Oncology Therapeutics Targeting the Metabolism of Amino Acids


Abstract
:1. Introduction
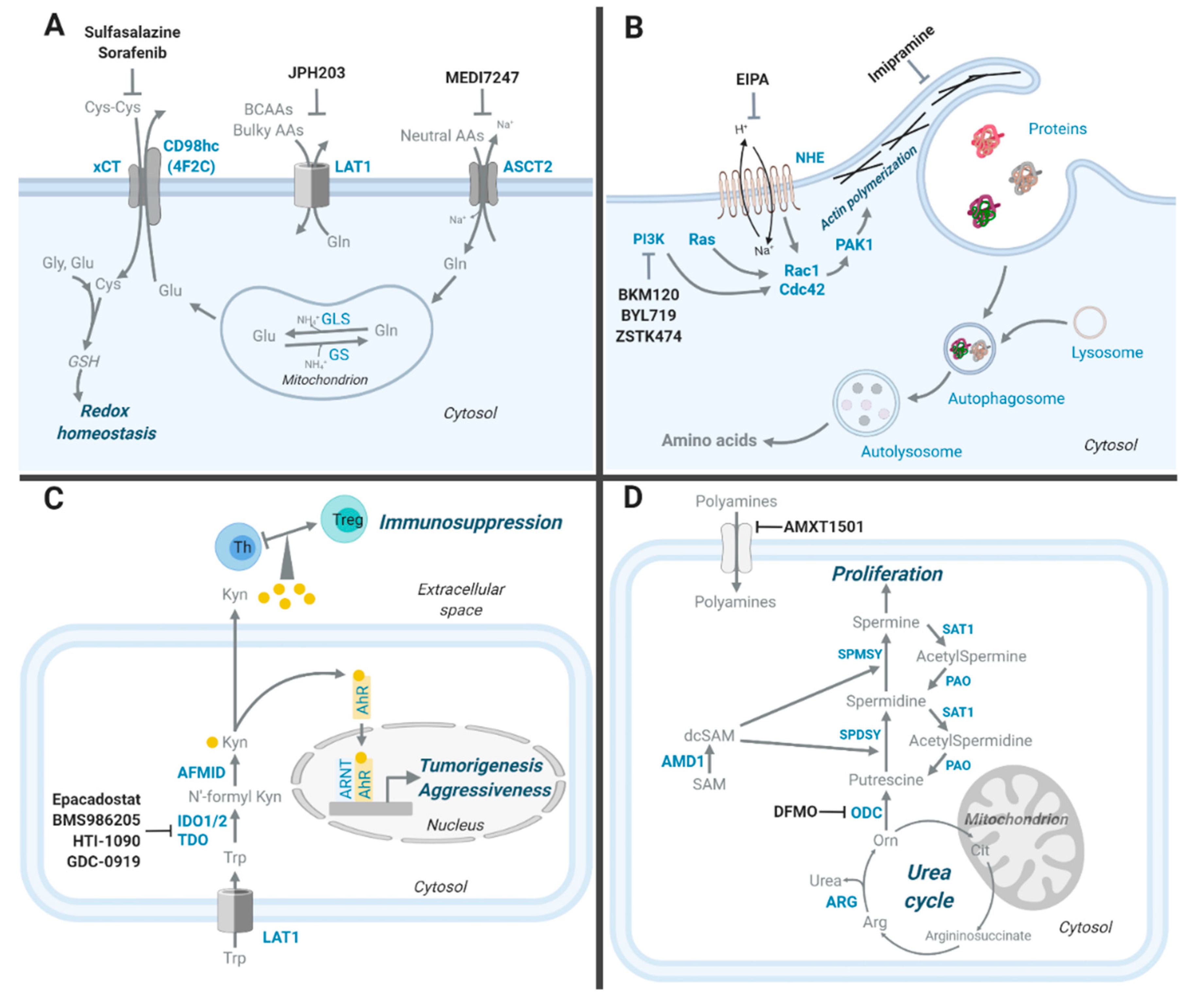
2. Amino Acid Transporters
3. Macropinocytosis, an Alternative Way to Obtain Amino Acids
4. Transaminase, a Key Mechanism of NEAA Synthesis
5. Serine and the One Carbon Metabolism
5.1. Serine De Novo Synthesis
5.2. Folate Metabolism Pathway
6. Methionine Metabolism
6.1. Methionine Cycle
6.2. SAM and Epigenetic Regulation
6.3. Methionine Salvage Pathway
7. Glutathione, a Crucial Metabolite against ROS
8. Nucleotide Synthesis
8.1. Pyrimidine De Novo Synthesis
8.2. Purine De Novo Synthesis
9. Kynurenine, a Tryptophan-Derived Oncometabolite
10. Polyamines, Arginine-Derived Oncometabolites
11. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [Green Version]
- Vazquez, A.; Kamphorst, J.J.; Markert, E.K.; Schug, Z.T.; Tardito, S.; Gottlieb, E. Cancer metabolism at a glance. J. Cell Sci. 2016, 129, 3367–3373. [Google Scholar] [CrossRef] [Green Version]
- Frezza, C. Addicted to serine. Nat. Chem. Biol. 2016, 12, 389–390. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Ye, J.; Kamphorst, J.J.; Shlomi, T.; Thompson, C.B.; Rabinowitz, J.D. Quantitative flux analysis reveals folate-dependent NADPH production. Nature 2014, 510, 298–302. [Google Scholar] [CrossRef] [Green Version]
- Platten, M.; von Knebel Doeberitz, N.; Oezen, I.; Wick, W.; Ochs, K. Cancer Immunotherapy by Targeting IDO1/TDO and Their Downstream Effectors. Front. Immunol. 2014, 5, 673. [Google Scholar] [CrossRef]
- Celano, P.; Baylin, S.B.; Casero, R.A., Jr. Polyamines differentially modulate the transcription of growth-associated genes in human colon carcinoma cells. J. Boil. Chem. 1989, 264, 8922–8927. [Google Scholar]
- Casero, R.A.; Stewart, T.M.; Pegg, A.E. Polyamine metabolism and cancer: Treatments, challenges and opportunities. Nat. Rev. Cancer 2018, 18, 681–695. [Google Scholar] [CrossRef]
- Lieu, E.L.; Nguyen, T.; Rhyne, S.; Kim, J. Amino acids in cancer. Exp. Mol. Med. 2020, 52, 15–30. [Google Scholar] [CrossRef]
- Choi, B.-H.; Coloff, J.L. The Diverse Functions of Non-Essential Amino Acids in Cancer. Cancers 2019, 11, 675. [Google Scholar] [CrossRef] [Green Version]
- Kandasamy, P.; Gyimesi, G.; Kanai, Y.; Hediger, M.A. Amino acid transporters revisited: New views in health and disease. Trends Biochem. Sci. 2018, 43, 752–789. [Google Scholar] [CrossRef]
- Pochini, L.; Scalise, M.; Galluccio, M.; Indiveri, C. Membrane transporters for the special amino acid glutamine: Structure/function relationships and relevance to human health. Front. Chem. 2014, 2, 61. [Google Scholar] [CrossRef] [Green Version]
- Scalise, M.; Pochini, L.; Console, L.; Losso, M.A.; Indiveri, C. The Human SLC1A5 (ASCT2) Amino Acid Transporter: From Function to Structure and Role in Cell Biology. Front. Cell. Dev. Biol. 2018, 6, 96. [Google Scholar] [CrossRef]
- Toda, K.; Nishikawa, G.; Iwamoto, M.; Itatani, Y.; Takahashi, R.; Sakai, Y.; Kawada, K. Clinical Role of ASCT2 (SLC1A5) in KRAS-Mutated Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 1632. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Hardie, R.-A.; Hoy, A.J.; Van Geldermalsen, M.; Gao, D.; Fazli, L.; Sadowski, M.C.; Balaban, S.; Schreuder, M.; Nagarajah, R.; et al. Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. J. Pathol. 2015, 236, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, K.; Kaira, K.; Tomizawa, Y.; Sunaga, N.; Kawashima, O.; Oriuchi, N.; Tominaga, H.; Nagamori, S.; Kanai, Y.; Yamada, M.; et al. ASC amino-acid transporter 2 (ASCT2) as a novel prognostic marker in non-small cell lung cancer. Br. J. Cancer 2014, 110, 2030–2039. [Google Scholar] [CrossRef] [Green Version]
- van Geldermalsen, M.; Wang, Q.; Nagarajah, R.; Marshall, A.D.; Thoeng, A.; Gao, D.; Ritchie, W.; Feng, Y.; Bailey, C.G.; Deng, N.; et al. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene 2016, 35, 3201–3208. [Google Scholar] [CrossRef] [Green Version]
- Kasai, N.; Sasakawa, A.; Hosomi, K.; Poh, T.W.; Chua, B.L.; Yong, W.P.; So, J.; Chan, S.L.; Soong, R.; Kono, K.; et al. Anti-tumor efficacy evaluation of a novel monoclonal antibody targeting neutral amino acid transporter ASCT2 using patient-derived xenograft mouse models of gastric cancer. Am. J. Transl. Res. 2017, 9, 3399–3410. [Google Scholar]
- Schifferli, K.P.; Monks, N.R.; Tammali, R.; Borrok, M.J.; Flynn, M.G.; Hurt, E.M.; Coats, S.R.; Herbst, R.; Tice, D.A.; Pore, N. Abstract LB-298: MEDI7247: A first in class antibody drug conjugate targeting ASCT2 in a range of solid tumors. Cancer Res. 2018, 78, LB-298. [Google Scholar]
- Schulte, M.L.; Fu, A.; Zhao, P.; Li, J.; Geng, L.; Smith, S.T.; Kondo, J.; Coffey, R.J.; Johnson, M.O.; Rathmell, J.C.; et al. Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat. Med. 2018, 24, 194–202. [Google Scholar] [CrossRef]
- Grewer, C.; Grabsch, E. New inhibitors for the neutral amino acid transporter ASCT2 reveal its Na+-dependent anion leak. J. Physiol. 2004, 557, 747–759. [Google Scholar] [CrossRef]
- van Geldermalsen, M.; Quek, L.-E.; Turner, N.; Freidman, N.; Pang, A.; Guan, Y.F.; Krycer, J.R.; Ryan, R.M.; Wang, Q.; Holst, J. Benzylserine inhibits breast cancer cell growth by disrupting intracellular amino acid homeostasis and triggering amino acid response pathways. BMC Cancer 2018, 18, 689. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.J.; Meng, L.-Y.; Shen, Y.; Zhu, Y.-Z.; Liu, H.-R. S-benzyl-cysteine-mediated cell cycle arrest and apoptosis involving activation of mitochondrial-dependent caspase cascade through the p53 pathway in human gastric cancer SGC-7901 cells. Asian Pac. J. Cancer Prev. 2013, 14, 6379–6384. [Google Scholar] [CrossRef]
- Marshall, A.D.; Van Geldermalsen, M.; Otte, N.J.; Lum, T.; Vellozzi, M.; Thoeng, A.; Pang, A.; Nagarajah, R.; Zhang, B.; Wang, Q.; et al. ASCT2 regulates glutamine uptake and cell growth in endometrial carcinoma. Oncogenesis 2017, 6, e367. [Google Scholar] [CrossRef]
- Chiu, M.; Sabino, C.; Taurino, G.; Bianchi, M.G.; Andreoli, R.; Giuliani, N.; Bussolati, O. GPNA inhibits the sodium-independent transport system L for neutral amino acids. Amino. Acids 2017, 49, 1365–1372. [Google Scholar] [CrossRef]
- Hassanein, M.; Qian, J.; Hoeksema, M.D.; Wang, J.; Jacobovitz, M.; Ji, X.; Harris, F.T.; Harris, B.K.; Boyd, K.L.; Chen, H.; et al. Targeting SLC1a5-mediated glutamine dependence in non-small cell lung cancer. Int. J. Cancer 2015, 137, 1587–1597. [Google Scholar] [CrossRef] [Green Version]
- Yue, M.; Jiang, J.; Gao, P.; Liu, H.; Qing, G. Oncogenic MYC Activates a Feedforward Regulatory Loop Promoting Essential Amino Acid Metabolism and Tumorigenesis. Cell. Rep. 2017, 21, 3819–3832. [Google Scholar] [CrossRef] [Green Version]
- Furuya, M.; Horiguchi, J.; Nakajima, H.; Kanai, Y.; Oyama, T. Correlation of L-type amino acid transporter 1 and CD98 expression with triple negative breast cancer prognosis. Cancer Sci. 2012, 103, 382–389. [Google Scholar] [CrossRef]
- Takeuchi, K.; Ogata, S.; Nakanishi, K.; Ozeki, Y.; Hiroi, S.; Tominaga, S.; Aida, S.; Matsuo, H.; Sakata, T.; Kawai, T. LAT1 expression in non-small-cell lung carcinomas: Analyses by semiquantitative reverse transcription-PCR (237 cases) and immunohistochemistry (295 cases). Lung Cancer 2010, 68, 58–65. [Google Scholar] [CrossRef]
- Kaira, K.; Oriuchi, N.; Imai, H.; Shimizu, K.; Yanagitani, N.; Sunaga, N.; Hisada, T.; Ishizuka, T.; Kanai, Y.; Endou, H.; et al. Prognostic significance of L-type amino acid transporter 1 (LAT1) and 4F2 heavy chain (CD98) expression in early stage squamous cell carcinoma of the lung. Cancer Sci. 2009, 100, 248–254. [Google Scholar] [CrossRef]
- Okano, N.; Naruge, D.; Kawai, K.; Kobayashi, T.; Nagashima, F.; Endou, H.; Furuse, J. First-in-human phase I study of JPH203, an L-type amino acid transporter 1 inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2020. [Google Scholar] [CrossRef] [PubMed]
- Kongpracha, P.; Nagamori, S.; Wiriyasermkul, P.; Tanaka, Y.; Kaneda, K.; Okuda, S.; Ohgaki, R.; Kanai, Y. Structure-activity relationship of a novel series of inhibitors for cancer type transporter L-type amino acid transporter 1 (LAT1). J. Pharmacol. Sci. 2017, 133, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Häfliger, P.; Graff, J.; Rubin, M.; Stooss, A.; Dettmer, M.S.; Altmann, K.-H.; Gertsch, J.; Charles, R.-P. The LAT1 inhibitor JPH203 reduces growth of thyroid carcinoma in a fully immunocompetent mouse model. J. Exp. Clin. Cancer Res. 2018, 37, 234. [Google Scholar] [CrossRef] [PubMed]
