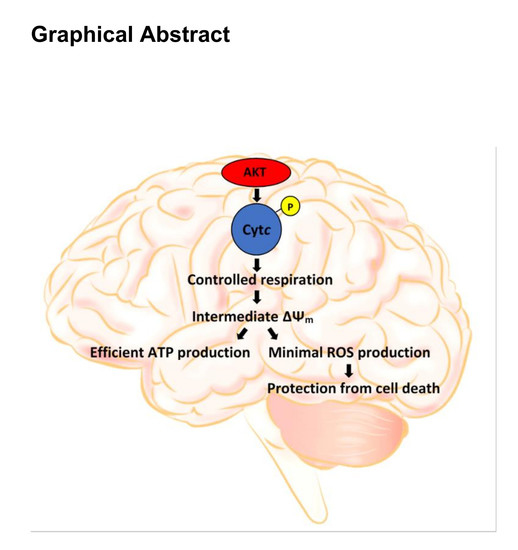
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Stable Transfection of Cytc Constructs
2.2. Gel Electrophoresis and Western Blotting
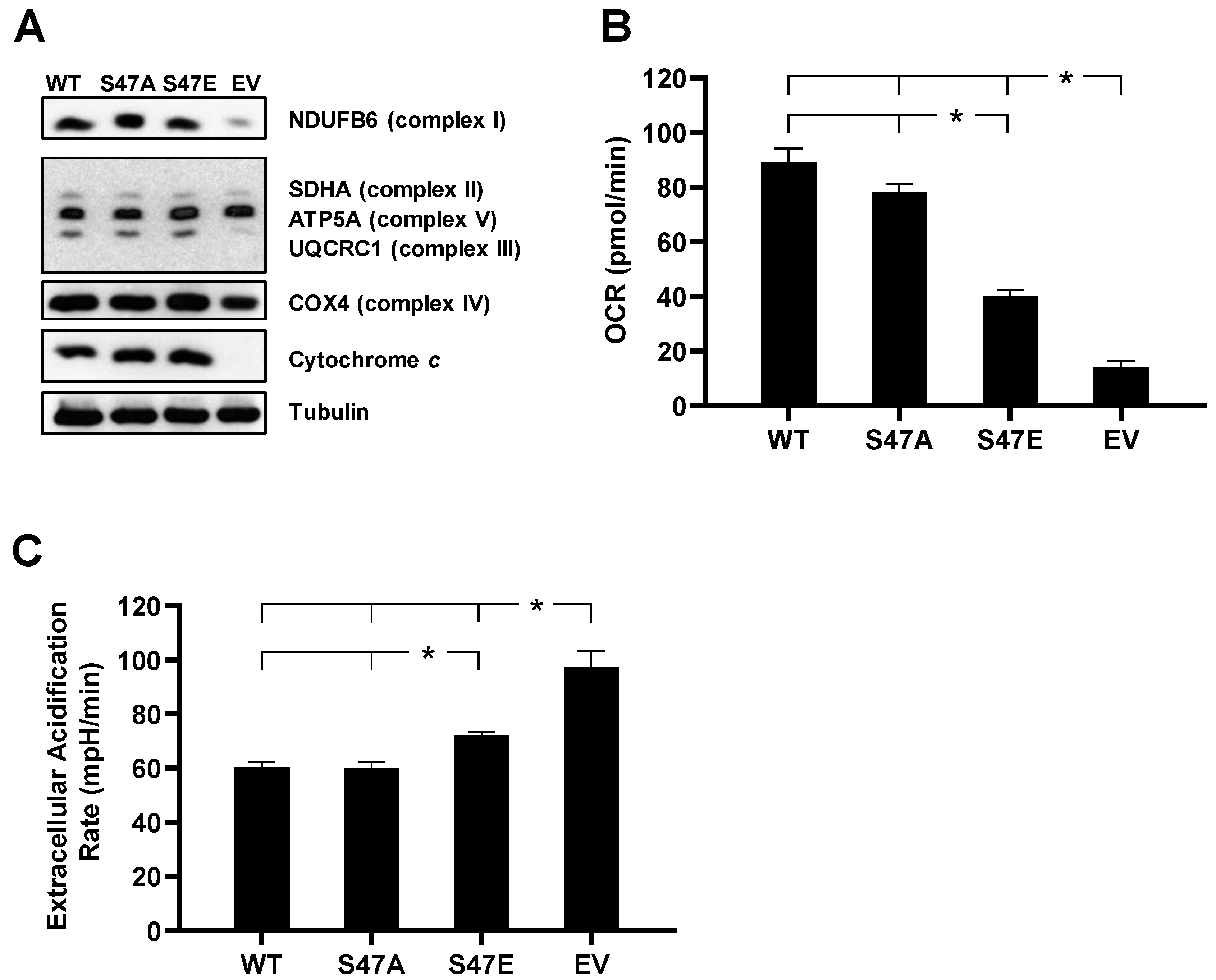
2.3. Measurement of Oxygen Consumption and Extracellular Acidification Rate
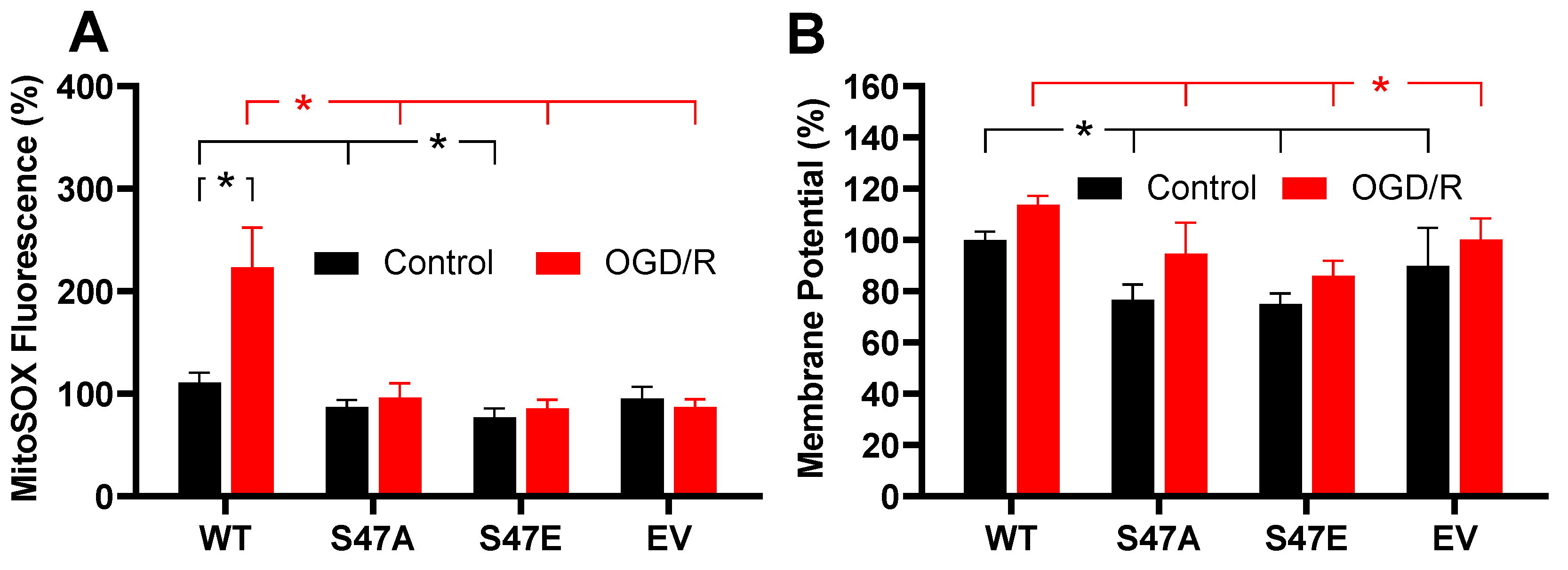
2.4. Measurement of Membrane Potential
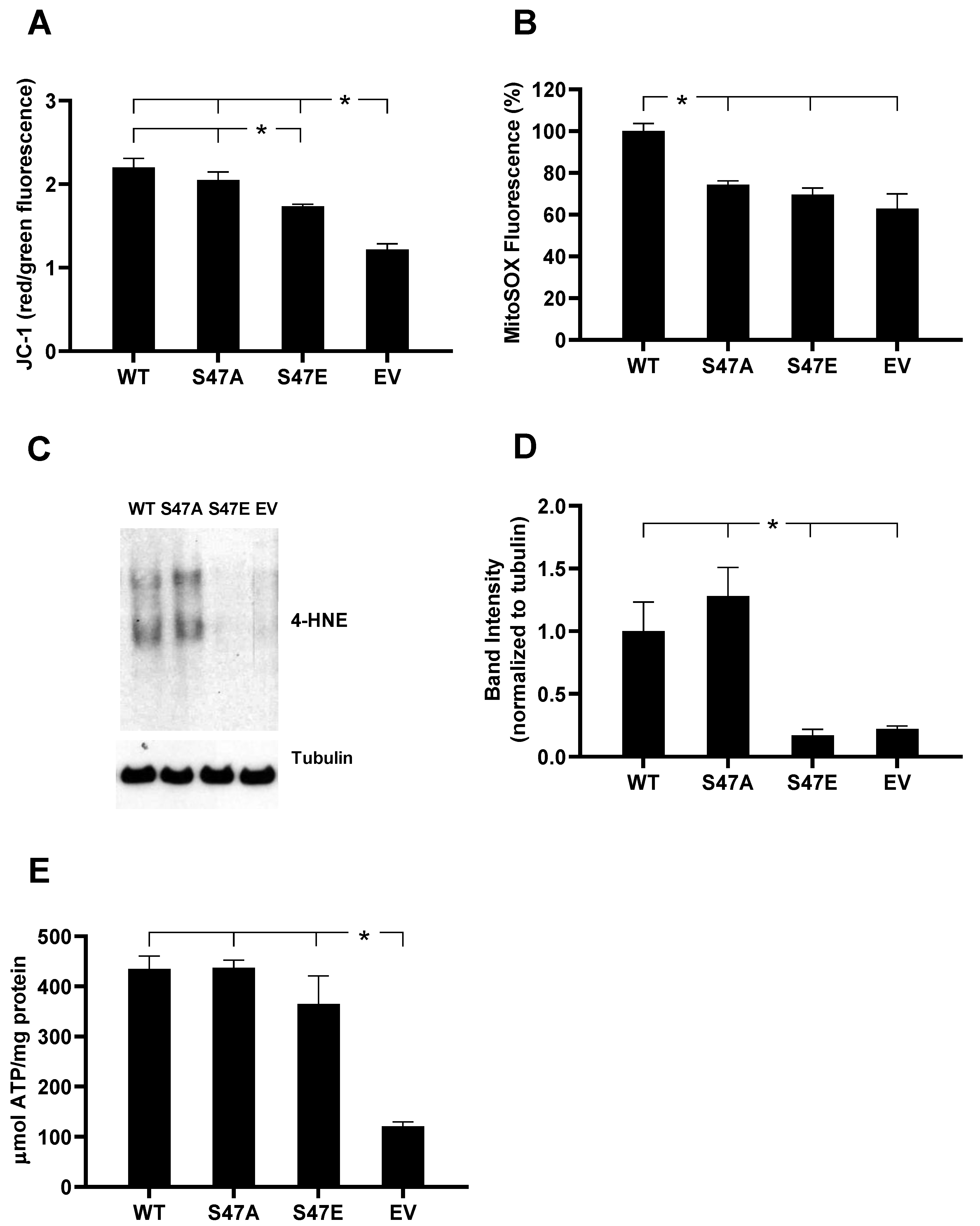
2.5. Measurement of Mitochondrial ROS Production
2.6. Measurement of ATP Levels
2.7. Oxygen-Glucose Deprivation/Reoxygenation (OGD/R) Experiments
2.8. Measurement of Cell Death Using Annexin V/PI Staining
2.9. Akt Kinase Assay
2.10. Tissue Treatment with Pharmacological Compounds and Cytochrome c Purification
2.11. Mitochondrial Subfractionation
2.12. Recombinant Cytc Purification and Cytc Oxidation by Hydrogen Peroxide
2.13. Statistical Analyses
3. Results
3.1. Overexpression of WT, S47A, and S47E Cytc in Cytc Double-Knockout Cells Restores Expression of Complex I, III, and IV
3.2. S47E Cytc Expression Results in Lower Mitochondrial Respiration and Higher Glycolysis Rates
3.3. S47E Cytc Expressing Cells Have a Lower Mitochondrial Membrane Potential (ΔΨm) That Corresponds to Lower ROS Production
3.4. Phosphomimetic S47E Cytc Attenuates ROS Production upon Oxygen-Glucose Deprivation/Reoxygenation (OGD/R)
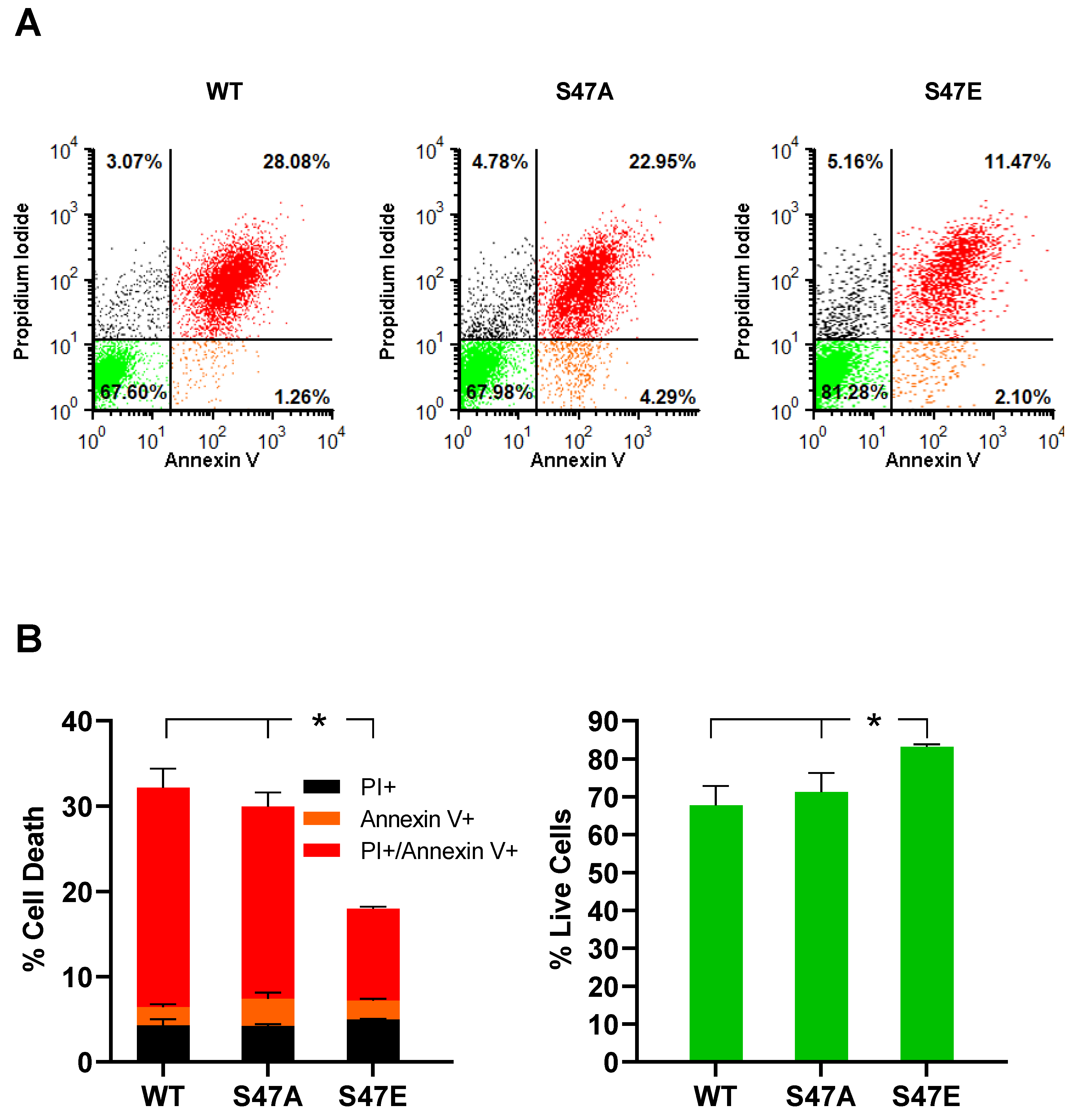
3.5. Phosphomimetic S47E Cytc Protects against Cell Death upon OGD/R and H2O2 Treatment
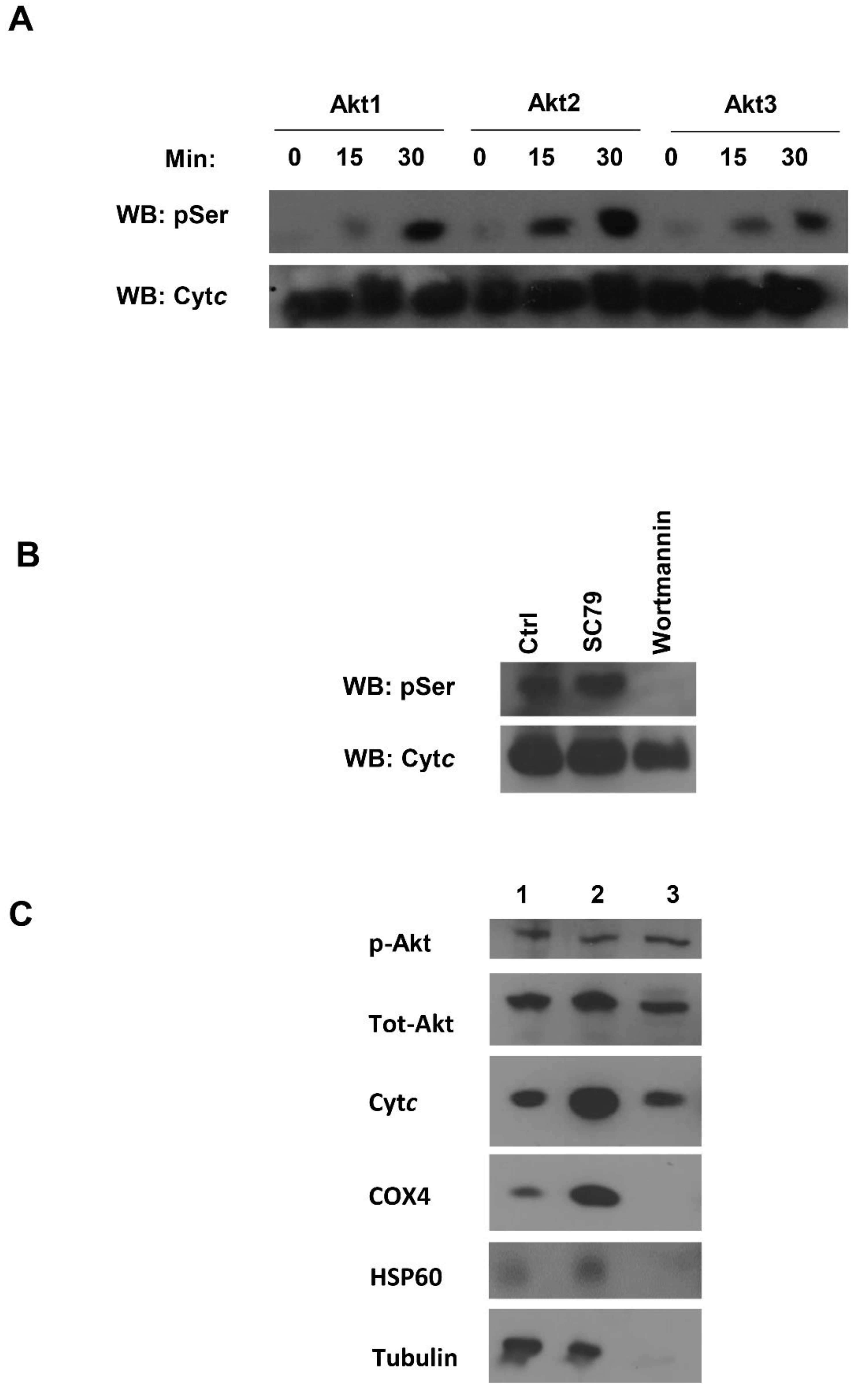
3.6. Akt Is a Potential Kinase Mediating S47 Phosphorylation of Cytc
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| COX | cytochrome c oxidase |
| Cytc | cytochrome c |
| ΔΨm | mitochondrial membrane potential |
| ETC | electron transport chain |
| OGD/R | oxygen-glucose deprivation/reoxygenation |
| OxPhos | oxidative phosphorylation |
| ROS | reactive oxygen species |
References
- Hüttemann, M.; Pecina, P.; Rainbolt, M.; Sanderson, T.H.; Kagan, V.E.; Samavati, L.; Doan, J.W.; Lee, I. The multiple functions of cytochrome c and their regulation in life and death decisions of the mammalian cell: From respiration to apoptosis. Mitochondrion 2011, 11, 369–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalpage, H.A.; Wan, J.; Morse, P.T.; Zurek, M.P.; Turner, A.A.; Khobeir, A.; Yazdi, N.; Hakim, L.; Liu, J.; Vaishnav, A.; et al. Cytochrome c phosphorylation: Control of mitochondrial electron transport chain flux and apoptosis. Int. J. Biochem. Cell Biol. 2020, 121, 105704. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalpage, H.A.; Bazylianska, V.; Recanati, M.A.; Fite, A.; Liu, J.; Wan, J.; Mantena, N.; Malek, M.H.; Podgorski, I.; Heath, E.I.; et al. Tissue-specific regulation of cytochrome c by post-translational modifications: Respiration, the mitochondrial membrane potential, ROS, and apoptosis. FASEB J. 2019, 33, 1540–1553. [Google Scholar] [CrossRef] [PubMed]
- Dalmonte, M.E.; Forte, E.; Genova, M.L.; Giuffre, A.; Sarti, P.; Lenaz, G. Control of respiration by cytochrome c oxidase in intact cells: Role of the membrane potential. J. Biol. Chem. 2009, 284, 32331–32335. [Google Scholar] [CrossRef] [Green Version]
- Kunz, W.S.; Kudin, A.; Vielhaber, S.; Elger, C.E.; Attardi, G.; Villani, G. Flux control of cytochrome c oxidase in human skeletal muscle. J. Biol. Chem. 2000, 275, 27741–27745. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.; Salomon, A.R.; Yu, K.; Doan, J.W.; Grossman, L.I.; Hüttemann, M. New prospects for an old enzyme: Mammalian cytochrome c is tyrosine-phosphorylated in vivo. Biochemistry 2006, 45, 9121–9128. [Google Scholar] [CrossRef] [Green Version]
- Sanderson, T.H.; Mahapatra, G.; Pecina, P.; Ji, Q.; Yu, K.; Sinkler, C.; Varughese, A.; Kumar, R.; Bukowski, M.J.; Tousignant, R.N.; et al. Cytochrome c is tyrosine 97 phosphorylated by neuroprotective insulin treatment. PLoS ONE 2013, 8, e78627. [Google Scholar] [CrossRef]
- Yu, H.; Lee, I.; Salomon, A.R.; Yu, K.; Hüttemann, M. Mammalian liver cytochrome c is tyrosine-48 phosphorylated in vivo, inhibiting mitochondrial respiration. Biochim. Et Biophys. Acta 2008, 1777, 1066–1071. [Google Scholar] [CrossRef] [Green Version]
- Mahapatra, G.; Varughese, A.; Ji, Q.; Lee, I.; Liu, J.; Vaishnav, A.; Sinkler, C.; Kapralov, A.A.; Moraes, C.T.; Sanderson, T.H.; et al. Phosphorylation of Cytochrome c Threonine 28 Regulates Electron Transport Chain Activity in Kidney: IMPLICATIONS FOR AMP KINASE. J. Biol. Chem. 2017, 292, 64–79. [Google Scholar] [CrossRef] [Green Version]
