Diverse Roles of Annexin A6 in Triple-Negative Breast Cancer Diagnosis, Prognosis and EGFR-Targeted Therapies

 , ,
, ,  ,
,  and
and 
Abstract
:1. Introduction
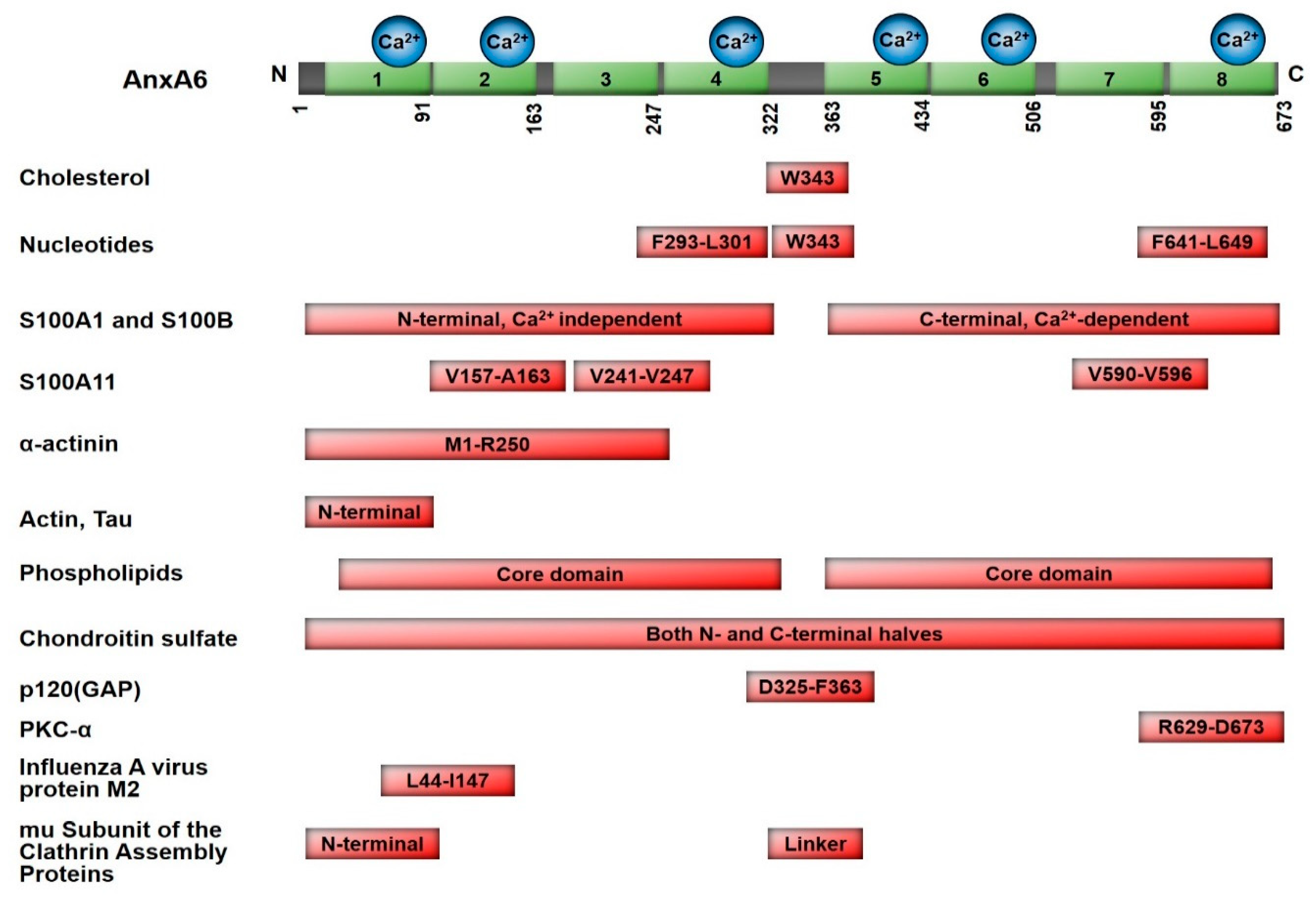
2. Molecular Characteristics and AnxA6-Mediated Functions
2.1. Ca2+-Dependent Interaction with Cellular Membranes
2.2. Cholesterol Binding and Subcellular Localization
2.3. Nucleotide-Binding Characteristic of Annexins
2.4. Scaffolding Functions of AnxA6
3. The Multiple and Diverse Roles of AnxA6 in Tumor Cell Growth and Motility
3.1. Altered Expression of AnxA6 in Tumor Cell Proliferation
3.2. Tumor Cell Motility and Invasiveness Mediated by AnxA6
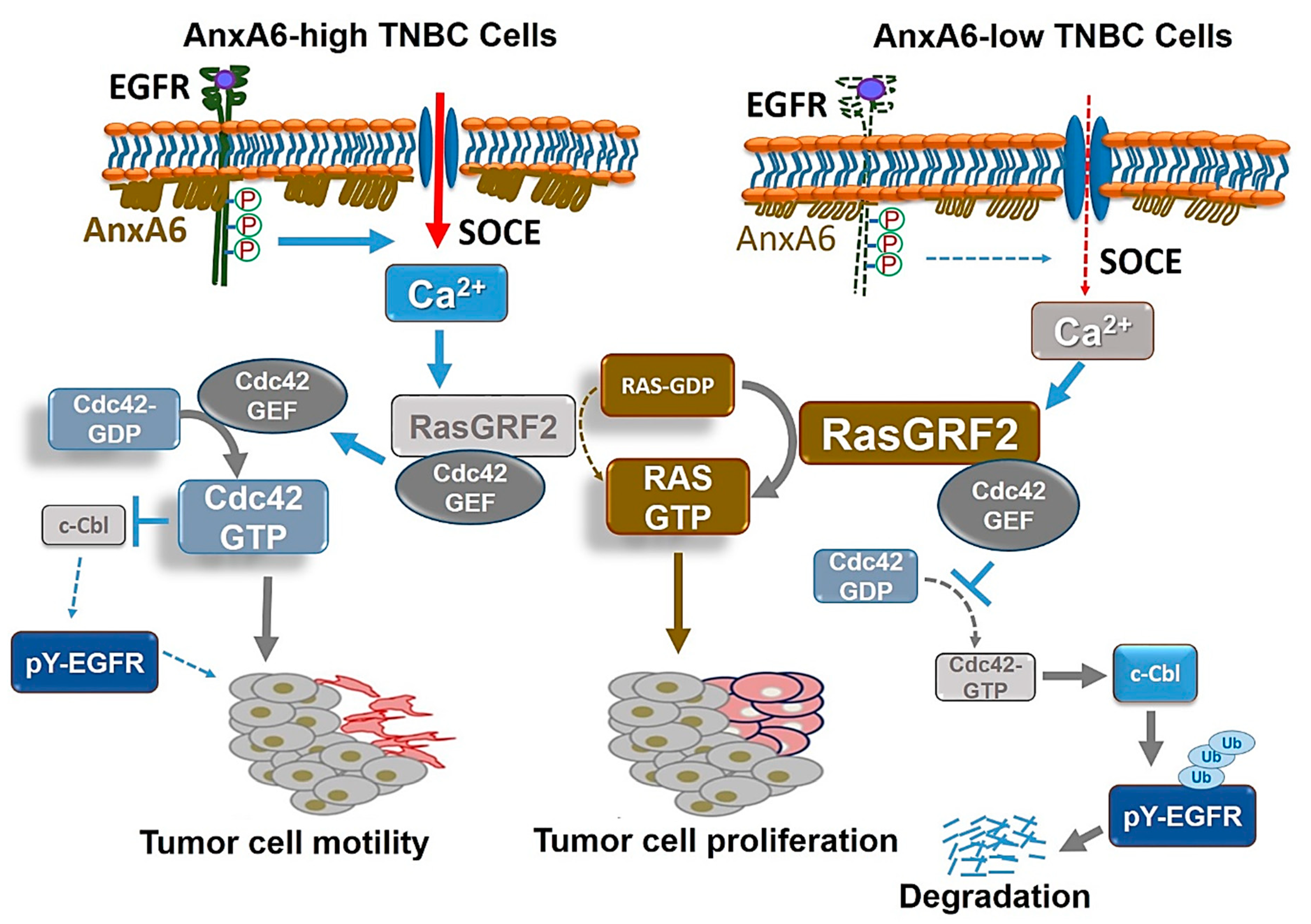
3.3. Modulation of the Effector Functions of Ca2+-Activated Ras Guanine Nucleotide Releasing Factor 2 (RasGRF2) by AnxA6
4. Relevance of Annexin A6 in Diagnosis, Prognosis and Therapeutic Interventions
4.1. AnxA6 as a Biomarker for Cancer Progression
4.2. Lack of Efficacy of EGFR-Targeted Therapies in the Treatment of TNBC
4.3. AnxA6 as a Predictor of Breast Cancer Recurrence and Response to Therapy
4.4. Upregulation of AnxA6 Expression and the Development of Acquired Resistance
5. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- DeSantis, C.E.; Ma, J.; Bryan, L.; Jemal, A. Breast cancer statistics. CA A Cancer J. Clin. 2013, 64, 52–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perou, C.M. Molecular Stratification of Triple-Negative Breast Cancers. Oncologist 2011, 16, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prat, A.; Adamo, B.; Cheang, M.C.U.; Anders, C.K.; Carey, L.A.; Perou, C.M. Molecular Characterization of Basal-Like and Non-Basal-Like Triple-Negative Breast Cancer. Oncologist 2013, 18, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [Green Version]
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Covington, K.R.; Contreras, A.; Fuqua, S.A.; Savage, M.I.; Osborne, C.K.; Hilsenbeck, S.G.; Chang, J.C.; et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin. Cancer Res. 2014, 21, 1688–1698. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, B.D.; Jovanović, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef]
- Liu, Y.-R.; Jiang, Y.-Z.; Xu, X.-E.; Yu, K.-D.; Jin, X.; Hu, X.; Zuo, W.-J.; Hao, S.; Wu, J.; Liu, G.-Y.; et al. Comprehensive transcriptome analysis identifies novel molecular subtypes and subtype-specific RNAs of triple-negative breast cancer. Breast Cancer Res. 2016, 18, 33. [Google Scholar] [CrossRef] [Green Version]
- Carey, L.A.; Perou, C.M.; Livasy, C.A.; Dressler, L.G.; Cowan, D.; Conway, K.; Karaca, G.; Troester, M.A.; Tse, C.K.; Edmiston, S.; et al. Race, Breast Cancer Subtypes, and Survival in the Carolina Breast Cancer Study. JAMA 2006, 295, 2492–2502. [Google Scholar] [CrossRef] [Green Version]
- Cheang, M.C.U.; Voduc, D.; Bajdik, C.; Leung, S.; McKinney, S.; Chia, S.K.; Perou, C.M.; Nielsen, T.O. Basal-Like Breast Cancer Defined by Five Biomarkers Has Superior Prognostic Value than Triple-Negative Phenotype. Clin. Cancer Res. 2008, 14, 1368–1376. [Google Scholar] [CrossRef] [Green Version]
- Rakha, E.A.; Reis-Filho, J.S. Basal-like breast carcinoma: From expression profiling to routine practice. Arch. Pathol. Lab. Med. 2009, 133, 1041–1063. [Google Scholar]
- Doody, J.F.; Wang, Y.; Patel, S.N.; Joynes, C.; Lee, S.P.; Gerlak, J.; Rolser, R.L.; Li, Y.; Steiner, P.; Bassi, R.; et al. Inhibitory activity of cetuximab on epidermal growth factor receptor mutations in non small cell lung cancers. Mol. Cancer Ther. 2007, 6, 2642–2651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baselga, J.; Albanell, J. Epithelial growth factor receptor interacting agents. Hematol. Clin. N. Am. 2002, 16, 1041–1063. [Google Scholar] [CrossRef]
- Carey, K.D.; Garton, A.J.; Romero, M.S.; Kahler, J.; Thomson, S.; Ross, S.; Park, F.; Haley, J.D.; Gibson, N.; Sliwkowski, M.X. Kinetic analysis of epidermal growth factor receptor somatic mutant proteins shows increased sensitivity to the epidermal growth factor receptor tyrosine kinase inhibitor, erlotinib. Cancer Res. 2006, 66, 8163–8171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arteaga, C.L. EGF Receptor As a Therapeutic Target: Patient Selection and Mechanisms of Resistance to Receptor-Targeted Drugs. J. Clin. Oncol. 2003, 21, 289–291. [Google Scholar] [CrossRef] [PubMed]
- Nakai, K.; Hung, M.C.; Yamaguchi, H. A perspective on anti-EGFR therapies targeting triple-negative breast cancer. Am. J. Cancer Res. 2016, 6, 1609–1623. [Google Scholar]
- Enrich, C.; Rentero, C.; De Muga, S.V.; Reverter, M.; Mulay, V.; Wood, P.; Köse, M.; Grewal, T. Annexin A6—Linking Ca2+ signaling with cholesterol transport. Biochim. Biophys. Acta. 2011, 1813, 935–947. [Google Scholar] [CrossRef] [Green Version]
- Enrich, C.; Rentero, C.; Grewal, T. Annexin A6 in the liver: From the endocytic compartment to cellular physiology. Biochim. Biophys. Acta. 2017, 1864, 933–946. [Google Scholar] [CrossRef]
- Vila de Muga, S.; Timpson, P.; Cubells, L.; Evans, R.; Hayes, T.E.; Rentero, C.; Hegemann, A.; Reverter, M.; Leschner, J.; Pol, A.; et al. Annexin A6 inhibits Ras signalling in breast cancer cells. Oncogene 2009, 28, 363–377. [Google Scholar] [CrossRef] [Green Version]