- Yun, D.W.; Lee, S.A.; Park, M.-G.; Kim, J.; Yu, S.-K.; Park, M.-R.; Kim, S.G.; Oh, J.-S.; Kim, C.S.; Kim, H.-J.; et al. JPH203, an L-type amino acid transporter 1-selective compound, induces apoptosis of YD-38 human oral cancer cells. J. Pharmacol. Sci. 2014, 124, 208–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, D.W.; Kim, K.; Kanai, Y.; Wempe, M.F.; Endou, H.; Kim, J.-K. JPH203, a selective L-type amino acid transporter 1 inhibitor, induces mitochondria-dependent apoptosis in Saos2 human osteosarcoma cells. Korean J. Physiol. Pharmacol. 2017, 21, 599–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Z.; Cho, H.T.; Williams, L.; Zhu, A.; Liang, K.; Huang, K.; Wu, H.; Jiang, C.; Hong, S.; Crowe, R.; et al. Potential Biomarker of L-type Amino Acid Transporter 1 in Breast Cancer Progression. Nucl. Med. Mol. Imaging 2011, 45, 93–102. [Google Scholar] [CrossRef] [Green Version]
- Imai, H.; Kaira, K.; Oriuchi, N.; Shimizu, K.; Tominaga, H.; Yanagitani, N.; Sunaga, N.; Ishizuka, T.; Nagamori, S.; Promchan, K.; et al. Inhibition of L-type amino acid transporter 1 has antitumor activity in non-small cell lung cancer. Anticancer Res. 2010, 30, 4819–4828. [Google Scholar]
- Wang, Q.; Bailey, C.G.; Ng, C.; Tiffen, J.; Thoeng, A.; Minhas, V.; Lehman, M.L.; Hendy, S.C.; Buchanan, G.; Nelson, C.C.; et al. Androgen receptor and nutrient signaling pathways coordinate the demand for increased amino acid transport during prostate cancer progression. Cancer Res. 2011, 71, 7525–7536. [Google Scholar] [CrossRef] [Green Version]
- Kaira, K.; Sunose, Y.; Ohshima, Y.; Ishioka, N.S.; Arakawa, K.; Ogawa, T.; Sunaga, N.; Shimizu, K.; Tominaga, H.; Oriuchi, N.; et al. Clinical significance of L-type amino acid transporter 1 expression as a prognostic marker and potential of new targeting therapy in biliary tract cancer. BMC Cancer 2013, 13, 482. [Google Scholar] [CrossRef] [Green Version]
- Ohshima, Y.; Kaira, K.; Yamaguchi, A.; Oriuchi, N.; Tominaga, H.; Nagamori, S.; Kanai, Y.; Yokobori, T.; Miyazaki, T.; Asao, T.; et al. Efficacy of system l amino acid transporter 1 inhibition as a therapeutic target in esophageal squamous cell carcinoma. Cancer Sci. 2016, 107, 1499–1505. [Google Scholar] [CrossRef]
- Timmerman, L.A.; Holton, T.; Yuneva, M.; Louie, R.J.; Padró, M.; Daemen, A.; Hu, M.; Chan, D.A.; Ethier, S.P.; van‘T Veer, L.J.; et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell. 2013, 24, 450–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Dai, Z.; Barbacioru, C.; Sadee, W. Cystine-glutamate transporter SLC7A11 in cancer chemosensitivity and chemoresistance. Cancer Res. 2005, 65, 7446–7454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Blower, P.E.; Pham, A.-N.; Fang, J.; Dai, Z.; Wise, C.; Green, B.; Teitel, C.H.; Ning, B.; Ling, W.; et al. Cystine-glutamate transporter SLC7A11 mediates resistance to geldanamycin but not to 17-(allylamino)-17-demethoxygeldanamycin. Mol. Pharmacol. 2007, 72, 1637–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, W.J.; Lyons, S.A.; Nelson, G.M.; Hamza, H.; Gladson, C.L.; Gillespie, G.Y.; Sontheimer, H. Inhibition of cystine uptake disrupts the growth of primary brain tumors. J. Neurosci. 2005, 25, 7101–7110. [Google Scholar] [CrossRef]
- Ji, X.; Qian, J.; Rahman, S.M.J.; Siska, P.J.; Zou, Y.; Harris, B.K.; Hoeksema, M.D.; Trenary, I.A.; Heidi, C.; Eisenberg, R.; et al. xCT (SLC7A11)-mediated metabolic reprogramming promotes non-small cell lung cancer progression. Oncogene 2018, 37, 5007–5019. [Google Scholar] [CrossRef]
- Dai, L.; Cao, Y.; Chen, Y.-H.; Parsons, C.; Qin, Z. Targeting xCT, a cystine-glutamate transporter induces apoptosis and tumor regression for KSHV/HIV-associated lymphoma. J. Hematol. Oncol. 2014, 7, 30. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Yagoda, N.; Von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef] [Green Version]
- Sato, M.; Kusumi, R.; Hamashima, S.; Kobayashi, S.; Sasaki, S.; Komiyama, Y.; Izumikawa, T.; Conrad, M.; Bannai, S.; Sato, H. The ferroptosis inducer erastin irreversibly inhibits system x (c)- and synergizes with cisplatin to increase cisplatin’s cytotoxicity in cancer cells. Sci. Rep. 2018, 8, 968. [Google Scholar] [CrossRef] [Green Version]
- Codenotti, S.; Poli, M.; Asperti, M.; Zizioli, D.; Marampon, F.; Fanzani, A. Cell growth potential drives ferroptosis susceptibility in rhabdomyosarcoma and myoblast cell lines. J. Cancer Res. Clin. Oncol. 2018, 144, 1717–1730. [Google Scholar] [CrossRef]
- Torii, S.; Shintoku, R.; Kubota, C.; Yaegashi, M.; Torii, R.; Sasaki, M.; Suzuki, T.; Mori, M.; Yoshimoto, Y.; Takeuchi, T.; et al. An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem. J. 2016, 473, 769–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; E Gleason, C.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 2014, 3, e02523. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; De Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Eisen, T.; Stadler, W.M.; Szczylik, C.; Oudard, S.; Siebels, M.; Négrier, S.; Chevreau, C.; Solska, E.; Desai, A.A.; et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 125–134. [Google Scholar] [CrossRef]
- Kloos, R.T.; Ringel, M.D.; Knopp, M.V.; Hall, N.C.; King, M.; Stevens, R.; Liang, J.; Wakely, P.E.; Vasko, V.V.; Saji, M.; et al. Phase II trial of sorafenib in metastatic thyroid cancer. J. Clin. Oncol. 2009, 27, 1675–1684. [Google Scholar] [CrossRef]
- Gout, P.W.; Buckley, A.R.; Simms, C.R.; Bruchovsky, N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: A new action for an old drug. Leukemia 2001, 15, 1633–1640. [Google Scholar] [CrossRef] [Green Version]
- Wahl, C.; Liptay, S.; Adler, G.; Schmid, R.M. Sulfasalazine: A potent and specific inhibitor of nuclear factor kappa B. J. Clin. Investig. 1998, 101, 1163–1174. [Google Scholar] [CrossRef] [Green Version]
- Chidley, C.; Haruki, H.; Pedersen, M.G.; Muller, E.; Johnsson, K. A yeast-based screen reveals that sulfasalazine inhibits tetrahydrobiopterin biosynthesis. Nat. Chem. Biol. 2011, 7, 375–383. [Google Scholar] [CrossRef]
- Jansen, G.; Van Der Heijden, J.; Oerlemans, R.; Lems, W.F.; Ifergan, I.; Scheper, R.J.; Assaraf, Y.G.; Dijkmans, B.A.C. Sulfasalazine is a potent inhibitor of the reduced folate carrier: Implications for combination therapies with methotrexate in rheumatoid arthritis. Arthritis Rheum. 2004, 50, 2130–2139. [Google Scholar] [CrossRef]
- Rao, A.D.; McArthur, G.A. Chapter 11—Targeting Metabolic Vulnerabilities in RAS-Mutant Cells, in Conquering RAS, 1st ed.; Azmi, A.S., Ed.; Academic Press: Boston, MA, USA, 2017; pp. 193–212. [Google Scholar]
- Commisso, C.; Davidson, S.M.; Soydaner-Azeloglu, R.G.; Parker, S.J.; Kamphorst, J.J.; Hackett, S.; Grabocka, E.; Nofal, M.; Drebin, J.A.; Thompson, C.B.; et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 2013, 497, 633–637. [Google Scholar] [CrossRef] [Green Version]
- Davidson, S.M.; Jonas, O.; Keibler, M.A.; Hou, H.W.; Luengo, A.; Mayers, J.R.; Wyckoff, J.; Del Rosario, A.M.; Whitman, M.; Chin, C.R.; et al. Direct evidence for cancer-cell-autonomous extracellular protein catabolism in pancreatic tumors. Nat. Med. 2017, 23, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Veithen, A.; Cupers, P.; Baudhuin, P.; Courtoy, P.J. v-Src induces constitutive macropinocytosis in rat fibroblasts. J. Cell. Sci. 1996, 109, 2005–2012. [Google Scholar] [PubMed]
- Bar-Sagi, D.; Feramisco, J.R. Induction of membrane ruffling and fluid-phase pinocytosis in quiescent fibroblasts by ras proteins. Science 1986, 233, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Redelman-Sidi, G.; Iyer, G.; Solit, D.B.; Glickman, M.S. Oncogenic activation of Pak1-dependent pathway of macropinocytosis determines BCG entry into bladder cancer cells. Cancer Res. 2013, 73, 1156–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seguin, L.; Camargo, M.F.; Wettersten, H.I.; Kato, S.; Desgrosellier, J.S.; Von Schalscha, T.; Elliott, K.C.; Cosset, E.; Lesperance, J.; Weis, S.M.; et al. Galectin-3, a Druggable Vulnerability for KRAS-Addicted Cancers. Cancer Discov. 2017, 7, 1464–1479. [Google Scholar] [CrossRef] [Green Version]
- Redelman-Sidi, G.; Binyamin, A.; Gaeta, I.; Palm, W.; Thompson, C.B.; Romesser, P.B.; Lowe, S.W.; Bagul, M.; Doench, J.G.; Root, D.E.; et al. The Canonical Wnt Pathway Drives Macropinocytosis in Cancer. Cancer Res. 2018, 78, 4658–4670. [Google Scholar] [CrossRef] [Green Version]