- Kalpage, H.A.; Vaishnav, A.; Liu, J.; Varughese, A.; Wan, J.; Turner, A.A.; Ji, Q.; Zurek, M.P.; Kapralov, A.A.; Kagan, V.E.; et al. Serine-47 phosphorylation of cytochrome c in the mammalian brain regulates cytochrome c oxidase and caspase-3 activity. FASEB J. 2019, 33, 13503–13514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, J.; Kalpage, H.A.; Vaishnav, A.; Liu, J.; Lee, I.; Mahapatra, G.; Turner, A.A.; Zurek, M.P.; Ji, Q.; Moraes, C.T.; et al. Regulation of Respiration and Apoptosis by Cytochrome c Threonine 58 Phosphorylation. Sci. Rep. 2019, 9, 15815. [Google Scholar] [CrossRef] [PubMed]
- Pecina, P.; Borisenko, G.G.; Belikova, N.A.; Tyurina, Y.Y.; Pecinova, A.; Lee, I.; Samhan-Arias, A.K.; Przyklenk, K.; Kagan, V.E.; Hüttemann, M. Phosphomimetic substitution of cytochrome c tyrosine 48 decreases respiration and binding to cardiolipin and abolishes ability to trigger downstream caspase activation. Biochemistry 2010, 49, 6705–6714. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Wang, J.; Feng, J. The neuroprotective effects of curcumin are associated with the regulation of the reciprocal function between autophagy and HIF-1alpha in cerebral ischemia-reperfusion injury. Drug Des. Dev. Ther. 2019, 13, 1135–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigo, R.; Fernandez-Gajardo, R.; Gutierrez, R.; Matamala, J.M.; Carrasco, R.; Miranda-Merchak, A.; Feuerhake, W. Oxidative stress and pathophysiology of ischemic stroke: Novel therapeutic opportunities. CNS Neurol. Disord. Drug Targets 2013, 12, 698–714. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, T.H.; Reynolds, C.A.; Kumar, R.; Przyklenk, K.; Hüttemann, M. Molecular mechanisms of ischemia-reperfusion injury in brain: Pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol. Neurobiol. 2013, 47, 9–23. [Google Scholar] [CrossRef] [Green Version]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef] [Green Version]
- Angelova, P.R.; Abramov, A.Y. Role of mitochondrial ROS in the brain: From physiology to neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef]
- Sun, Y.; Zhu, W.; Zhou, S.; Wang, Z.; Chen, X.; Jia, L. Exploring the model of PC12 apoptosis induced by OGSD/R through in vitro experiments. Oncotarget 2017, 8, 90176–90184. [Google Scholar] [CrossRef] [Green Version]