- Whalen, D.S.; Widatalla, S.E.; Korolkova, O.Y.; Nangami, G.S.; Beasley, H.K.; Williams, S.D.; Virgous, C.; Lehmann, B.D.; Ochieng, J.; Sakwe, A.M. Implication of calcium activated RasGRF2 in Annexin A6-mediated breast tumor cell growth and motility. Oncotarget 2019, 10, 133–151. [Google Scholar] [CrossRef] [Green Version]
- Widatalla, S.E.; Korolkova, O.Y.; Whalen, D.S.; Goodwin, J.S.; Williams, K.P.; Ochieng, J.; Sakwe, A.M. Lapatinib-induced annexin A6 upregulation as an adaptive response of triple-negative breast cancer cells to EGFR tyrosine kinase inhibitors. Carcinogenesis 2019, 40, 998–1009. [Google Scholar] [CrossRef] [Green Version]
- Sakwe, A.M.; Koumangoye, R.; Guillory, B.; Ochieng, J. Annexin A6 contributes to the invasiveness of breast carcinoma cells by influencing the organization and localization of functional focal adhesions. Exp. Cell Res. 2011, 317, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Monastyrskaya, K.; Babiychuk, E.B.; Hostettler, A.; Wood, P.; Grewal, T.; Draeger, A. Plasma Membrane-associated Annexin A6 Reduces Ca2+Entry by Stabilizing the Cortical Actin Cytoskeleton. J. Boil. Chem. 2009, 284, 17227–17242. [Google Scholar] [CrossRef] [Green Version]
- Strzelecka-Kiliszek, A.; Buszewska, M.E.; Podszywalow-Bartnicka, P.; Pikula, S.; Otulak, K.; Buchet, R.; Bandorowicz-Pikula, J. Calcium- and pH-dependent localization of annexin A6 isoforms in Balb/3T3 fibroblasts reflecting their potential participation in vesicular transport. J. Cell Biochem. 2008, 104, 418–434. [Google Scholar] [CrossRef]
- Koese, M.; Rentero, C.; Kota, B.P.; Hoque, M.; Cairns, R.; Wood, P.; De Muga, S.V.; Reverter, M.; Alvarez-Guaita, A.; Monastyrskaya, K.; et al. Annexin A6 is a scaffold for PKCα to promote EGFR inactivation. Oncogene 2012, 32, 2858–2872. [Google Scholar] [CrossRef] [Green Version]
- Seedorf, K.; Sherman, M.; Ullrich, A. Protein kinase C mediates short- and long-term effects on receptor tyrosine kinases. Regulation of tyrosine phosphorylation and degradation. Ann. N. Y. Acad. Sci. 1995, 11, 18953–18960. [Google Scholar] [CrossRef] [Green Version]
- Cubells, L.; De Muga, S.V.; Tebar, F.; Wood, P.; Evans, R.; Ingelmo-Torres, M.; Calvo, M.; Gaus, K.; Pol, A.; Grewal, T.; et al. Annexin A6-Induced Alterations in Cholesterol Transport and Caveolin Export from the Golgi Complex. Traffic 2007, 8, 1568–1589. [Google Scholar] [CrossRef]
- García-Melero, A.; Reverter, M.; Hoque, M.; Meneses-Salas, E.; Köse, M.; Conway, J.R.W.; Johnsen, C.H.; Alvarez-Guaita, A.; Morales-Paytuví, F.; Elmaghrabi, Y.A.; et al. Annexin A6 and Late Endosomal Cholesterol Modulate Integrin Recycling and Cell Migration. J. Boil. Chem. 2015, 291, 1320–1335. [Google Scholar] [CrossRef] [Green Version]
- Koumangoye, R.B.; Nangami, G.; Thompson, P.D.; Agboto, V.; Ochieng, J.; Sakwe, A.M. Reduced annexin A6 expression promotes the degradation of activated epidermal growth factor receptor and sensitizes invasive breast cancer cells to EGFR-targeted tyrosine kinase inhibitors. Mol. Cancer 2013, 12, 167. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Guaita, A.; De Muga, S.V.; Owen, D.M.; Williamson, D.J.; Magenau, A.; García-Melero, A.; Reverter, M.; Hoque, M.; Cairns, R.; Cornely, R.; et al. Evidence for annexin A6-dependent plasma membrane remodelling of lipid domains. Br. J. Pharmacol. 2015, 172, 1677–1690. [Google Scholar] [CrossRef] [Green Version]
- Babiychuk, E.B.; Monastyrskaya, K.; Potez, S.; Draeger, A. Intracellular Ca2+ operates a switch between repair and lysis of streptolysin O-perforated cells. Cell Death Differ. 2009, 16, 1126–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boye, T.L.; Maeda, K.; Pezeshkian, W.; Sønder, S.L.; Haeger, S.C.; Gerke, V.; Simonsen, A.C.; Nylandsted, J. Annexin A4 and A6 induce membrane curvature and constriction during cell membrane repair. Nat. Commun. 2017, 8, 1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demonbreun, A.R.; Fallon, K.S.; Oosterbaan, C.C.; Bogdanovic, E.; Warner, J.L.; Sell, J.J.; Page, P.G.; Quattrocelli, M.; Barefield, D.Y.; McNally, E.M. Recombinant annexin A6 promotes membrane repair and protects against muscle injury. J. Clin. Investig. 2019, 129, 4657–4670. [Google Scholar] [CrossRef] [PubMed]
- Krautbauer, S.; Haberl, E.M.; Eisinger, K.; Pohl, R.; Rein-Fischboeck, L.; Rentero, C.; Alvarez-Guaita, A.; Enrich, C.; Grewal, T.; Buechler, C.; et al. Annexin A6 regulates adipocyte lipid storage and adiponectin release. Mol. Cell. Endocrinol. 2017, 439, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Hulce, J.J.; Cognetta, A.B.; Niphakis, M.J.; Tully, S.E.; Cravatt, B.F. Proteome-wide mapping of cholesterol-interacting proteins in mammalian cells. Nat. Methods 2013, 10, 259–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayala-Sanmartin, J. Cholesterol enhances phospholipid binding and aggregation of annexins by their core domain. Biochem. Biophys. Res. 2001, 283, 72–79. [Google Scholar] [CrossRef]
- Domon, M.M.; Matar, G.; Strzelecka-Kiliszek, A.; Bandorowicz-Pikula, J.; Pikula, S.; Besson, F. Interaction of annexin A6 with cholesterol rich membranes is pH-dependent and mediated by the sterol OH. J. Colloid Interface Sci. 2010, 346, 436–441. [Google Scholar] [CrossRef]
- De Diego, I.; Schwartz, F.; Siegfried, H.; Dauterstedt, P.; Heeren, J.; Beisiegel, U.; Enrich, C.; Grewal, T. Cholesterol Modulates the Membrane Binding and Intracellular Distribution of Annexin 6. J. Boil. Chem. 2002, 277, 32187–32194. [Google Scholar] [CrossRef] [Green Version]