- Koivusalo, M.; Welch, C.; Hayashi, H.; Scott, C.C.; Kim, M.; Alexander, T.; Touret, N.; Hahn, K.M.; Grinstein, S. Amiloride inhibits macropinocytosis by lowering submembranous pH and preventing Rac1 and Cdc42 signaling. J. Cell Biol. 2010, 188, 547–563. [Google Scholar] [CrossRef] [Green Version]
- Ridley, A.J.; Hall, A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 1992, 70, 389–399. [Google Scholar] [CrossRef]
- Campa, C.C.; Ciraolo, E.; Ghigo, A.; Germena, G.; Hirsch, E. Crossroads of PI3K and Rac pathways. Small GTPases 2015, 6, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Dharmawardhane, S.; Sanders, L.C.; Martin, S.S.; Daniels, R.H.; Bokoch, G.M. Localization of p21-activated kinase 1 (PAK1) to pinocytic vesicles and cortical actin structures in stimulated cells. J. Cell Biol. 1997, 138, 1265–1278. [Google Scholar] [CrossRef] [Green Version]
- Dharmawardhane, S.; Schurmann, A.; Sells, M.A.; Chernoff, J.; Schmid, S.L.; Bokoch, G.M. Regulation of macropinocytosis by p21-activated kinase-1. Mol. Biol. Cell 2000, 11, 3341–3352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardel, M.L.; Schneider, I.C.; Aratyn-Schaus, Y.; Waterman, C.M. Mechanical integration of actin and adhesion dynamics in cell migration. Annu. Rev. Cell Dev. Biol. 2010, 26, 315–333. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.M.; Nguyen, T.T.; Ravi, A.; Kubiniok, P.; Finicle, B.T.; Jayashankar, V.; Malacrida, L.; Hou, J.; Robertson, J.; Gao, D.; et al. PTEN Deficiency and AMPK Activation Promote Nutrient Scavenging and Anabolism in Prostate Cancer Cells. Cancer Discov. 2018, 8, 866–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodakoski, C.; Hopkins, B.D.; Zhang, G.; Su, T.; Cheng, Z.; Morris, R.; Rhee, K.Y.; Goncalves, M.D.; Cantley, L.C. Rac-Mediated Macropinocytosis of Extracellular Protein Promotes Glucose Independence in Non-Small Cell Lung Cancer. Cancers 2019, 11, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.P.; Singla, B.; Ghoshal, P.; Faulkner, J.L.; Cherian-Shaw, M.; She, J.-X.; De Chantemèle, E.J.B.; Csanyi, G.; O’Connor, P.M. Identification of novel macropinocytosis inhibitors using a rational screen of Food and Drug Administration-approved drugs. Br. J. Pharmacol. 2018, 175, 3640–3655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Beuster, G.; Zarse, K.; Kaleta, C.; Thierbach, R.; Kiehntopf, M.; Steinberg, P.; Schuster, S.; Ristow, M. Inhibition of alanine aminotransferase in silico and in vivo promotes mitochondrial metabolism to impair malignant growth. J. Biol. Chem. 2011, 286, 22323–22330. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Jia, Y.; Cao, Y.; Wu, S.; Jiang, H.; Sun, X.; Ma, J.; Yin, X.; Mao, A.; Shang, M. Overexpression of Phosphoserine Aminotransferase 1 (PSAT1) Predicts Poor Prognosis and Associates with Tumor Progression in Human Esophageal Squamous Cell Carcinoma. Cell. Physiol. Biochem. 2016, 39, 395–406. [Google Scholar] [CrossRef]
- Vié, N.; Copois, V.; Bascoul-Mollevi, C.; Denis, V.; Bec, N.; Robert, B.; Fraslon, C.; Conseiller, E.; Molina, F.; Larroque, C.; et al. Overexpression of phosphoserine aminotransferase PSAT1 stimulates cell growth and increases chemoresistance of colon cancer cells. Mol. Cancer 2008, 7, 14. [Google Scholar] [CrossRef] [Green Version]
- Pollari, S.; Käkönen, S.-M.; Edgren, H.; Wolf, M.; Kohonen, P.; Sara, H.; Guise, T.; Nees, M.; Kallioniemi, O. Enhanced serine production by bone metastatic breast cancer cells stimulates osteoclastogenesis. Breast Cancer Res. Treat. 2011, 125, 421–430. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Wu, J.; Cai, J.; He, Z.; Yuan, J.; Zhu, X.; Li, Y.; Li, M.; Guan, H. PSAT1 regulates cyclin D1 degradation and sustains proliferation of non-small cell lung cancer cells. Int. J. Cancer 2015, 136, E39–E50. [Google Scholar] [CrossRef] [PubMed]
- Li, J.T.; Yin, M.; Wang, D.; Wang, J.; Lei, M.-Z.; Zhang, Y.; Liu, Y.; Zhang, L.; Zou, S.-W.; Hu, L.-P.; et al. BCAT2-mediated BCAA catabolism is critical for development of pancreatic ductal adenocarcinoma. Nat. Cell Biol. 2020, 22, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Ananieva, E.A.; Wilkinson, A.C. Branched-chain amino acid metabolism in cancer. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 64–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panosyan, E.H.; Lin, H.J.; Koster, J.; Lasky, J. In search of druggable targets for GBM amino acid metabolism. BMC Cancer 2017, 17, 162. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.Q.; Faddaoui, A.; Bachvarova, M.; Plante, M.; Gregoire, J.; Renaud, M.-C.; Sebastianelli, A.; Guillemette, C.; Gobeil, S.; Macdonald, E.; et al. BCAT1 expression associates with ovarian cancer progression: Possible implications in altered disease metabolism. Oncotarget 2015, 6, 31522–31543. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.-G.; Luan, S.; Qi, C.; Tong, Q.; Yan, S.; Li, H.; Zhang, Y. Aspulvinone O, a natural inhibitor of GOT1 suppresses pancreatic ductal adenocarcinoma cells growth by interfering glutamine metabolism. Cell Commun. Signal. 2019, 17, 111. [Google Scholar] [CrossRef] [Green Version]
- Anglin, J.; Zavareh, R.B.; Sander, P.N.; Haldar, S.; Mullarky, E.; Cantley, L.C.; Kimmelman, A.C.; Lyssiotis, C.A.; Lairson, L.L. Discovery and optimization of aspartate aminotransferase 1 inhibitors to target redox balance in pancreatic ductal adenocarcinoma. Bioorg. Med. Chem. Lett. 2018, 28, 2675–2678. [Google Scholar] [CrossRef]
- Yoshida, T.; Yamasaki, S.; Kaneko, O.; Taoka, N.; Tomimoto, Y.; Namatame, I.; Yahata, T.; Kuromitsu, S.; Cantley, L.C.; Lyssiotis, C.A. A covalent small molecule inhibitor of glutamate-oxaloacetate transaminase 1 impairs pancreatic cancer growth. Biochem. Biophys. Res. Commun. 2020, 522, 633–638. [Google Scholar] [CrossRef]
- Henderson, J.L.; Sawant-Basak, A.; Tuttle, J.B.; Dounay, A.B.; McAllister, L.A.; Pandit, J.; Rong, S.; Hou, X.; Bechle, B.M.; Kim, J.-Y.; et al. Discovery of hydroxamate bioisosteres as KAT II inhibitors with improved oral bioavailability and pharmacokinetics. Med. Chem. Comm. 2013, 4, 125–129. [Google Scholar] [CrossRef]
- Chaneton, B.; Hillmann, P.; Zheng, L.; Martin, A.C.L.; Maddocks, O.D.K.; Chokkathukalam, A.; Coyle, J.E.; Jankevics, A.; Holding, F.P.; Vousden, K.H.; et al. Serine is a natural ligand and allosteric activator of pyruvate kinase M2. Nature 2012, 491, 458–462. [Google Scholar] [CrossRef] [Green Version]
- Jing, Z.; Heng, W.; Aiping, D.; Yafei, Q.; Shulan, Z. Expression and clinical significance of phosphoglycerate dehydrogenase and squamous cell carcinoma antigen in cervical cancer. Int. J. Gynecol. Cancer 2013, 23, 1465–1469. [Google Scholar] [CrossRef] [PubMed]
- Possemato, R.; Marks, K.M.; Shaul, Y.D.; Pacold, M.E.; Kim, D.; Birsoy, K.; Sethumadhavan, S.; Woo, H.-K.; Jang, H.G.; Jha, A.K.; et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 2011, 476, 346–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locasale, J.W.; Grassian, A.R.; Melman, T.; Lyssiotis, C.A.; Mattaini, K.R.; Bass, A.J.; Heffron, G.; Metallo, C.M.; Muranen, T.; Sharfi, H.; et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 2011, 43, 869–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullarky, E.; Lucki, N.C.; Zavareh, R.B.; Anglin, J.L.; Gomes, A.P.; Nicolay, B.N.; Wong, J.C.Y.; Christen, S.; Takahashi, H.; Singh, P.K.; et al. Identification of a small molecule inhibitor of 3-phosphoglycerate dehydrogenase to target serine biosynthesis in cancers. Proc. Natl. Acad. Sci. USA 2016, 113, 1778–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacold, M.E.; Brimacombe, K.R.; Chan, S.H.; Rohde, J.M.; Lewis, C.A.; Swier, L.J.; Possemato, R.; Chen, W.W.; Sullivan, L.B.; Fiske, B.P.; et al. A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat. Chem. Biol. 2016, 12, 452–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, L.; Lee, D.; Law, C.-T.; Zhang, M.S.; Shen, J.; Chin, D.W.-C.; Zhang, A.; Tsang, F.H.-C.; Wong, C.L.-S.; Ng, I.O.; et al. Genome-wide CRISPR/Cas9 library screening identified PHGDH as a critical driver for Sorafenib resistance in HCC. Nat. Commun. 2019, 10, 4681. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Liberti, M.V.; Liu, P.; Deng, X.; Liu, Y.; Locasale, J.W.; Lai, L. Rational Design of Selective Allosteric Inhibitors of PHGDH and Serine Synthesis with Anti-tumor Activity. Cell Chem. Biol. 2017, 24, 55–65. [Google Scholar] [CrossRef]