- Vempati, U.D.; Diaz, F.; Barrientos, A.; Narisawa, S.; Mian, A.M.; Millan, J.L.; Boise, L.H.; Moraes, C.T. Role of cytochrome C in apoptosis: Increased sensitivity to tumor necrosis factor alpha is associated with respiratory defects but not with lack of cytochrome C release. Mol. Cell. Biol. 2007, 27, 1771–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- TeSlaa, T.; Teitell, M.A. Techniques to monitor glycolysis. Methods Enzymol. 2014, 542, 91–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hüttemann, M.; Lee, I.; Pecinova, A.; Pecina, P.; Przyklenk, K.; Doan, J.W. Regulation of oxidative phosphorylation, the mitochondrial membrane potential, and their role in human disease. J. Bioenerg. Biomembr. 2008, 40, 445–456. [Google Scholar] [CrossRef]
- Breitzig, M.; Bhimineni, C.; Lockey, R.; Kolliputi, N. 4-Hydroxy-2-nonenal: A critical target in oxidative stress? Am. J. Physiol. Cell Physiol. 2016, 311, C537–C543. [Google Scholar] [CrossRef]
- Kalogeris, T.; Bao, Y.; Korthuis, R.J. Mitochondrial reactive oxygen species: A double edged sword in ischemia/reperfusion vs preconditioning. Redox Biol. 2014, 2, 702–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryou, M.G.; Mallet, R.T. An In Vitro Oxygen-Glucose Deprivation Model for Studying Ischemia-Reperfusion Injury of Neuronal Cells. Methods Mol. Biol. (Clifton N. J.) 2018, 1717, 229–235. [Google Scholar] [CrossRef]
- Crowley, L.C.; Marfell, B.J.; Scott, A.P.; Waterhouse, N.J. Quantitation of Apoptosis and Necrosis by Annexin V Binding, Propidium Iodide Uptake, and Flow Cytometry. Cold Spring Harb. Protoc. 2016. [Google Scholar] [CrossRef]
- Lecoeur, H. Nuclear apoptosis detection by flow cytometry: Influence of endogenous endonucleases. Exp. Cell Res. 2002, 277, 1–14. [Google Scholar] [CrossRef]
- Park, W.H. H(2)O(2) inhibits the growth of human pulmonary fibroblast cells by inducing cell death, GSH depletion and G1 phase arrest. Mol. Med. Rep. 2013, 7, 1235–1240. [Google Scholar] [CrossRef] [Green Version]
- Blom, N.; Sicheritz-Ponten, T.; Gupta, R.; Gammeltoft, S.; Brunak, S. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 2004, 4, 1633–1649. [Google Scholar] [CrossRef]