- Meneses-Salas, E.; García-Melero, A.; Blanco-Muñoz, P.; Jose, J.; Brenner, M.-S.; Lu, A.; Tebar, F.; Rentero, C.; Rentero, C.; Enrich, C. Selective Degradation Permits a Feedback Loop Controlling Annexin A6 and Cholesterol Levels in Endolysosomes of NPC1 Mutant Cells. Cells 2020, 9, 1152. [Google Scholar] [CrossRef]
- Reverter, M.; Rentero, C.; de Muga, S.V.; Alvarez-Guaita, A.; Mulay, V.; Cairns, R.; Wood, P.; Monastyrskaya, K.; Pol, A.; Tebar, F.; et al. Cholesterol transport from late endosomes to the Golgi regulates t-SNARE trafficking, assembly, and function. Mol. Biol Cell 2011, 22, 4108–4123. [Google Scholar] [CrossRef]
- Kuhnl, A.; Musiol, A.; Heitzig, N.; Johnson, D.E.; Ehrhardt, C.; Grewal, T.; Gerke, V.; Ludwig, S.; Rescher, U. Late Endosomal/Lysosomal Cholesterol Accumulation Is a Host Cell-Protective Mechanism Inhibiting Endosomal Escape of Influenza A Virus. mBio 2018, 24. [Google Scholar] [CrossRef] [Green Version]
- Musiol, A.; Gran, S.; Ehrhardt, C.; Ludwig, S.; Grewal, T.; Gerke, V.; Rescher, U. Annexin A6-Balanced Late Endosomal Cholesterol Controls Influenza A Replication and Propagation. mBio 2013, 4, e00608–e00613. [Google Scholar] [CrossRef] [Green Version]
- Creutz, C.E.; Hira, J.K.; Gee, V.E.; Eaton, J.M. Protection of the membrane permeability barrier by annexins. Biochemistry 2012, 51, 9966–9983. [Google Scholar]
- Potez, S.; Luginbühl, M.; Monastyrskaya, K.; Hostettler, A.; Draeger, A.; Babiychuk, E.B. Tailored Protection against Plasmalemmal Injury by Annexins with Different Ca2+ Sensitivities. J. Boil. Chem. 2011, 286, 17982–17991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandorowicz-Pikula, J.; Danieluk, M.; Wrzosek, A.; Bus, R.; Buchet, R.; Pikula, S. Annexin VI: An intracellular target for ATP. Acta Biochim Pol. 1999, 46, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Danieluk, M.; Pikula, S.; Bandorowicz-Pikula, J. Annexin VI interacts with adenine nucleotides and their analogs. Biochimie 1999, 81, 717–726. [Google Scholar] [CrossRef]
- Kirilenko, A.; Golczak, M.; Pikuła, S.; Bandorowicz-Pikuła, J. GTP-binding properties of the membrane-bound form of porcine liver annexin VI. Acta Biochim. Pol. 2001, 48, 851–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandorowicz-Pikula, J.; Kirilenko, A.; van Deursen, R.; Golczak., M.; Kuhnel, M.; Lancelin, J.M.; Pikula, S.; Buchet, R. A putative consensus sequence for the nucleotide-binding site of annexin A6. Biochemistry 2003, 42, 9137–9146. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Meng, J.; Huang, Y.; Wu, J.; Wang, B.; Ibrahim, M.M.; Tang, J. Guanine nucleotide-binding protein subunit beta-2-like 1, a new Annexin A7 interacting protein. Biochem. Biophys. Res. Commun. 2014, 445, 58–63. [Google Scholar] [CrossRef]
- Kirilenko, A.; Pikula, S.; Bandorowicz-Pikula, J. Effects of Mutagenesis of W343 in Human Annexin A6 Isoform 1 on Its Interaction with GTP: Nucleotide-Induced Oligomer Formation and Ion Channel Activity. Biochemistry 2006, 45, 4965–4973. [Google Scholar] [CrossRef]
- Mishra, S.; Chander, V.; Banerjee, P.; Oh, J.G.; Lifirsu, E.; Park, W.J.; Kim, D.H.; Bandyopadhyay, A. Interaction of annexin A6 with alpha actinin in cardiomyocytes. BMC Cell Biol. 2011, 12, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, J.K.; Nylandsted, J. S100 and annexin proteins identify cell membrane damage as the Achilles heel of metastatic cancer cells. Cell Cycle 2015, 14, 502–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nedjadi, T.; Kitteringham, N.; Campbell, F.; Jenkins, R.E.; Park, B.K.; Navarro, P.; Ashcroft, F.; Tepikin, A.V.; Neoptolemos, J.; Costello, E. S100A6 binds to annexin 2 in pancreatic cancer cells and promotes pancreatic cancer cell motility. Br. J. Cancer 2009, 101, 1145–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bode, G.; Luken, A.; Kerkhoff, C.; Roth, J.; Ludwig, S.; Nacken, W. Interaction between S100A8/A9 and annexin A6 is involved in the calcium-induced cell surface exposition of S100A8/A9. J. Biol. Chem. 2008, 283, 31776–31784. [Google Scholar] [CrossRef] [Green Version]
- Chang, N.; Sutherland, C.; Hesse, E.; Winkfein, R.; Wiehler, W.B.; Pho, M.; Veillette, C.; Li, S.; Wilson, D.P.; Kiss, E.; et al. Identification of a novel interaction between the Ca2+-binding protein S100A11 and the Ca2+- and phospholipid-binding protein annexin A6. Am. J. Physiol. Physiol. 2007, 292, C1417–C1430. [Google Scholar] [CrossRef] [Green Version]
- Garbuglia, M.; Verzini, M.; Donato, R. Annexin VI binds S100A1 and S100B and blocks the ability of S100A1 and S100B to inhibit desmin and GFAP assemblies into intermediate filaments. Cell Calcium 1998, 24, 177–191. [Google Scholar] [CrossRef]
- Gauthier-Kemper, A.; Alonso, M.S.; Sündermann, F.; Niewidok, B.; Fernández, M.-P.; Bakota, L.; Heinisch, J.J.; Brandt, R. Annexins A2 and A6 interact with the extreme N terminus of tau and thereby contribute to tau’s axonal localization. J. Boil. Chem. 2018, 293, 8065–8076. [Google Scholar] [CrossRef] [Green Version]
- Matrone, M.A.; Whipple, R.A.; Thompson, K.; Cho, E.; Vitolo, M.I.; Balzer, E.M.; Yoon, J.R.; Ioffe, O.B.; Tuttle, K.C.; Tan, M.; et al. Metastatic breast tumors express increased tau, which promotes microtentacle formation and the reattachment of detached breast tumor cells. Oncogene 2010, 29, 3217–3227. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, S.; Nohara, T.; Iwamoto, M.; Sumiyoshi, K.; Kimura, K.; Takahashi, Y.; Tanigawa, N. Tau Expression and Efficacy of Paclitaxel Treatment in Metastatic Breast Cancer. Poster Sess. Abstr. 2009, 69, 1138. [Google Scholar] [CrossRef]