- Ducker, G.S.; Ghergurovich, J.M.; Mainolfi, N.; Suri, V.; Jeong, S.K.; Li, S.H.-J.; Friedman, A.; Manfredi, M.G.; Gitai, Z.; Kim, H.; et al. Human SHMT inhibitors reveal defective glycine import as a targetable metabolic vulnerability of diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. USA 2017, 114, 11404–11409. [Google Scholar] [CrossRef] [Green Version]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Farber, S.; Diamond, L.K. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. N. Engl. J. Med. 1948, 238, 787–793. [Google Scholar] [CrossRef]
- Osborn, M.J.; Freeman, M.; Huennekens, F.M. Inhibition of dihydrofolic reductase by aminopterin and amethopterin. Proc. Soc. Exp. Biol. Med. 1958, 97, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Loughran, T.P., Jr.; Kidd, P.G.; Starkebaum, G. Treatment of large granular lymphocyte leukemia with oral low-dose methotrexate. Blood 1994, 84, 2164–2170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knoop, A.S.; Knudsen, H.; Balslev, E.; Rasmussen, B.B.; Overgaard, J.; Nielsen, K.V.; Schonau, A.; Gunnarsdóttir, K.; Olsen, K.E.; Mouridsen, H.; et al. Retrospective Analysis of Topoisomerase IIa Amplifications and Deletions As Predictive Markers in Primary Breast Cancer Patients Randomly Assigned to Cyclophosphamide, Methotrexate, and Fluorouracil or Cyclophosphamide, Epirubicin, and Fluorouracil: Danish Breast Cancer Cooperative Group. J. Clin. Oncol. 2005, 23, 7483–7490. [Google Scholar] [PubMed]
- Prodduturi, P.; Bierman, P.J. Current and emerging pharmacotherapies for primary CNS lymphoma. Clin. Med. Insights Oncol. 2012, 6, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, S.; Moran, R.G.; Goldman, I.D. Pemetrexed: Biochemical and cellular pharmacology, mechanisms, and clinical applications. Mol. Cancer Ther. 2007, 6, 404–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Racanelli, A.C.; Rothbart, S.B.; Heyer, C.L.; Moran, R.G. Therapeutics by cytotoxic metabolite accumulation: Pemetrexed causes ZMP accumulation, AMPK activation, and mammalian target of rapamycin inhibition. Cancer Res. 2009, 69, 5467–5474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daidone, F.; Florio, R.; Rinaldo, S.; Contestabile, R.; Di Salvo, M.L.; Cutruzzolà, F.; Bossa, F.; Paiardini, A. In silico and in vitro validation of serine hydroxymethyltransferase as a chemotherapeutic target of the antifolate drug pemetrexed. Eur. J. Med. Chem. 2011, 46, 1616–1621. [Google Scholar] [CrossRef]
- Beardsley, G.P.; A Moroson, B.; Taylor, E.C.; Moran, R.G. A new folate antimetabolite, 5,10-dideaza-5,6,7,8-tetrahydrofolate is a potent inhibitor of de novo purine synthesis. J. Biol. Chem. 1989, 264, 328–333. [Google Scholar]
- Laohavinij, S.; Wedge, S.R.; Lind, M.J.; Bailey, N.; Humphreys, A.; Proctor, M.; Chapman, F.; Simmons, D.; Oakley, A.; Robson, L.; et al. A phase I clinical study of the antipurine antifolate lometrexol (DDATHF) given with oral folic acid. Investig. New Drugs 1996, 14, 325–335. [Google Scholar] [CrossRef]
- Scaletti, E.; Jemth, A.-S.; Helleday, T.; Stenmark, P. Structural basis of inhibition of the human serine hydroxymethyltransferase SHMT2 by antifolate drugs. FEBS Lett. 2019, 593, 1863–1873. [Google Scholar] [CrossRef]
- Mecham, J.O.; Rowitch, D.H.; Wallace, C.; Stern, P.H.; Hoffman, R.M. The metabolic defect of methionine dependence occurs frequently in human tumor cell lines. Biochem. Biophys. Res. Commun. 1983, 117, 429–434. [Google Scholar] [CrossRef]
- Chello, P.L.; Bertino, J.R. Dependence of 5-methyltetrahydrofolate utilization by L5178Y murine leukemia cells in vitro on the presence of hydroxycobalamin and transcobalamin II. Cancer Res. 1973, 33, 1898–1904. [Google Scholar] [PubMed]
- Kawaguchi, K.; Han, Q.; Li, S.; Tan, Y.; Igarashi, K.; Murakami, T.; Unno, M.; Hoffman, R.M. Efficacy of Recombinant Methioninase (rMETase) on Recalcitrant Cancer Patient-Derived Orthotopic Xenograft (PDOX) Mouse Models: A Review. Cells 2019, 8, 410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreis, W.; Hession, C. Isolation and purification of L-methionine-alpha-deamino-gamma-mercaptomethane-lyase (L-methioninase) from Clostridium sporogenes. Cancer Res. 1973, 33, 1862–1865. [Google Scholar] [PubMed]
- Kawaguchi, K.; Igarashi, K.; Li, S.; Han, Q.; Tan, Y.; Miyake, K.; Kiyuna, T.; Miyake, M.; Murakami, T.; Chmielowski, B.; et al. Recombinant methioninase (rMETase) is an effective therapeutic for BRAF-V600E-negative as well as -positive melanoma in patient-derived orthotopic xenograft (PDOX) mouse models. Oncotarget 2018, 9, 915–923. [Google Scholar] [CrossRef]
- Murakami, T.; Li, S.; Han, Q.; Tan, Y.; Kiyuna, T.; Igarashi, K.; Kawaguchi, K.; Hwang, H.K.; Miyake, K.; Singh, A.S.; et al. Recombinant methioninase effectively targets a Ewing’s sarcoma in a patient-derived orthotopic xenograft (PDOX) nude-mouse model. Oncotarget 2017, 8, 35630–35638. [Google Scholar] [CrossRef] [Green Version]
- Igarashi, K.; Kawaguchi, K.; Kiyuna, T.; Miyake, K.; Murakami, T.; Yamamoto, N.; Hayashi, K.; Kimura, H.; Miwa, S.; Tsuchiya, H.; et al. Effective Metabolic Targeting of Human Osteosarcoma Cells in Vitro and in Orthotopic Nude-mouse Models with Recombinant Methioninase. Anticancer Res. 2017, 37, 4807–4812. [Google Scholar]
- Hoffman, R.M.; Tan, Y.; Li, S.; Han, Q.; Zavala, J. Pilot Phase I Clinical Trial of Methioninase on High-Stage Cancer Patients: Rapid Depletion of Circulating Methionine. Methods Mol. Biol. 2019, 1866, 231–242. [Google Scholar]
- Sanderson, S.M.; Gao, X.; Dai, Z.; Locasale, J.W. Methionine metabolism in health and cancer: A nexus of diet and precision medicine. Nat. Rev. Cancer 2019, 19, 625–637. [Google Scholar] [CrossRef]
- Chiang, P.K.; Gordon, R.K.; Tal, J.; Zeng, G.C.; Doctor, B.P.; Pardhasaradhi, K.; McCann, P.P. S-Adenosylmethionine and methylation. FASEB J. 1996, 10, 471–480. [Google Scholar] [CrossRef] [Green Version]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef]
- Teodoridis, J.M.; Strathdee, G.; Brown, R. Epigenetic silencing mediated by CpG island methylation: Potential as a therapeutic target and as a biomarker. Drug Resist. Updates 2004, 7, 267–278. [Google Scholar] [CrossRef]
- Baylin, S.B.; Höppener, J.W.; De Bustros, A.; Steenbergh, P.H.; Lips, C.J.; Nelkin, B.D. DNA methylation patterns of the calcitonin gene in human lung cancers and lymphomas. Cancer Res. 1986, 46, 2917–2922. [Google Scholar]
- Okada, Y.; Feng, Q.; Lin, Y.; Jiang, Q.; Li, Y.; Coffield, V.M.; Su, L.; Xu, G.; Zhang, Y. hDOT1L links histone methylation to leukemogenesis. Cell 2005, 121, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Thienpont, B.; Steinbacher, J.; Zhao, H.; D’Anna, F.; Kuchnio, A.; Ploumakis, A.; Ghesquière, B.; Van Dyck, L.; Boeckx, B.; Schoonjans, L.; et al. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature 2016, 537, 63–68. [Google Scholar] [CrossRef]
- Saunderson, E.A.; Stepper, P.; Gomm, J.J.; Hoa, L.; Morgan, A.; Allen, M.; Jones, J.L.; Gribben, J.G.; Jurkowski, T.P.; Ficz, G. Hit-and-run epigenetic editing prevents senescence entry in primary breast cells from healthy donors. Nat. Commun. 2017, 8, 1450. [Google Scholar] [CrossRef]
- Wolff, E.M.; Byun, H.-M.; Han, H.F.; Sharma, S.; Nichols, P.W.; Siegmund, K.D.; Yang, A.S.; Jones, P.A.; Liang, G. Hypomethylation of a LINE-1 promoter activates an alternate transcript of the MET oncogene in bladders with cancer. PLoS Genet. 2010, 6, e1000917. [Google Scholar] [CrossRef]
- Ulanovskaya, O.A.; Zuhl, A.M.; Cravatt, B.F. NNMT promotes epigenetic remodeling in cancer by creating a metabolic methylation sink. Nat. Chem. Biol. 2013, 9, 300–306. [Google Scholar] [CrossRef] [Green Version]
- Roessler, M. Identification of nicotinamide N-methyltransferase as a novel serum tumor marker for colorectal cancer. Clin Cancer Res. 2005, 11, 6550–6557. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Siadaty, M.S.; Berens, M.E.; Hampton, G.M.; Theodorescu, D. Overlapping gene expression profiles of cell migration and tumor invasion in human bladder cancer identify metallothionein 1E and nicotinamide N-methyltransferase as novel regulators of cell migration. Oncogene 2008, 27, 6679–6689. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Hong, S.J.; Lim, E.K.; Yu, Y.S.; Kim, S.W.; Roh, J.H.; Do, I.G.; Joh, J.-W.; Kim, D.S. Expression of nicotinamide N-methyltransferase in hepatocellular carcinoma is associated with poor prognosis. J. Exp. Clin. Cancer Res. 2009, 28, 20. [Google Scholar] [CrossRef] [Green Version]
- Tomida, M.; Mikami, I.; Takeuchi, S.; Nishimura, H.; Akiyama, H. Serum levels of nicotinamide N-methyltransferase in patients with lung cancer. J. Cancer Res. Clin. Oncol. 2009, 135, 1223–1229. [Google Scholar] [CrossRef]