- Jo, H.; Mondal, S.; Tan, D.; Nagata, E.; Takizawa, S.; Sharma, A.K.; Hou, Q.; Shanmugasundaram, K.; Prasad, A.; Tung, J.K.; et al. Small molecule-induced cytosolic activation of protein kinase Akt rescues ischemia-elicited neuronal death. Proc. Natl. Acad. Sci. USA 2012, 109, 10581–10586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powis, G.; Bonjouklian, R.; Berggren, M.M.; Gallegos, A.; Abraham, R.; Ashendel, C.; Zalkow, L.; Matter, W.F.; Dodge, J.; Grindey, G.; et al. Wortmannin, a potent and selective inhibitor of phosphatidylinositol-3-kinase. Cancer Res. 1994, 54, 2419–2423. [Google Scholar] [PubMed]
- Ng, S.S.; Tsao, M.S.; Nicklee, T.; Hedley, D.W. Wortmannin inhibits pkb/akt phosphorylation and promotes gemcitabine antitumor activity in orthotopic human pancreatic cancer xenografts in immunodeficient mice. Clin. Cancer Res. 2001, 7, 3269–3275. [Google Scholar] [PubMed]
- Bijur, G.N.; Jope, R.S. Rapid accumulation of Akt in mitochondria following phosphatidylinositol 3-kinase activation. J. Neurochem. 2003, 87, 1427–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchi, S.; Corricelli, M.; Branchini, A.; Vitto, V.A.M.; Missiroli, S.; Morciano, G.; Perrone, M.; Ferrarese, M.; Giorgi, C.; Pinotti, M.; et al. Akt-mediated phosphorylation of MICU1 regulates mitochondrial Ca(2+) levels and tumor growth. EMBO J. 2019, 38. [Google Scholar] [CrossRef]
- Chae, Y.C.; Vaira, V.; Caino, M.C.; Tang, H.Y.; Seo, J.H.; Kossenkov, A.V.; Ottobrini, L.; Martelli, C.; Lucignani, G.; Bertolini, I.; et al. Mitochondrial Akt Regulation of Hypoxic Tumor Reprogramming. Cancer Cell 2016, 30, 257–272. [Google Scholar] [CrossRef] [Green Version]
- Santi, S.A.; Lee, H. The Akt isoforms are present at distinct subcellular locations. Am. J. Physiol. 2010, 298, C580–C591. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, J.C.; Siegelin, M.D.; Vaira, V.; Faversani, A.; Tavecchio, M.; Chae, Y.C.; Lisanti, S.; Rampini, P.; Giroda, M.; Caino, M.C.; et al. Adaptive mitochondrial reprogramming and resistance to PI3K therapy. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [Green Version]
- Narisawa, S.; Hecht, N.B.; Goldberg, E.; Boatright, K.M.; Reed, J.C.; Millan, J.L. Testis-specific cytochrome c-null mice produce functional sperm but undergo early testicular atrophy. Mol. Cell. Biol. 2002, 22, 5554–5562. [Google Scholar] [CrossRef] [Green Version]