- Takagi, H.; Asano, Y.; Yamakawa, N.; Matsumoto, I.; Kimata, K. Annexin 6 is a putative cell surface receptor for chondroitin sulfate chains. J. Cell Sci. 2002, 115. [Google Scholar]
- Ma, H.; Kien, F.; Manière, M.; Zhang, Y.; Lagarde, N.; Tse, K.S.; Poon, L.L.M.; Nal, B. Human Annexin A6 Interacts with Influenza A Virus Protein M2 and Negatively Modulates Infection. J. Virol. 2011, 86, 1789–1801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creutz, C.E.; Snyder, S.L. Interactions of annexins with the mu subunits of the clathrin assembly proteins. Biochemistry 2005, 44, 13795–13806. [Google Scholar] [PubMed]
- Gunteski-Hamblin, A.M.; Song, G.; Walsh, R.A.; Frenzke, M.; Boivin, G.P.; Dorn, G.W.; Kaetzel, M.A.; Horseman, N.D.; Dedman, J.R. Annexin VI overexpression targeted to heart alters cardiomyocyte function in transgenic mice. Am. J. Physiol. Circ. Physiol. 1996, 270, H1091–H1100. [Google Scholar] [CrossRef] [PubMed]
- Hazarika, P.; Sheldon, A.; Kaetzel, M.A.; Diaz-Munoz, M.; Hamilton, S.L.; Dedman, J.R. Regulation of the sarcoplasmic reticulum Ca2+-release channel requires intact annexin VI. J. Cell. Biochem. 1991, 46, 86–93. [Google Scholar] [CrossRef]
- Song, G.; Harding, S.E.; Duchen, M.R.; Tunwell, R.; O’Gara, P.; Hawkins, T.E.; Moss, S.E. Altered mechanical properties and intracellular calcium signaling in cardiomyocytes from annexin 6 null-mutant mice. FASEB J. 2002, 16, 622–624. [Google Scholar] [CrossRef] [Green Version]
- Schmitz-Peiffer, C.; Browne, C.L.; Walker, J.H.; Biden, T.J. Activated protein kinase C alpha associates with annexin VI from skeletal muscle. Biochem. J. 1998, 330, 675–681. [Google Scholar] [CrossRef]
- Davis, A.J.; Butt, J.T.; Walker, J.H.; Moss, S.E.; Gawler, D. The Ca2+-dependent lipid binding domain of P120GAP mediates protein-protein interactions with Ca2+-dependent membrane-binding proteins. Evidence for a direct interaction between annexin VI and P120GAP. J. Boil. Chem. 1996, 271, 24333–24336. [Google Scholar] [CrossRef] [Green Version]
- Grewal, T.; Evans, R.; Rentero, C.; Tebar, F.; Cubells, L.; De Diego, I.; Kirchhoff, M.F.; Hughes, W.E.; Heeren, J.; Rye, K.-A.; et al. Annexin A6 stimulates the membrane recruitment of p120GAP to modulate Ras and Raf-1 activity. Oncogene 2005, 24, 5809–5820. [Google Scholar] [CrossRef] [Green Version]
- Pons, M.; Grewal, T.; Rius, E.; Schnitgerhans, T.; Jäckle, S.; Enrich, C. Evidence for the Involvement of Annexin 6 in the Trafficking between the Endocytic Compartment and Lysosomes. Exp. Cell Res. 2001, 269, 13–22. [Google Scholar] [CrossRef]
- Campbell, K.A.; Minashima, T.; Zhang, Y.; Hadley, S.; Lee, Y.J.; Giovinazzo, J.; Quirno, M.; Kirsch, T. Annexin A6 interacts with p65 and stimulates NF-kappaB activity and catabolic events in articular chondrocytes. Arthritis Rheum. 2013, 65, 3120–3129. [Google Scholar] [CrossRef]
- Meneses-Salas, E.; Garcia-Melero, A.; Kanerva, K.; Blanco-Munoz, P.; Morales-Paytuvi, F.; Bonjoch, J.; Casas, J.; Egert, A.; Beevi, S.S.; Jose, J.; et al. Annexin A6 modulates TBC1D15/Rab7/StARD3 axis to control endosomal cholesterol export in NPC1 cells. Cell Mol. Life Sci. 2019, 77, 2839–2857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, R.; Römisch, J.; Paques, E.P. The crystal and molecular structure of human annexin V, an anticoagulant protein that binds to calcium and membranes. EMBO J. 1990, 9, 3867–3874. [Google Scholar] [CrossRef] [PubMed]
- Santamaria-Kisiel, L.; Rintala-Dempsey, A.C.; Shaw, G.S. Calcium-dependent and -independent interactions of the S100 protein family. Biochem. J. 2006, 396, 201–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locate, S.; Colyer, J.; Gawler, D.J.; Walker, J.H. Annexin A6 at the cardiac myocyte sarcolemma – Evidence for self-association and binding to actin. Cell Boil. Int. 2008, 32, 1388–1396. [Google Scholar] [CrossRef]
- Chow, A.; Gawler, D.J. Mapping the site of interaction between annexin VI and the p120GAP C2 domain. FEBS Lett. 1999, 460, 166–172. [Google Scholar] [CrossRef] [Green Version]
- Minashima, T.; Small, W.; Moss, S.E.; Kirsch, T. Intracellular Modulation of Signaling Pathways by Annexin A6 Regulates Terminal Differentiation of Chondrocytes. J. Boil. Chem. 2012, 287, 14803–14815. [Google Scholar] [CrossRef] [Green Version]
- Qi, H.; Liu, S.; Guo, C.; Wang, J.; Greenaway, F.T.; Sun, M.-Z. Role of annexin A6 in cancer. Oncol. Lett. 2015, 10, 1947–1952. [Google Scholar] [CrossRef] [Green Version]
- Cornely, R.; Pollock, A.H.; Rentero, C.; Norris, S.E.; Alvarez-Guaita, A.; Grewal, T.; Mitchell, T.; Enrich, C.; Moss, S.E.; Parton, R.G.; et al. Annexin A6 regulates interleukin-2-mediated T-cell proliferation. Immunol. Cell Boil. 2016, 94, 543–553. [Google Scholar] [CrossRef]
- Theobald, J.; Smith, P.D.; Jacob, S.M.; Moss, S.E. Expression of annexin VI in A431 carcinoma cells suppresses proliferation: A possible role for annexin VI in cell growth regulation. Biochim. Biophys. Acta 1994, 1223, 383–390. [Google Scholar] [CrossRef]