- Luo, J.; Li, Y.-N.; Wang, F.; Zhang, W.-M.; Geng, X. S-adenosylmethionine inhibits the growth of cancer cells by reversing the hypomethylation status of c-myc and H-ras in human gastric cancer and colon cancer. Int. J. Biol. Sci. 2010, 6, 784–795. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Sun, Z.; Szyf, M. S-adenosyl-methionine (SAM) alters the transcriptome and methylome and specifically blocks growth and invasiveness of liver cancer cells. Oncotarget 2017, 8, 111866–111881. [Google Scholar] [CrossRef]
- Lin, D.W.; Chung, B.P.; Kaiser, P. S-adenosylmethionine limitation induces p38 mitogen-activated protein kinase and triggers cell cycle arrest in G1. J. Cell. Sci. 2014, 127, 50–59. [Google Scholar] [CrossRef] [Green Version]
- Albers, E. Metabolic characteristics and importance of the universal methionine salvage pathway recycling methionine from 5′-methylthioadenosine. IUBMB Life 2009, 61, 1132–1142. [Google Scholar] [CrossRef]
- Chu, Y.D.; Lai, H.-Y.; Pai, L.-M.; Huang, Y.-H.; Lin, Y.-H.; Liang, K.-H.; Yeh, C.-T. The methionine salvage pathway-involving ADI1 inhibits hepatoma growth by epigenetically altering genes expression via elevating S-adenosylmethionine. Cell Death Dis. 2019, 10, 240. [Google Scholar] [CrossRef] [Green Version]
- Oram, S.W.; Ai, J.; Pagani, G.M.; Hitchens, M.R.; A Stern, J.; Eggener, S.; Pins, M.; Xiao, W.; Cai, X.; Haleem, R.; et al. Expression and function of the human androgen-responsive gene ADI1 in prostate cancer. Neoplasia 2007, 9, 643–651. [Google Scholar] [CrossRef] [Green Version]
- Schmid, M.; Malicki, D.; Nobori, T.; Rosenbach, M.D.; Campbell, K.; A Carson, D.; Carrera, C.J. Homozygous deletions of methylthioadenosine phosphorylase (MTAP) are more frequent than p16INK4A (CDKN2) homozygous deletions in primary non-small cell lung cancers (NSCLC). Oncogene 1998, 17, 2669–2675. [Google Scholar] [CrossRef] [Green Version]
- M’Soka, T.J.; Nishioka, J.; Taga, A.; Kato, K.; Kawasaki, H.; Yamada, Y.; Yu, A.L.; Komada, Y.; Nobori, T. Detection of methylthioadenosine phosphorylase (MTAP) and p16 gene deletion in T cell acute lymphoblastic leukemia by real-time quantitative PCR assay. Leukemia 2000, 14, 935–940. [Google Scholar] [CrossRef] [Green Version]
- Brat, D.J.; James, C.D.; Jedlicka, A.E.; Connolly, D.C.; Chang, E.; Castellani, R.J.; Schmid, M.; Schiller, M.; Carson, D.A.; Burger, P.C. Molecular Genetic Alterations in Radiation-Induced Astrocytomas. Am. J. Pathol. 1999, 154, 1431–1438. [Google Scholar] [CrossRef] [Green Version]
- Marjon, K.; Cameron, M.J.; Quang, P.; Clasquin, M.F.; Mandley, E.; Kunii, K.; McVay, M.; Choe, S.; Kernytsky, A.; Gross, S.; et al. MTAP Deletions in Cancer Create Vulnerability to Targeting of the MAT2A/PRMT5/RIOK1 Axis. Cell. Rep. 2016, 15, 574–587. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, C.L.; E Kaiser, S.; Bolaños, B.; Nowlin, D.; Grantner, R.; Karlicek-Bryant, S.; Feng, J.L.; Jenkinson, S.; Freeman-Cook, K.; Dann, S.G.; et al. Targeting S-adenosylmethionine biosynthesis with a novel allosteric inhibitor of Mat2A. Nat. Chem. Biol. 2017, 13, 785–792. [Google Scholar] [CrossRef]
- Zhang, W.; Sviripa, V.M.; Chen, X.; Shi, J.; Yu, T.; Hamza, A.; Ward, N.D.; Kril, L.M.; Kooi, C.W.V.; Zhan, C.-G.; et al. Fluorinated N,N-dialkylaminostilbenes repress colon cancer by targeting methionine S-adenosyltransferase 2A. ACS Chem. Biol. 2013, 8, 796–803. [Google Scholar] [CrossRef] [Green Version]
- Fendt, S.M.; Lunt, S.Y. Dynamic ROS Regulation by TIGAR: Balancing Anti-cancer and Pro-metastasis Effects. Cancer Cell. 2020, 37, 141–142. [Google Scholar] [CrossRef]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Aspects Med. 2009, 30, 42–59. [Google Scholar] [CrossRef] [Green Version]
- Oakley, A. Glutathione transferases: A structural perspective. Drug Metab. Rev. 2011, 43, 138–151. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.H.; Kuo, M.T. Role of glutathione in the regulation of Cisplatin resistance in cancer chemotherapy. Met. Based Drugs 2010, 2010, 430939. [Google Scholar] [CrossRef]
- Godwin, A.K.; Meister, A.; O’Dwyer, P.J.; Huang, C.S.; Hamilton, T.C.; Anderson, M. E High resistance to cisplatin in human ovarian cancer cell lines is associated with marked increase of glutathione synthesis. Proc. Natl. Acad. Sci. USA 1992, 89, 3070–3074. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.F.; Wung, C.-H.; Chen, M.-S.; Chen, C.-F.; Yin, P.-H.; Yeh, T.-S.; Chang, Y.-L.; Chou, Y.-C.; Hung, H.; Lee, H.-C. Activated Integrated Stress Response Induced by Salubrinal Promotes Cisplatin Resistance in Human Gastric Cancer Cells via Enhanced xCT Expression and Glutathione Biosynthesis. Int. J. Mol. Sci. 2018, 19, 3389. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Stokes, W.; Chater, E.; Roy, R.; De Bruin, E.; Hu, Y.; Liu, Z.; Smit, E.F.; Heynen, G.J.; Downward, J.; et al. Decreased glutathione biosynthesis contributes to EGFR T790M-driven erlotinib resistance in non-small cell lung cancer. Cell Discov. 2016, 2, 16031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tagde, A.; Singh, H.; Kang, M.H.; Reynolds, C.P. The glutathione synthesis inhibitor buthionine sulfoximine synergistically enhanced melphalan activity against preclinical models of multiple myeloma. Blood Cancer J. 2014, 4, e229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, C.P.; Reynolds, C.P. Synergistic cytotoxicity of buthionine sulfoximine (BSO) and intensive melphalan (L-PAM) for neuroblastoma cell lines established at relapse after myeloablative therapy. Bone Marrow Transplant. 2002, 30, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Lien, E.C.; Lyssiotis, C.A.; Juvekar, A.; Hu, H.; Asara, J.M.; Cantley, L.C.; Toker, A. Glutathione biosynthesis is a metabolic vulnerability in PI(3)K/Akt-driven breast cancer. Nat. Cell Biol. 2016, 18, 572–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villablanca, J.G.; Volchenboum, S.L.; Cho, H.; Kang, M.H.; Cohn, S.L.; Anderson, C.P.; Marachelian, A.; Groshen, S.G.; Tsao-Wei, D.; Matthay, K.K.; et al. A Phase I New Approaches to Neuroblastoma Therapy Study of Buthionine Sulfoximine and Melphalan With Autologous Stem Cells for Recurrent/Refractory High-Risk Neuroblastoma. Pediatr. Blood Cancer 2016, 63, 1349–1356. [Google Scholar] [CrossRef]
- Moscow, J.A.; Townsend, A.J.; Cowan, K.H. Elevation of pi class glutathione S-transferase activity in human breast cancer cells by transfection of the GST pi gene and its effect on sensitivity to toxins. Mol. Pharmacol. 1989, 36, 22–28. [Google Scholar]
- Goto, S.; Iida, T.; Cho, S.; Oka, M.; Kohno, S.; Kondo, T. Overexpression of glutathione S-transferase pi enhances the adduct formation of cisplatin with glutathione in human cancer cells. Free Radic. Res. 1999, 31, 549–558. [Google Scholar] [CrossRef]
- Li, T.; Liu, G.; Li, H.; Yang, X.; Jing, Y.; Zhao, G. The synthesis of ethacrynic acid thiazole derivatives as glutathione S-transferase pi inhibitors. Bioorg. Med. Chem. 2012, 20, 2316–2322. [Google Scholar] [CrossRef]
- Harris, M.J.; Coggan, M.; Langton, L.; Wilson, S.R.; Board, P.G. Polymorphism of the Pi class glutathione S-transferase in normal populations and cancer patients. Pharmacogenetics 1998, 8, 27–31. [Google Scholar] [CrossRef]
- Gate, L.; Majumdar, R.S.; Lunk, A.; Tew, K.D. Increased myeloproliferation in glutathione S-transferase pi-deficient mice is associated with a deregulation of JNK and Janus kinase/STAT pathways. J. Biol. Chem. 2004, 279, 8608–8616. [Google Scholar] [CrossRef] [Green Version]
- Punganuru, S.R.; Mostofa, A.; Madala, H.R.; Basak, D.; Srivenugopal, K.S. Potent anti-proliferative actions of a non-diuretic glucosamine derivative of ethacrynic acid. Bioorg. Med. Chem. Lett. 2016, 26, 2829–2833. [Google Scholar] [CrossRef] [PubMed]
- Mignani, S.; El Brahmi, N.; El Kazzouli, S.; Eloy, L.; Courilleau, D.; Caron, J.; Bousmina, M.; Caminade, A.-M.; Cresteil, T.; Majoral, J.-P. A novel class of ethacrynic acid derivatives as promising drug-like potent generation of anticancer agents with established mechanism of action. Eur. J. Med. Chem. 2016, 122, 656–673. [Google Scholar] [CrossRef] [PubMed]