- Limbach, K.J.; Wu, R. Characterization of a mouse somatic cytochrome c gene and three cytochrome c pseudogenes. Nucleic Acids Res. 1985, 13, 617–630. [Google Scholar] [CrossRef] [Green Version]
- Sousa, J.S.; D’Imprima, E.; Vonck, J. Mitochondrial Respiratory Chain Complexes. Sub Cell. Biochem. 2018, 87, 167–227. [Google Scholar] [CrossRef]
- Perez-Mejias, G.; Guerra-Castellano, A.; Diaz-Quintana, A.; De la Rosa, M.A.; Diaz-Moreno, I. Cytochrome c: Surfing Off of the Mitochondrial Membrane on the Tops of Complexes III and IV. Comput. Struct. Biotechnol. 2019, 17, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Vempati, U.D.; Han, X.; Moraes, C.T. Lack of cytochrome c in mouse fibroblasts disrupts assembly/stability of respiratory complexes I and IV. J. Biol. Chem. 2009, 284, 4383–4391. [Google Scholar] [CrossRef] [Green Version]
- Kokhan, O.; Wraight, C.A.; Tajkhorshid, E. The binding interface of cytochrome c and cytochrome c(1) in the bc(1) complex: Rationalizing the role of key residues. Biophys J. 2010, 99, 2647–2656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaim, G.; Dimroth, P. ATP synthesis by F-type ATP synthase is obligatorily dependent on the transmembrane voltage. EMBO J. 1999, 18, 4118–4127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.S. Cooperation of a "reactive oxygen cycle" with the Q cycle and the proton cycle in the respiratory chain--superoxide generating and cycling mechanisms in mitochondria. J. Bioenerg. Biomembr. 1999, 31, 367–376. [Google Scholar] [CrossRef]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef] [Green Version]
- Miklos, Z.; Ivanics, T.; Roemen, T.H.; van der Vusse, G.J.; Dezsi, L.; Szekeres, M.; Kemecsei, P.; Toth, A.; Kollai, M.; Ligeti, L. Time related changes in calcium handling in the isolated ischemic and reperfused rat heart. Mol. Cell. Biochem. 2003, 250, 115–124. [Google Scholar] [CrossRef]
- Bull, R.; Finkelstein, J.P.; Galvez, J.; Sanchez, G.; Donoso, P.; Behrens, M.I.; Hidalgo, C. Ischemia enhances activation by Ca2+ and redox modification of ryanodine receptor channels from rat brain cortex. J. Neurosci. 2008, 28, 9463–9472. [Google Scholar] [CrossRef]