- Hoque, M.; Elmaghrabi, Y.A.; Köse, M.; Beevi, S.S.; Jose, J.; Meneses-Salas, E.; Blanco-Muñoz, P.; Conway, J.R.W.; Swarbrick, A.; Timpson, P.; et al. Annexin A6 improves anti-migratory and anti-invasive properties of tyrosine kinase inhibitors in EGFR overexpressing human squamous epithelial cells. FEBS J. 2019. [Google Scholar] [CrossRef]
- Fleet, A.; Ashworth, R.; Kubista, H.; Edwards, H.; Bolsover, S.; Mobbs, P.; Moss, S.E. Inhibition of EGF-Dependent Calcium Influx by Annexin VI is Splice Form-Specific. Biochem. Biophys. Res. Commun. 1999, 260, 540–546. [Google Scholar] [CrossRef]
- Alvarez-Guaita, A.; Blanco-Muñoz, P.; Meneses-Salas, E.; Wahba, M.; Pollock, A.H.; Bosch, M.; Gaus, K.; Lu, A.; Pol, A.; Tebar, F.; et al. Annexin A6 is critical to maintain glucose homeostasis and survival during liver regeneration. Hepatology 2020. [Google Scholar] [CrossRef]
- Cairns, R.; Alvarez-Guaita, A.; Martínez-Saludes, I.; Wason, S.J.; Hanh, J.; Nagarajan, S.; Hosseini-Beheshti, E.; Monastyrskaya, K.; Hoy, A.J.; Buechler, C.; et al. Role of hepatic Annexin A6 in fatty acid-induced lipid droplet formation. Exp. Cell Res. 2017, 358, 397–410. [Google Scholar] [CrossRef]
- Monastyrskaya, K.; Tschumi, F.; Babiychuk, E.B.; Stroka, D.; Draeger, A. Annexins sense changes in intracellular pH during hypoxia. Biochem. J. 2007, 409, 65–75. [Google Scholar] [CrossRef]
- Chlystun, M.; Campanella, M.; Law, A.-L.; Duchen, M.; Fatimathas, L.; Levine, T.P.; Gerke, V.; Moss, S.E. Regulation of Mitochondrial Morphogenesis by Annexin A6. PLoS ONE 2013, 8, e53774. [Google Scholar] [CrossRef] [Green Version]
- Pfander, D.; Swoboda, B.; Kirsch, T. Expression of Early and Late Differentiation Markers (Proliferating Cell Nuclear Antigen, Syndecan-3, Annexin VI, and Alkaline Phosphatase) by Human Osteoarthritic Chondrocytes. Am. J. Pathol. 2001, 159, 1777–1783. [Google Scholar] [CrossRef] [Green Version]
- Shah, A.; Taneyhill, L.A. Differential expression pattern of Annexin A6 in chick neural crest and placode cells during cranial gangliogenesis. Gene Expr. Patterns 2015, 18, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.Y.; Taneyhill, L.A. Annexin a6 modulates chick cranial neural crest cell emigration. PLoS ONE 2012, 7, e44903. [Google Scholar] [CrossRef] [Green Version]
- Ridley, A.J. Rho GTPases and cell migration. J. Cell Sci. 2001, 114, 2713–2722. [Google Scholar]
- Koumangoye, R.B.; Sakwe, A.M.; Goodwin, J.S.; Patel, T.; Ochieng, J. Detachment of Breast Tumor Cells Induces Rapid Secretion of Exosomes Which Subsequently Mediate Cellular Adhesion and Spreading. PLoS ONE 2011, 6, e24234. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Weaver, A.M. Extracellular vesicles: Important collaborators in cancer progression. Essays Biochem. 2018, 62, 149–163. [Google Scholar]
- Ochieng, J.; Pratap, S.; Khatua, A.K.; Sakwe, A.M. Anchorage-independent growth of breast carcinoma cells is mediated by serum exosomes. Exp. Cell Res. 2009, 315, 1875–1888. [Google Scholar] [CrossRef] [Green Version]
- Kundranda, M.N.; Ray, S.; Saria, M.; Friedman, D.; Matrisian, L.M.; Lukyanov, P.; Ochieng, J. Annexins expressed on the cell surface serve as receptors for adhesion to immobilized fetuin-A. Biochim. Biophys. Acta (BBA) Bioenerg. 2004, 1693, 111–123. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, D.; Dowling, P.; Joyce, H.; McAuley, E.; McCann, A.; Henry, M.; McGovern, B.; Barham, P.; Kelleher, F.C.; Murphy, J.; et al. A novel inhibitory anti-invasive MAb isolated using phenotypic screening highlights AnxA6 as a functionally relevant target protein in pancreatic cancer. Br. J. Cancer 2017, 117, 1326–1335. [Google Scholar] [CrossRef]
- Minashima, T.; Kirsch, T. Annexin A6 regulates catabolic events in articular chondrocytes via the modulation of NF-kappaB and Wnt/ss-catenin signaling. PLoS ONE 2018, 13, e0197690. [Google Scholar] [CrossRef] [Green Version]
- Leca, J.; Martinez, S.; Lac, S.; Nigri, J.; Secq, V.; Rubis, M.; Bressy, C.; Sergé, A.; Lavaut, M.-N.; Dusetti, N.; et al. Cancer-associated fibroblast-derived annexin A6+ extracellular vesicles support pancreatic cancer aggressiveness. J. Clin. Investig. 2016, 126, 4140–4156. [Google Scholar] [CrossRef] [Green Version]
- Keklikoglou, I.; Cianciaruso, C.; Güç, E.; Squadrito, M.L.; Spring, L.M.; Tazzyman, S.; Lambein, L.; Poissonnier, A.; Ferraro, G.B.; Baer, C.; et al. Chemotherapy elicits pro-metastatic extracellular vesicles in breast cancer models. Nat. Cell Biol. 2018, 21, 190–202. [Google Scholar] [CrossRef] [Green Version]
- Calvo, F.; Sanz-Moreno, V.; Agudo-Ibáñez, L.; Wallberg, F.; Sahai, E.; Marshall, C.J.; Crespo, P. RasGRF suppresses Cdc42-mediated tumour cell movement, cytoskeletal dynamics and transformation. Nat. Cell Biol. 2011, 13, 819–826. [Google Scholar] [CrossRef]
- Ma, X.; Espana-Serrano, L.; Kim, W.J.; Thayele Purayil, H.; Nie, Z.; Daaka, Y. betaArrestin1 regulates the guanine nucleotide exchange factor RasGRF2 expression and the small GTPase Rac-mediated formation of membrane protrusion and cell motility. J. Biol. Chem. 2014, 289, 13638–13650. [Google Scholar] [CrossRef] [Green Version]
- Anborgh, P.H.; Qian, X.; Papageorge, A.G.; Vass, W.C.; DeClue, J.E.; Lowy, D.R. Ras-Specific Exchange Factor GRF: Oligomerization through Its Dbl Homology Domain and Calcium-Dependent Activation of Raf. Mol. Cell. Boil. 1999, 19, 4611–4622. [Google Scholar] [CrossRef] [Green Version]