- Harshbarger, W.; Gondi, S.; Ficarro, S.B.; Hunter, J.; Udayakumar, D.; Gurbani, D.; Singer, W.D.; Liu, Y.; Li, L.; Marto, J.A.; et al. Structural and Biochemical Analyses Reveal the Mechanism of Glutathione S-Transferase Pi 1 Inhibition by the Anti-cancer Compound Piperlongumine. J. Biol. Chem. 2017, 292, 112–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bezerra, D.P.; Moura, D.J.; Rosa, R.M.; Vasconcellos, M.; E Silva, A.C.R.; De Moraes, M.O.; Silveira, E.R.; Lima, M.A.S.; Henriques, J.A.P.; Costa-Lotufo, L.V.; et al. Evaluation of the genotoxicity of piplartine, an alkamide of Piper tuberculatum, in yeast and mammalian V79 cells. Mutat. Res. 2008, 652, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Kong, E.H.; Kim, Y.-J.; Kim, Y.-J.; Cho, H.-J.; Yu, S.-N.; Kim, K.-Y.; Chang, J.-H.; Ahn, S.-C. Piplartine induces caspase-mediated apoptosis in PC-3 human prostate cancer cells. Oncol. Rep. 2008, 20, 785–792. [Google Scholar] [PubMed]
- Jafri, A.; Siddiqui, S.; Rais, J.; Ahmad, S.; Kumar, S.; Jafar, T.; Afzal, M. Induction of apoptosis by piperine in human cervical adenocarcinoma via ROS mediated mitochondrial pathway and caspase-3 activation. EXCLI J. 2019, 18, 154–164. [Google Scholar] [PubMed]
- Lai, L.H.; Fu, Q.-H.; Liu, Y.; Jiang, K.; Guo, Q.-M.; Chen, Q.-Y.; Yan, B.; Wang, Q.-Q.; Shen, J.-G. Piperine suppresses tumor growth and metastasis in vitro and in vivo in a 4T1 murine breast cancer model. Acta Pharmacol. Sin. 2012, 33, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Chowdhury, N.; Doddapaneni, R.; Boakye, C.H.A.; Godugu, C.; Singh, M. Piperlongumine for Enhancing Oral Bioavailability and Cytotoxicity of Docetaxel in Triple-Negative Breast Cancer. J. Pharm. Sci. 2015, 104, 4417–4426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricci, G.; De Maria, F.; Antonini, G.; Turella, P.; Bullo, A.; Stella, L.; Filomeni, G.; Federici, G.; Caccuri, A.M. 7-Nitro-2,1,3-benzoxadiazole derivatives, a new class of suicide inhibitors for glutathione S-transferases. Mechanism of action of potential anticancer drugs. J. Biol. Chem. 2005, 280, 26397–26405. [Google Scholar] [CrossRef] [Green Version]
- Pellizzari Tregno, F.; Sau, A.; Pezzola, S.; Geroni, C.; Lapenta, C.; Spada, M.; Filomeni, G.; Bonanno, E.; Federici, G.; Caccuri, A.M. In vitro and in vivo efficacy of 6-(7-nitro-2,1,3-benzoxadiazol-4-ylthio)hexanol (NBDHEX) on human melanoma. Eur. J. Cancer 2009, 45, 2606–2617. [Google Scholar] [CrossRef]
- Turella, P.; Filomeni, G.; Dupuis, M.L.; Ciriolo, M.R.; Molinari, A.; De Maria, F.; Tombesi, M.; Cianfriglia, M.; Federici, G.; Ricci, G.; et al. A strong glutathione S-transferase inhibitor overcomes the P-glycoprotein-mediated resistance in tumor cells. 6-(7-Nitro-2,1,3-benzoxadiazol-4-ylthio)hexanol (NBDHEX) triggers a caspase-dependent apoptosis in MDR1-expressing leukemia cells. J. Biol. Chem. 2006, 281, 23725–23732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filomeni, G.; Turella, P.; Dupuis, M.L.; Forini, O.; Ciriolo, M.R.; Cianfriglia, M.; Pezzola, S.; Federici, G.; Caccuri, A.M. 6-(7-Nitro-2,1,3-benzoxadiazol-4-ylthio)hexanol, a specific glutathione S-transferase inhibitor, overcomes the multidrug resistance (MDR)-associated protein 1-mediated MDR in small cell lung cancer. Mol. Cancer Ther. 2008, 7, 371–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wadler, S.; Horowitz, R.; Rao, J.; Mao, X.; Schlesinger, K.; Schwartz, E.L. Interferon augments the cytotoxicity of hydroxyurea without enhancing its activity against the M2 subunit of ribonucleotide reductase: Effects in wild-type and resistant human colon cancer cells. Cancer Chemother Pharmacol. 1996, 38, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Federici, L.; Sterzo, C.L.; Pezzola, S.; Di Di Matteo, A.; Scaloni, F.; Federici, G.; Caccuri, A.M. Structural basis for the binding of the anticancer compound 6-(7-nitro-2,1,3-benzoxadiazol-4-ylthio)hexanol to human glutathione s-transferases. Cancer Res. 2009, 69, 8025–8034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Luca, A.; Carpanese, D.; Rapanotti, M.C.; Viguria, T.M.S.; Forgione, M.A.; Rotili, D.; Fulci, C.; Iorio, E.; Quintieri, L.; Chimenti, S.; et al. The nitrobenzoxadiazole derivative MC3181 blocks melanoma invasion and metastasis. Oncotarget 2017, 8, 15520–15538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moffatt, B.A.; Ashihara, H. Purine and pyrimidine nucleotide synthesis and metabolism. Arab. Book 2002, 1, e0018. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Hu, Z.; Cai, L.; Li, K.; Choi, E.; Faubert, B.; Bezwada, D.; Rodriguez-Canales, J.; Villalobos, P.; Lin, Y.-F.; et al. CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nature 2017, 546, 168–172. [Google Scholar] [CrossRef]
- Liu, Y.; Marks, K.; Cowley, G.S.; Carretero, J.; Liu, Q.; Nieland, T.J.; Xu, C.; Cohoon, T.J.; Gao, P.; Zhang, Y.; et al. Metabolic and functional genomic studies identify deoxythymidylate kinase as a target in LKB1-mutant lung cancer. Cancer Discov. 2013, 3, 870–879. [Google Scholar] [CrossRef] [Green Version]
- Koundinya, M.; Sudhalter, J.; Courjaud, A.; Lionne, B.; Touyer, G.; Bonnet, L.; Menguy, I.; Schreiber, I.; Perrault, C.; Vougier, S.; et al. Dependence on the Pyrimidine Biosynthetic Enzyme DHODH Is a Synthetic Lethal Vulnerability in Mutant KRAS-Driven Cancers. Cell Chem. Biol. 2018, 25, 705.e11–717.e11. [Google Scholar] [CrossRef] [Green Version]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef]
- Ser, Z.; Gao, X.; Johnson, C.; Mehrmohamadi, M.; Liu, X.; Li, S.; Locasale, J.W. Targeting One Carbon Metabolism with an Antimetabolite Disrupts Pyrimidine Homeostasis and Induces Nucleotide Overflow. Cell. Rep. 2016, 15, 2367–2376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaferian, S.; Negahdari, B.; Eatemadi, A. Colon cancer targeting using conjugates biomaterial 5-flurouracil. Biomed. Pharmacother. 2016, 84, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Wang, Y.; Collins, M.; Mitchell, B.S.; Graves, L.M. A77 1726 induces differentiation of human myeloid leukemia K562 cells by depletion of intracellular CTP pools. Mol. Pharmacol. 2002, 62, 463–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sykes, D.B.; Kfoury, Y.S.; Mercier, F.E.; Wawer, M.J.; Law, J.M.; Haynes, M.K.; Lewis, T.A.; Schajnovitz, A.; Jain, E.; Lee, D.; et al. Inhibition of Dihydroorotate Dehydrogenase Overcomes Differentiation Blockade in Acute Myeloid Leukemia. Cell 2016, 167, 171–186.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buettner, R.; Morales, C.; Wu, X.; Sanchez, J.F.; Li, H.; Melstrom, L.G.; Rosen, S.T. Leflunomide Synergizes with Gemcitabine in Growth Inhibition of PC Cells and Impairs c-Myc Signaling through PIM Kinase Targeting. Mol. Ther. Oncolytics 2019, 14, 149–158. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, E.F.; Saili, K.S.; Koch, D.C.; Kopparapu, P.R.; Farrer, D.; Bisson, W.H.; Mathew, L.K.; Sengupta, S.; Kerkvliet, N.I.; Tanguay, R.L.; et al. The anti-inflammatory drug leflunomide is an agonist of the aryl hydrocarbon receptor. PLoS ONE 2010, 5, e13128. [Google Scholar] [CrossRef] [PubMed]
- Cody, R.; Stewart, D.; Deforni, M.; Moore, M.; Dallaire, B.; Azarnia, N.; Gyves, J. Multicenter phase II study of brequinar sodium in patients with advanced breast cancer. Am J. Clin. Oncol. 1993, 16, 526–528. [Google Scholar] [CrossRef]
- Dodion, P.F.; Wagener, T.; Stoter, G.; Drozd, A.; Lev, L.; Skovsgaard, T.; Renard, J.; Cavalli, F. Phase II trial with Brequinar (DUP-785, NSC 368390) in patients with metastatic colorectal cancer: A study of the Early Clinical Trials Group of the EORTC. Ann. Oncol. 1990, 1, 79–80. [Google Scholar] [CrossRef]
- Urba, S.; Doroshow, J.; Cripps, C.; Robert, F.; Velez-Garcia, E.; Dallaire, B.; Adams, D.; Carlson, R.; Grillo-López, A.; Gyves, J. Multicenter phase II trial of brequinar sodium in patients with advanced squamous-cell carcinoma of the head and neck. Cancer Chemother Pharmacol. 1992, 31, 167–169. [Google Scholar] [CrossRef]
- Okesli-Armlovich, A.; Gupta, A.; Jimenez, M.; Auld, D.; Liu, Q.; Bassik, M.C.; Khosla, C. Discovery of small molecule inhibitors of human uridine-cytidine kinase 2 by high-throughput screening. Bioorg. Med. Chem. Lett. 2019, 29, 2559–2564. [Google Scholar] [CrossRef]
- Hu, C.M.; Yeh, M.-T.; Tsao, N.; Chen, C.-W.; Gao, Q.-Z.; Chang, C.-Y.; Lee, M.-H.; Fang, J.-M.; Sheu, S.-Y.; Lin, C.-J.; et al. Tumor cells require thymidylate kinase to prevent dUTP incorporation during DNA repair. Cancer Cell. 2012, 22, 36–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.H.; Hsu, H.-Y.; Yeh, M.-T.; Chen, C.-C.; Huang, C.-Y.; Chung, Y.-H.; Chang, Z.-F.; Kuo, W.-C.; Chan, N.-L.; Weng, J.-H.; et al. Chemical Inhibition of Human Thymidylate Kinase and Structural Insights into the Phosphate Binding Loop and Ligand-Induced Degradation. J. Med. Chem. 2016, 59, 9906–9918. [Google Scholar] [PubMed]