- Robb-Gaspers, L.D.; Burnett, P.; Rutter, G.A.; Denton, R.M.; Rizzuto, R.; Thomas, A.P. Integrating cytosolic calcium signals into mitochondrial metabolic responses. EMBO J. 1998, 17, 4987–5000. [Google Scholar] [CrossRef] [Green Version]
- McCormack, J.G.; Denton, R.M. The role of intramitochondrial Ca2+ in the regulation of oxidative phosphorylation in mammalian tissues. Biochem. Soc. Trans. 1993, 21, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Hopper, R.K.; Carroll, S.; Aponte, A.M.; Johnson, D.T.; French, S.; Shen, R.F.; Witzmann, F.A.; Harris, R.A.; Balaban, R.S. Mitochondrial matrix phosphoproteome: Effect of extra mitochondrial calcium. Biochemistry 2006, 45, 2524–2536. [Google Scholar] [CrossRef] [Green Version]
- Guerra-Castellano, A.; Diaz-Moreno, I.; Velazquez-Campoy, A.; De la Rosa, M.A.; Diaz-Quintana, A. Structural and functional characterization of phosphomimetic mutants of cytochrome c at threonine 28 and serine 47. Biochim. Et Biophys. Acta 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luan, Q.; Pan, L.; He, D.; Gong, X.; Zhou, H. SC79, the AKT Activator Protects Cerebral Ischemia in a Rat Model of Ischemia/Reperfusion Injury. Med. Sci. Mon. Int. Med. J. Exp. Clin. Res. 2018, 24, 5391–5397. [Google Scholar] [CrossRef]
- Liu, X.; Kiss, G.K.; Mellender, S.J.; Weiss, H.R.; Chi, O.Z. Activation of Akt by SC79 decreased cerebral infarct in early cerebral ischemia-reperfusion despite increased BBB disruption. Neurosci. Lett. 2018, 681, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Weiss, H.R.; Chi, O.Z.; Kiss, G.K.; Liu, X.; Damito, S.; Jacinto, E. Akt activation improves microregional oxygen supply/consumption balance after cerebral ischemia-reperfusion. Brain Res. 2018, 1683, 48–54. [Google Scholar] [CrossRef]
- Sanderson, T.H.; Wider, J.M.; Lee, I.; Reynolds, C.A.; Liu, J.; Lepore, B.; Tousignant, R.; Bukowski, M.J.; Johnston, H.; Fite, A.; et al. Publisher Correction: Inhibitory modulation of cytochrome c oxidase activity with specific near-infrared light wavelengths attenuates brain ischemia/reperfusion injury. Sci. Rep. 2018, 8, 6729. [Google Scholar] [CrossRef] [Green Version]