- De Hoog, C.L.; Koehler, J.A.; Goldstein, M.D.; Taylor, P.; Figeys, D.; Moran, M.F. Ras Binding Triggers Ubiquitination of the Ras Exchange Factor Ras-GRF2. Mol. Cell. Boil. 2001, 21, 2107–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tebar, F.; Chavero, A.; Agell, N.; Lu, A.; Rentero, C.; Enrich, C.; Grewal, T. Pleiotropic Roles of Calmodulin in the Regulation of KRas and Rac1 GTPases: Functional Diversity in Health and Disease. Int. J. Mol. Sci. 2020, 21, 3680. [Google Scholar] [CrossRef] [PubMed]
- Korolkova, O.Y.; Widatalla, S.E.; Whalen, D.S.; Nangami, G.N.; Abimbola, A.; Williams, S.D.; Beasley, H.K.; Reisenbichler, E.; Washington, M.K.; Ochieng, J.; et al. Reciprocal expression of Annexin A6 and RasGRF2 discriminates rapidly growing from invasive triple negative breast cancer subsets. PLoS ONE 2020, 15, e0231711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, M.H.H.; Husnjak, K.; Szymkiewicz, I.; Haglund, K.; Dikic, I. Cbl escapes Cdc42-mediated inhibition by downregulation of the adaptor molecule betaPix. Oncogene 2006, 25, 3071–3078. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.J.; Tu, S.; Cerione, R.A. Activated Cdc42 Sequesters c-Cbl and Prevents EGF Receptor Degradation. Cell 2003, 114, 715–725. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, D.S.; Shen, Y.; Wu, W.J. Growth and Motility Inhibition of Breast Cancer Cells by Epidermal Growth Factor Receptor Degradation Is Correlated with Inactivation of Cdc42. Cancer Res. 2006, 66, 3523–3530. [Google Scholar] [CrossRef] [Green Version]
- Noreen, S.; Gardener, Q.A.; Fatima, I.; Sadaf, S.; Akhtar, M.W. Up-Regulated Expression of Calcium Dependent Annexin A6: A Potential Biomarker of Ovarian Carcinoma. Proteom. Clin. Appl. 2019, e1900078. [Google Scholar]
- Lee, H.-S.; Kang, Y.; Tae, K.; Bae, G.-U.; Park, J.Y.; Cho, Y.H.; Yang, M. Proteomic Biomarkers for Bisphenol A–Early Exposure and Women’s Thyroid Cancer. Cancer Res. Treat. 2018, 50, 111–117. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Zhang, J.; Deng, Q.; Li, J.; Li, Z.; Xiao, Y.; Hu, S.; Li, T.; Tan, Q.; Li, X.; et al. Proteomic Profiling for Identification of Novel Biomarkers Differentially Expressed in Human Ovaries from Polycystic Ovary Syndrome Patients. PLoS ONE 2016, 11, e0164538. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, A.H.; Gopalakrishnan, V.; Kasi, P.M.; Zeng, X.; Malhotra, U.; Balasubramanian, J.; Visweswaran, S.; Sun, M.; Flint, M.; Davison, J.M.; et al. Evaluation of a 4-protein serum biomarker panel-biglycan, annexin-A6, myeloperoxidase, and protein S100-A9 (B-AMP)-for the detection of esophageal adenocarcinoma. Cancer 2014, 120, 3902–3913. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Pan, Q.; Stow, P.; Behm, F.G.; Goorha, R.; Pui, C.H.; Neale, G. Quantification of minimal residual disease in T-lineage acute lymphoblastic leukemia with the TAL-1 deletion using a standardized real-time PCR assay. Leukemia 2001, 15, 166–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francia, G.; Mitchell, S.D.; Moss, S.E.; Hanby, A.M.; Marshall, J.F.; Hart, I.R. Identification by differential display of annexin-VI, a gene differentially expressed during melanoma progression. Cancer Res. 1996, 56, 3855–3858. [Google Scholar] [PubMed]
- Lomnytska, M.; Becker, S.; Bodin, I.; Olsson, A.; Hellman, K.; Hellström, A.-C.; Mints, M.; Hellman, U.; Auer, G.; Andersson, S. Differential expression of ANXA6, HSP27, PRDX2, NCF2, and TPM4 during uterine cervix carcinogenesis: Diagnostic and prognostic value. Br. J. Cancer 2010, 104, 110–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Zhang, S.; Zhang, J.; Lam, E.; Liu, X.; Sun, J.; Feng, L.; Lu, H.; Yu, J.; Jin, H. Annexin A6 is down-regulated through promoter methylation in gastric cancer. Am. J. Transl. Res. 2013, 5, 555–562. [Google Scholar]
- Meier, E.M.; Rein-Fischboeck, L.; Pohl, R.; Wanninger, J.; Hoy, A.J.; Grewal, T.; Eisinger, K.; Krautbauer, S.; Liebisch, G.; Weiss, T.; et al. Annexin A6 protein is downregulated in human hepatocellular carcinoma. Mol. Cell. Biochem. 2016, 418, 81–90. [Google Scholar] [CrossRef]
- Lomnytska, M.I.; Becker, S.; Hellman, K.; Hellstrom, A.C.; Souchelnytskyi, S.; Mints, M.; Hellman, U.; Andersson, S.; Auer, G. Diagnostic protein marker patterns in squamous cervical cancer. Proteomics Clin. Appl 2010, 4, 17–31. [Google Scholar] [CrossRef]
- Grewal, T.; Hoque, M.; Conway, J.R.W.; Reverter, M.; Wahba, M.; Beevi, S.S.; Timpson, P.; Enrich, C.; Rentero, C. Annexin A6—A multifunctional scaffold in cell motility. Cell Adhes. Migr. 2017, 11, 288–304. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-S.; Coustan-Smith, E.; Suzuki, T.; Neale, G.A.; Mihara, K.; Pui, C.-H.; Campana, D. Identification of novel markers for monitoring minimal residual disease in acute lymphoblastic leukemia. Blood 2001, 97, 2115–2120. [Google Scholar] [CrossRef] [Green Version]
- Croci, S.; Recktenwald, C.V.; Lichtenfels, R.; Nicoletti, G.; Dressler, S.P.; De Giovanni, C.; Astolfi, A.; Palladini, A.; Shin-ya, K.; Landuzzi, L.; et al. Proteomic and PROTEOMEX profiling of mammary cancer progression in a HER-2/neu oncogene-driven animal model system. Proteomics 2010, 10, 3835–3853. [Google Scholar] [CrossRef]