- Huang, F.; Ni, M.; Chalishazar, M.D.; Huffman, K.E.; Kim, J.; Cai, L.; Shi, X.; Cai, F.; Zacharias, L.G.; Ireland, A.S.; et al. Inosine Monophosphate Dehydrogenase Dependence in a Subset of Small Cell Lung Cancers. Cell Metab. 2018, 28, 369.e5–382.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deis, S.M.; Doshi, A.; Hou, Z.; Matherly, L.H.; Gangjee, A.; Dann, C.E. Structural and Enzymatic Analysis of Tumor-Targeted Antifolates That Inhibit Glycinamide Ribonucleotide Formyltransferase. Biochemistry 2016, 55, 4574–4582. [Google Scholar] [PubMed] [Green Version]
- Deng, Y.; Wang, Y.; Cherian, C.; Hou, Z.; Buck, S.A.; Matherly, L.H.; Gangjee, A. Synthesis and discovery of high affinity folate receptor-specific glycinamide ribonucleotide formyltransferase inhibitors with antitumor activity. J. Med. Chem. 2008, 51, 5052–5063. [Google Scholar] [PubMed] [Green Version]
- Deng, Y.; Zhou, X.; Desmoulin, S.K.; Wu, J.; Cherian, C.; Hou, Z.; Matherly, L.H.; Gangjee, A. Synthesis and biological activity of a novel series of 6-substituted thieno [2,3-d]pyrimidine antifolate inhibitors of purine biosynthesis with selectivity for high affinity folate receptors over the reduced folate carrier and proton-coupled folate transporter for cellular entry. J. Med. Chem. 2009, 52, 2940–2951. [Google Scholar]
- Elnakat, H.; Ratnam, M. Distribution, functionality and gene regulation of folate receptor isoforms: Implications in targeted therapy. Adv. Drug Deliv. Rev. 2004, 56, 1067–1084. [Google Scholar] [CrossRef]
- Bender, D.A. Biochemistry of tryptophan in health and disease. Mol. Aspects Med. 1983, 6, 101–197. [Google Scholar] [CrossRef]
- Chen, Y.; Guillemin, G.J. Kynurenine pathway metabolites in humans: Disease and healthy States. Int. J. Tryptophan Res. 2009, 2, 1–19. [Google Scholar] [CrossRef] [Green Version]
- McCreanor, G.M.; Bender, D.A. The metabolism of high intakes of tryptophan, nicotinamide and nicotinic acid in the rat. Br. J. Nutr. 1986, 56, 577–586. [Google Scholar] [CrossRef] [Green Version]
- Raison, C.L.; Dantzer, R.; Kelley, K.W.; Lawson, M.A.; Woolwine, B.J.; Vogt, G.; Spivey, J.R.; Saito, K.; Miller, A.H. CSF concentrations of brain tryptophan and kynurenines during immune stimulation with IFN-alpha: Relationship to CNS immune responses and depression. Mol. Psychiatry 2010, 15, 393–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maddison, D.C.; Giorgini, F. The kynurenine pathway and neurodegenerative disease. Semin. Cell Dev. Biol. 2015, 40, 134–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkateswaran, N.; Lafita-Navarro, M.C.; Hao, Y.-H.; Kilgore, J.A.; Perez-Castro, L.; Braverman, J.; Borenstein-Auerbach, N.; Kim, M.; Lesner, N.P.; Mishra, P.; et al. MYC promotes tryptophan uptake and metabolism by the kynurenine pathway in colon cancer. Genes Dev. 2019, 33, 1236–1251. [Google Scholar] [CrossRef] [Green Version]
- Huang, A.; Fuchs, D.; Widner, B.; Glover, C.; Henderson, D.C.; Allen-Mersh, T.G. Serum tryptophan decrease correlates with immune activation and impaired quality of life in colorectal cancer. Br. J. Cancer 2002, 86, 1691–1696. [Google Scholar] [CrossRef] [PubMed]
- Schroecksnadel, K.; Winkler, C.; Fuith, L.C.; Fuchs, D. Tryptophan degradation in patients with gynecological cancer correlates with immune activation. Cancer Lett. 2005, 223, 323–329. [Google Scholar] [CrossRef]
- Weinlich, G.; Murr, C.; Richardsen, L.; Winkler, C.; Fuchs, D. Decreased serum tryptophan concentration predicts poor prognosis in malignant melanoma patients. Dermatology 2007, 214, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Suda, T.; Furuhashi, K.; Suzuki, M.; Fujie, M.; Hahimoto, D.; Nakamura, Y.; Inui, N.; Nakamura, H.; Chida, K. Increased serum kynurenine/tryptophan ratio correlates with disease progression in lung cancer. Lung Cancer 2010, 67, 361–365. [Google Scholar] [CrossRef]
- Nakamura, T.; Shima, T.; Saeki, A.; Hidaka, T.; Nakashima, A.; Takikawa, O.; Saito, S. Expression of indoleamine 2, 3-dioxygenase and the recruitment of Foxp3-expressing regulatory T cells in the development and progression of uterine cervical cancer. Cancer Sci. 2007, 98, 874–881. [Google Scholar] [CrossRef]
- Platten, M.; Nollen, E.A.A.; Röhrig, U.F.; Fallarino, F.; Opitz, C.A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nature Rev. Drug Discov. 2019, 18, 379–401. [Google Scholar] [CrossRef]
- Denison, M.S.; Pandini, A.; Nagy, S.R.; Baldwin, E.P.; Bonati, L. Ligand binding and activation of the Ah receptor. Chem. Biol. Interact. 2002, 141, 3–24. [Google Scholar] [CrossRef] [Green Version]
- Vorderstrasse, B.A.; Kerkvliet, N.I. 2,3,7,8-Tetrachlorodibenzo-p-dioxin affects the number and function of murine splenic dendritic cells and their expression of accessory molecules. Toxicol. Appl. Pharmacol. 2001, 171, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, G.C.; Smith, C.; Thomas, S.; Mandik-Nayak, L.; Laury-Kleintop, L.; Metz, R.; Muller, A.J. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol. Immunother. 2014, 63, 721–735. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.F.; Goth, S.R.; Dong, B.; Pessah, I.N.; Matsumura, F. Aryl hydrocarbon receptor signaling mediates expression of indoleamine 2,3-dioxygenase. Biochem. Biophys. Res. Commun. 2008, 375, 331–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novikov, O.; Wang, Z.; Stanford, E.A.; Parks, A.J.; Ramirez-Cardenas, A.; Landesman, E.; Laklouk, I.; Sarita-Reyes, C.; Gusenleitner, D.; Li, A.; et al. An Aryl Hydrocarbon Receptor-Mediated Amplification Loop That Enforces Cell Migration in ER-/PR-/Her2- Human Breast Cancer Cells. Mol. Pharmacol. 2016, 90, 674–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witkiewicz, A.; Williams, T.K.; Cozzitorto, J.; Durkan, B.; Showalter, S.L.; Yeo, C.J.; Brody, J.R. Expression of indoleamine 2,3-dioxygenase in metastatic pancreatic ductal adenocarcinoma recruits regulatory T cells to avoid immune detection. J. Am. Coll. Surg. 2008, 206, 849–854. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Q.; Lu, K.; Feng, L.-L.; Ding, M.; Gao, J.-M.; Ge, X.-L.; Wang, X. Up-regulated expression of indoleamine 2,3-dioxygenase 1 in non-Hodgkin lymphoma correlates with increased regulatory T-cell infiltration. Leuk. Lymphoma 2014, 55, 405–414. [Google Scholar] [CrossRef]
- Curti, A.; Pandolfi, S.; Valzasina, B.; Aluigi, M.; Isidori, A.; Ferri, E.; Salvestrini, V.; Bonanno, G.; Rutella, S.; Durelli, I.; et al. Modulation of tryptophan catabolism by human leukemic cells results in the conversion of CD25- into CD25+ T regulatory cells. Blood 2007, 109, 2871–2877. [Google Scholar] [CrossRef]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Liang, X.; Dong, W.; Fang, Y.; Lv, J.; Zhang, T.; Fiskesund, R.; Xie, J.; Liu, J.; Yin, X.; et al. Tumor-Repopulating Cells Induce PD-1 Expression in CD8(+) T Cells by Transferring Kynurenine and AhR Activation. Cancer Cell. 2018, 33, 480.e7–494.e7. [Google Scholar] [CrossRef] [Green Version]
- Yue, E.W.; Sparks, R.; Polam, P.; Modi, D.; Douty, B.; Wayland, B.; Glass, B.; Takvorian, A.; Glenn, J.; Zhu, W.; et al. INCB24360 (Epacadostat), a Highly Potent and Selective Indoleamine-2,3-dioxygenase 1 (IDO1) Inhibitor for Immuno-oncology. ACS Med. Chem. Lett. 2017, 8, 486–491. [Google Scholar] [CrossRef]
- Richards, T.; Brin, E. Cell based functional assays for IDO1 inhibitor screening and characterization. Oncotarget 2018, 9, 30814–30820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prendergast, G.C.; Malachowski, W.P.; DuHadaway, J.B.; Muller, A.J. Discovery of IDO1 Inhibitors: From Bench to Bedside. Cancer Res. 2017, 77, 6795–6811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soliman, H.; Khambati, F.; Han, H.S.; Ismail-Khan, R.; Bui, M.M.; Sullivan, D.M.; Antonia, S. A phase-1/2 study of adenovirus-p53 transduced dendritic cell vaccine in combination with indoximod in metastatic solid tumors and invasive breast cancer. Oncotarget 2018, 9, 10110–10117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liquori, A.M.; Costantino, L.; Crescenzi, V.; Elia, V.; Giglio, E.; Puliti, R.; Savino, M.D.S.; Vitagliano, V. Complexes between DNA and polyamines: A molecular model. J. Mol. Biol. 1967, 24, 113–122. [Google Scholar] [CrossRef]
- Frydman, L.; Rossomando, P.C.; Fernandez, C.O.; Samejima, K.; Frydman, V.; Frydman, B. Interactions between natural polyamines and tRNA: An 15N NMR analysis. Proc. Natl. Acad. Sci. USA 1992, 89, 9186–9190. [Google Scholar] [CrossRef] [Green Version]