- Strubakos, C.D.; Malik, M.; Wider, J.M.; Lee, I.; Reynolds, C.A.; Mitsias, P.; Przyklenk, K.; Huttemann, M.; Sanderson, T.H. Non-invasive treatment with near-infrared light: A novel mechanisms-based strategy that evokes sustained reduction in brain injury after stroke. J. Cereb. Blood Flow Metab. 2020, 40, 833–844. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cytc Function | WT | S47A | S47E/S47D | EV | Reference |
|---|---|---|---|---|---|
| Brain cytochrome c oxidase (COX) activity | 100% | ~82% | ~46% | - | [12] |
| Caspase-3 activity | 100% | ~38% | ~35% | - | [12] |
| 100% | ~50% | ~30% | [53] | ||
| Cardiolipin peroxidase activity (cardiolipin: Cytc) | 100% (10:1) | ~37% (10:1) | ~50% (10:1) | - | [12] |
| 100% (20:1) | ~54% (20:1) | ~51% (20:1) | |||
| 100% (100:1) | ~60% (100:1) | ~80% (100:1) | [53] | ||
| Intact cell respiration | 100% | ~81% | ~42% | ~15% | This study |
| Mitochondrial membrane potential | 100% | ~93% | ~79% | ~55% | This study |
| Mitochondrial ROS production | 100% | ~74% | ~70% | ~63% | This study |
| ATP production | 100% | ~100% | ~84% | ~28% | This study |
| Cell death (after OGD/R) | ~32% | ~30% | ~18% | - | This study |
| Cell death (after H2O2 treatment) | ~25% | ~13% | ~11% | - | This study |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalpage, H.A.; Wan, J.; Morse, P.T.; Lee, I.; Hüttemann, M. Brain-Specific Serine-47 Modification of Cytochrome c Regulates Cytochrome c Oxidase Activity Attenuating ROS Production and Cell Death: Implications for Ischemia/Reperfusion Injury and Akt Signaling. Cells 2020, 9, 1843. https://doi.org/10.3390/cells9081843
Kalpage HA, Wan J, Morse PT, Lee I, Hüttemann M. Brain-Specific Serine-47 Modification of Cytochrome c Regulates Cytochrome c Oxidase Activity Attenuating ROS Production and Cell Death: Implications for Ischemia/Reperfusion Injury and Akt Signaling. Cells. 2020; 9(8):1843. https://doi.org/10.3390/cells9081843
Chicago/Turabian StyleKalpage, Hasini A., Junmei Wan, Paul T. Morse, Icksoo Lee, and Maik Hüttemann. 2020. "Brain-Specific Serine-47 Modification of Cytochrome c Regulates Cytochrome c Oxidase Activity Attenuating ROS Production and Cell Death: Implications for Ischemia/Reperfusion Injury and Akt Signaling" Cells 9, no. 8: 1843. https://doi.org/10.3390/cells9081843