- Carey, L.A.; Rugo, H.S.; Marcom, P.K.; Mayer, E.L.; Esteva, F.J.; Ma, C.X.; Liu, M.C.; Storniolo, A.M.; Rimawi, M.F.; Forero-Torres, A.; et al. TBCRC 001: Randomized phase II study of cetuximab in combination with carboplatin in stage IV triple-negative breast cancer. J Clin Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 2615–2623. [Google Scholar] [CrossRef] [Green Version]
- Ferraro, D.A.; Gaborit, N.; Maron, R.; Cohen-Dvashi, H.; Porat, Z.; Pareja, F.; Lavi, S.; Lindzen, M.; Ben-Chetrit, N.; Sela, M.; et al. Inhibition of triple-negative breast cancer models by combinations of antibodies to EGFR. Proc. Natl. Acad. Sci. USA 2013, 110, 1815–1820. [Google Scholar] [CrossRef] [Green Version]
- Burness, M.L.; Grushko, T.A.; Olopade, O.I. Epidermal growth factor receptor in triple-negative and basal-like breast cancer: Promising clinical target or only a marker? Cancer J. 2010, 16, 23–32. [Google Scholar] [CrossRef]
- Baselga, J.; Gomez, P.; Greil, R.; Braga, S.; Climent, M.A.; Wardley, A.M.; Kaufman, B.; Stemmer, S.M.; Pego, A.; Chan, A.; et al. Randomized phase II study of the anti-epidermal growth factor receptor monoclonal antibody cetuximab with cisplatin versus cisplatin alone in patients with metastatic triple-negative breast cancer. J. Clin. Oncol. 2013, 31, 2586–2592. [Google Scholar] [CrossRef]
- Inomata, M.; Shukuya, T.; Takahashi, T.; Ono, A.; Nakamura, Y.; Tsuya, A.; Tanigawara, Y.; Naito, T.; Murakami, H.; Harada, H.; et al. Continuous administration of EGFR-TKIs following radiotherapy after disease progression in bone lesions for non-small cell lung cancer. Anticancer. Res. 2011, 31. [Google Scholar]
- Shukuya, T.; Takahashi, T.; Naito, T.; Kaira, R.; Ono, A.; Nakamura, Y.; Tsuya, A.; Tanigawara, Y.; Murakami, H.; Harada, H.; et al. Continuous EGFR-TKI administration following radiotherapy for non-small cell lung cancer patients with isolated CNS failure. Lung Cancer 2011, 74, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Soh, J.; Toyooka, S.; Ichihara, S.; Suehisa, H.; Kobayashi, N.; Ito, S.; Yamane, M.; Aoe, M.; Sano, Y.; Kiura, K.; et al. EGFR mutation status in pleural fluid predicts tumor responsiveness and resistance to gefitinib. Lung Cancer 2007, 56, 445–448. [Google Scholar] [CrossRef]
- Zhang, Q.; Ke, E.; Niu, F.; Deng, W.; Chen, Z.; Xu, C.; Zhang, X.-C.; Zhao, N.; Su, J.; Yang, J.; et al. The role of T790M mutation in EGFR-TKI re-challenge for patients with EGFR-mutant advanced lung adenocarcinoma. Oncotarget 2017, 8, 4994–5002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fountzilas, C.; Chhatrala, R.; Khushalani, N.; Hutson, A.; Tucker, C.; Ma, W.W.; Warren, G.; Boland, P.; Tan, W.; LeVea, C.; et al. A phase II trial of erlotinib monotherapy in advanced pancreatic cancer as a first- or second-line agent. Cancer Chemother. Pharmacol. 2017, 80, 497–505. [Google Scholar] [CrossRef]
- Moore, M.J.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A.; et al. Erlotinib Plus Gemcitabine Compared with Gemcitabine Alone in Patients With Advanced Pancreatic Cancer: A Phase III Trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2007, 25, 1960–1966. [Google Scholar] [CrossRef]
- Bernier, J. Drug Insight: Cetuximab in the treatment of recurrent and metastatic squamous cell carcinoma of the head and neck. Nat. Clin. Pract. Oncol. 2008, 5, 705–713. [Google Scholar] [CrossRef]
- Yu, H.; Riely, G.J.; Lovly, C.M. Therapeutic strategies utilized in the setting of acquired resistance to EGFR tyrosine kinase inhibitors. Clin. Cancer Res. 2014, 20, 5898–5907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012, 486, 532–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jhawer, M.; Goel, S.; Wilson, A.J.; Montagna, C.; Ling, Y.-H.; Byun, -S.; Nasser, S.; Arango, D.; Shin, J.; Klampfer, L.; et al. PIK3CA mutation/PTEN expression status predicts response of colon cancer cells to the epidermal growth factor receptor inhibitor cetuximab. Cancer Res. 2008, 68, 1953–1961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berg, M.; Søreide, K. EGFR and downstream genetic alterations in KRAS/BRAF and PI3K/AKT pathways in colorectal cancer: Implications for targeted therapy. Discov. Med. 2012, 14. [Google Scholar]
- Grob, T.J.; Heilenkötter, U.; Geist, S.; Paluchowski, P.; Wilke, C.; Jaenicke, F.; Quaas, A.; Wilczak, W.; Choschzick, M.; Sauter, G.; et al. Rare oncogenic mutations of predictive markers for targeted therapy in triple-negative breast cancer. Breast Cancer Res. Treat. 2012, 134, 561–567. [Google Scholar] [CrossRef]
- Hoque, M.; Lee, Y.E.; Kim, H.R.; Shin, M.-G. Potential biomarkers and antagonists for fluoranthene-induced cellular toxicity of bone marrow derived mesenchymal stem cells. Blood Res. 2019, 54, 253–261. [Google Scholar] [CrossRef] [Green Version]
- Houtman, R.; Krijgsveld, J.; Kool, M.; Romijn, E.P.; Redegeld, F.A.; Nijkamp, F.P.; Heck, A.J.; Humphery-Smith, I. Lung proteome alterations in a mouse model for nonallergic asthma. Proteomics 2003, 3, 2008–2018. [Google Scholar] [CrossRef]
- Johannsdottir, H.K.; Jonsson, G.; Johannesdottir, G.; Agnarsson, B.A.; Eerola, H.; Arason, A.; Heikkila, P.; Egilsson, V.; Olsson, H.; Johannsson, O.T.; et al. Chromosome 5 imbalance mapping in breast tumors from BRCA1 and BRCA2 mutation carriers and sporadic breast tumors. Int. J. Cancer 2006, 119, 1052–1060. [Google Scholar] [CrossRef]
- Loo, L.W.M. Array Comparative Genomic Hybridization Analysis of Genomic Alterations in Breast Cancer Subtypes. Cancer Res. 2004, 64, 8541–8549. [Google Scholar] [CrossRef] [Green Version]
- Pierga, J.-Y.; Reis-Filho, J.S.; Cleator, S.J.; Dexter, T.; Mackay, A.; Simpson, P.T.; Fenwick, K.; Iravani, M.; Salter, J.; Hills, M.; et al. Microarray-based comparative genomic hybridisation of breast cancer patients receiving neoadjuvant chemotherapy. Br. J. Cancer 2006, 96, 341–351. [Google Scholar] [CrossRef]
- Cruz, P.M.; Mo, H.; McConathy, W.J.; Sabnis, N.A.; Lacko, A.G. The role of cholesterol metabolism and cholesterol transport in carcinogenesis: A review of scientific findings, relevant to future cancer therapeutics. Front. Pharmacol. 2013, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poirot, M.; Silvente-Poirot, S.; Weichselbaum, R.R. Cholesterol metabolism and resistance to tamoxifen. Curr. Opin. Pharmacol. 2012, 12, 683–689. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Saha, S.T.; Thomas, J.; Kaur, M. Targeting cellular cholesterol for anticancer therapy. FEBS J. 2019, 286, 4192–4208. [Google Scholar] [CrossRef]
- Guillaumond, F.; Bidaut, G.; Ouaissi, M.; Servais, S.; Gouirand, V.; Olivares, O.; Lac, S.; Borge, L.; Roques, J.; Gayet, O.; et al. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2015, 112, 2473–2478. [Google Scholar] [CrossRef] [Green Version]
- Vasseur, S.; Guillaumond, F. LDL Receptor: An open route to feed pancreatic tumor cells. Mol. Cell. Oncol. 2015, 3, e1033586. [Google Scholar] [CrossRef] [Green Version]
- Rentero, C.; Blanco-Muñoz, P.; Meneses-Salas, E.; Grewal, T.; Enrich, C. Annexins—Coordinators of Cholesterol Homeostasis in Endocytic Pathways. Int. J. Mol. Sci. 2018, 19, 1444. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Cancer Type | AnxA6 Expression Status | Diagnostic, Prognostic or Therapeutic Value | Citation |
|---|---|---|---|
| Ovarian carcinoma | Markedly increased in advanced-stage tumors vs. benign controls | Diagnosis of advanced ovarian cancer stages | [107] |
| Pancreatic cancer | High expression in pancreatic cancer and lung squamous cancer vs. normal tissues | Monoclonal antibody 9E1 as a therapeutic option for invasive cancers | [94] |
| Pancreatic ductal adenocarcinoma | AnxA6 enriched in EVs from cancer-associated fibroblasts and following chemotherapy | Biomarker and therapeutic target | [96,97] |
| Esophageal adenocarcinoma | AnxA6 is a component of a 4-protein serum biomarker panel | Noninvasive detection of early tumor stages in patient serum | [110] |
| Squamous cervical cancer | Expression is increased in cervical intraepithelial neoplasia and microinvasive cervical cancer vs. squamous cervical cancer precursor lesions. | Diagnosis of cervical cancer progression | [113,116] |
| Acute lymphoblastic leukemia | Highly expressed in B-lineage acute lymphoblastic leukemia vs. normal B-cell progenitors | Diagnosis of B-lineage acute lymphoblastic leukemia | [118] |
| Breast cancer | Downregulated in EGFR-overexpressing and estrogen receptor (ER)-negative breast cancer cells | Biomarker for EGFR-overexpressing, ER-negative breast cancer | [19] |
| Reduced expression in breast cancer tissues, but elevated in invasive breast cancer phenotypes | Biomarker for invasive breast cancer phenotypes | [22] | |
| Expression status significantly associated with the survival of patients with basal-like breast cancer | Predictive biomarker for basal-like breast cancer patient survival | [29] | |
| Elevated expression associated with acquired resistance to lapatinib in TNBC. | Predictive biomarker for response to EGFR-targeted therapies | [21] | |
| Loss of AnxA6 associated with the early onset and rapid growth of xenograft TNBC tumors in mice | Biomaker for TNBC progression | [20] | |
| HER-2/neu-driven mammary tumor | Associated with tumor progression | Biomarker for rapidly growing breast cancer | [119] |
| Melanoma | Decrease or loss of expression as melanomas progress from benign to malignant phenotypes | Detection of melanoma progression | [112] |
| Gastric cancer | Downregulated in gastric cancer cells and primary gastric carcinomas | Diagnosis of gastric cancer | [114] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korolkova, O.Y.; Widatalla, S.E.; Williams, S.D.; Whalen, D.S.; Beasley, H.K.; Ochieng, J.; Grewal, T.; Sakwe, A.M. Diverse Roles of Annexin A6 in Triple-Negative Breast Cancer Diagnosis, Prognosis and EGFR-Targeted Therapies. Cells 2020, 9, 1855. https://doi.org/10.3390/cells9081855
Korolkova OY, Widatalla SE, Williams SD, Whalen DS, Beasley HK, Ochieng J, Grewal T, Sakwe AM. Diverse Roles of Annexin A6 in Triple-Negative Breast Cancer Diagnosis, Prognosis and EGFR-Targeted Therapies. Cells. 2020; 9(8):1855. https://doi.org/10.3390/cells9081855
Chicago/Turabian StyleKorolkova, Olga Y., Sarrah E. Widatalla, Stephen D. Williams, Diva S. Whalen, Heather K. Beasley, Josiah Ochieng, Thomas Grewal, and Amos M. Sakwe. 2020. "Diverse Roles of Annexin A6 in Triple-Negative Breast Cancer Diagnosis, Prognosis and EGFR-Targeted Therapies" Cells 9, no. 8: 1855. https://doi.org/10.3390/cells9081855
APA StyleKorolkova, O. Y., Widatalla, S. E., Williams, S. D., Whalen, D. S., Beasley, H. K., Ochieng, J., Grewal, T., & Sakwe, A. M. (2020). Diverse Roles of Annexin A6 in Triple-Negative Breast Cancer Diagnosis, Prognosis and EGFR-Targeted Therapies. Cells, 9(8), 1855. https://doi.org/10.3390/cells9081855