- Matsufuji, S.; Matsufuji, T.; Miyazaki, Y.; Murakami, Y.; Atkins, J.F.; Gesteland, R.F.; Hayashi, S.-I. Autoregulatory frameshifting in decoding mammalian ornithine decarboxylase antizyme. Cell 1995, 80, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Murakami, Y.; Hayashi, S. Role of antizyme in degradation of ornithine decarboxylase in HTC cells. Biochem. J. 1985, 226, 893–896. [Google Scholar] [CrossRef] [Green Version]
- Park, M.H.; Wolff, E.C.; Folk, J.E. Hypusine: Its post-translational formation in eukaryotic initiation factor 5A and its potential role in cellular regulation. Biofactors 1993, 4, 95–104. [Google Scholar]
- Goyns, M.H. Polyamine content of a non-aqueously isolated chromosome preparation. Exp. Cell Res. 1979, 122, 377–380. [Google Scholar] [CrossRef]
- Sunkara, P.S.; Chang, C.C.; Prakash, N.J. Role of polyamines during chromosome condensation of mammalian cells. Cell. Biol. Int. Rep. 1983, 7, 455–465. [Google Scholar] [CrossRef]
- Tabor, H. The stabilization of Bacillus subtilis transforming principle by spermine. Biochem. Biophys. Res. Commun. 1961, 4, 228–231. [Google Scholar] [CrossRef]
- Brown, P.E. Some effects of binding agents on the x-irradiation of DNA. Radiat. Res. 1968, 34, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Soulet, D.; Gagnon, B.; Rivest, S.; Audette, M.; Poulin, R. A fluorescent probe of polyamine transport accumulates into intracellular acidic vesicles via a two-step mechanism. J. Biol. Chem. 2004, 279, 49355–49366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belting, M.; Mani, K.; Jönsson, M.; Cheng, F.; Sandgren, S.; Jonsson, S.; Ding, K.; Delcros, J.-G.; Fransson, L. Glypican-1 is a vehicle for polyamine uptake in mammalian cells: A pivital role for nitrosothiol-derived nitric oxide. J. Biol. Chem. 2003, 278, 47181–47189. [Google Scholar] [CrossRef] [Green Version]
- Gamble, L.D.; Purgato, S.; Murray, J.; Xiao, L.; Yu, D.M.T.; Hanssen, K.; Giorgi, F.M.; Carter, D.R.; Gifford, A.J.; Valli, E.; et al. Inhibition of polyamine synthesis and uptake reduces tumor progression and prolongs survival in mouse models of neuroblastoma. Sci. Transl. Med. 2019, 11, eaau1099. [Google Scholar] [CrossRef] [Green Version]
- Ozfiliz, P.; Kizilboga, T.; Demir, S.; Alkurt, G.; E Arisan, D.; Palavan-Unsal, N.; Dinler-Doganay, G. Bag-1 promotes cell survival through c-Myc-mediated ODC upregulation that is not preferred under apoptotic stimuli in MCF-7 cells. Cell Biochem. Funct. 2015, 33, 293–307. [Google Scholar] [CrossRef]
- Koomoa, D.L.; Geerts, D.; Lange, I.; Koster, J.; Pegg, A.E.; Feith, D.J.; Bachmann, A.S. DFMO/eflornithine inhibits migration and invasion downstream of MYCN and involves p27Kip1 activity in neuroblastoma. Int. J. Oncol. 2013, 42, 1219–1228. [Google Scholar] [CrossRef] [Green Version]
- Hogarty, M.D.; Norris, M.D.; Davis, K.; Liu, X.; Evageliou, N.F.; Hayes, C.S.; Pawel, B.; Guo, R.; Zhao, H.; Sekyere, E.; et al. ODC1 is a critical determinant of MYCN oncogenesis and a therapeutic target in neuroblastoma. Cancer Res. 2008, 68, 9735–9745. [Google Scholar] [CrossRef] [Green Version]
- Zabala-Letona, A.; Arruabarrena-Aristorena, A.; Martín-Martín, N.; Fernandez-Ruiz, S.; Sutherland, J.D.; Clasquin, M.; Tomas-Cortazar, J.; Jimenez, J.; Torres, I.; Quang, P.; et al. mTORC1-dependent AMD1 regulation sustains polyamine metabolism in prostate cancer. Nature 2017, 547, 109–113. [Google Scholar] [CrossRef]
- Peters, M.C.; Minton, A.; Iv, O.P.; Gilmour, S.K.; Phanstiel, O. A Novel Polyamine-Targeted Therapy for BRAF Mutant Melanoma Tumors. Med. Sci. 2018, 6, 3. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Li, P.; Bi, C.W.; Ma, R.; Yin, Y.; Bi, K.; Li, Q. Plasma N-acetylputrescine, cadaverine and 1,3-diaminopropane: Potential biomarkers of lung cancer used to evaluate the efficacy of anticancer drugs. Oncotarget 2017, 8, 88575–88585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Liu, R.; He, B.; Bi, C.W.; Bi, K.; Li, Q. Polyamine Metabolites Profiling for Characterization of Lung and Liver Cancer Using an LC-Tandem MS Method with Multiple Statistical Data Mining Strategies: Discovering Potential Cancer Biomarkers in Human Plasma and Urine. Molecules 2016, 21, 1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miolo, G.; Muraro, E.; Caruso, D.; Crivellari, D.; Ash, A.; Scalone, S.; Lombardi, D.; Rizzolio, F.; Giordano, A.; Corona, G. Pharmacometabolomics study identifies circulating spermidine and tryptophan as potential biomarkers associated with the complete pathological response to trastuzumab-paclitaxel neoadjuvant therapy in HER-2 positive breast cancer. Oncotarget 2016, 7, 39809–39822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, N.; Ignatenko, N.A.; Yamauchi, P.; Stringer, D.E.; Levenson, C.; Shannon, P.; Perrin, S.; Gerner, E.W. Inhibition of human ornithine decarboxylase activity by enantiomers of difluoromethylornithine. Biochem. J. 2003, 375, 465–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, V.A.; Hess, K.R.; Choucair, A.; Flynn, P.J.; Jaeckle, K.A.; Kyritsis, A.P.; Yung, W.K.; Prados, M.; Bruner, J.M.; Ictech, S.; et al. Phase III randomized study of postradiotherapy chemotherapy with combination alpha-difluoromethylornithine-PCV versus PCV for anaplastic gliomas. Clin. Cancer Res. 2003, 9, 981–990. [Google Scholar]
- Levin, V.A.; Uhm, J.H.; A Jaeckle, K.; Choucair, A.; Flynn, P.J.; Wka, Y.; Prados, M.D.; Bruner, J.M.; Chang, S.M.; Kyritsis, A.P.; et al. Phase III randomized study of postradiotherapy chemotherapy with alpha-difluoromethylornithine-procarbazine, N-(2-chloroethyl)-N′-cyclohexyl-N-nitrosurea, vincristine (DFMO-PCV) versus PCV for glioblastoma multiforme. Clin. Cancer Res. 2000, 6, 3878–3884. [Google Scholar]
- Corral, M.; Wallace, H.M. Upregulation of Polyamine Transport in Human Colorectal Cancer Cells. Biomolecules 2020, 10, 499. [Google Scholar] [CrossRef] [Green Version]
- Gitto, S.B.; Pandey, V.; Oyer, J.L.; Copik, A.J.; Hogan, F.C.; Phanstiel, O.; Altomare, D.A. Difluoromethylornithine Combined with a Polyamine Transport Inhibitor Is Effective against Gemcitabine Resistant Pancreatic Cancer. Mol. Pharm. 2018, 15, 369–376. [Google Scholar] [CrossRef]
- Burns, M.R.; Graminski, G.F.; Weeks, R.S.; Chen, Y.; O’Brien, T.G. Lipophilic lysine-spermine conjugates are potent polyamine transport inhibitors for use in combination with a polyamine biosynthesis inhibitor. J. Med. Chem. 2009, 52, 1983–1993. [Google Scholar] [CrossRef] [Green Version]
- Pleshkewych, A.; Kramer, D.L.; Kelly, E.; Porter, C.W. Independence of drug action on mitochondria and polyamines in L1210 leukemia cells treated with methylglyoxal-bis(guanylhydrazone). Cancer Res. 1980, 40, 4533–4540. [Google Scholar]
- Regenass, U.; Caravatti, G.; Mett, H.; Stanek, J.; Schneider, P.; Müller, M.; Matter, A.; Vertino, P.; Porter, C.W. New S-adenosylmethionine decarboxylase inhibitors with potent antitumor activity. Cancer Res. 1992, 52, 4712–4718. [Google Scholar] [PubMed]
- Millward, M.J.; Joshua, A.; Kefford, R.F.; Aamdal, S.; Thomson, D.; Hersey, P.; Toner, G.C.; Lynch, K. Multi-centre Phase II trial of the polyamine synthesis inhibitor SAM486A (CGP48664) in patients with metastatic melanoma. Investig. New Drugs 2005, 23, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Byers, T.L.; Wiest, L.; Wechter, R.S.; Pegg, A.E. Effects of chronic 5′-([(Z)-4-amino-2-butenyl]methylamino)-5′-deoxy-adenosine (AbeAdo) treatment on polyamine and eIF-5A metabolism in AbeAdo-sensitive and -resistant L1210 murine leukaemia cells. Biochem. J. 1993, 290, 115–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacchi, C.J.; Barker, R.H.; Rodriguez, A.; Hirth, B.; Rattendi, D.; Yarlett, N.; . Hendrick, C.L.; Sybertz, E. Trypanocidal activity of 8-methyl-5′-{[(Z)-4-aminobut-2-enyl]-(methylamino)}adenosine (Genz-644131), an adenosylmethionine decarboxylase inhibitor. Antimicrob. Agents Chemother. 2009, 53, 3269–3272. [Google Scholar] [CrossRef] [Green Version]
- Regenass, U.; Mett, H.; Stanek, J.; Mueller, M.; Kramer, D.; Porter, C.W. CGP 48664, a new S-adenosylmethionine decarboxylase inhibitor with broad spectrum antiproliferative and antitumor activity. Cancer Res. 1994, 54, 3210–3217. [Google Scholar]
- Murray-Stewart, T.R.; Woster, P.M.; Casero, R.A., Jr. Targeting polyamine metabolism for cancer therapy and prevention. Biochem. J. 2016, 473, 2937–2953. [Google Scholar] [CrossRef] [Green Version]
- Evageliou, N.F.; Haber, S.N.; Vu, A.; Laetsch, T.W.; Murray, J.; Gamble, L.D.; Cheng, N.C.; Liu, K.; Reese, M.; Corrigan, K.A.; et al. Polyamine Antagonist Therapies Inhibit Neuroblastoma Initiation and Progression. Clin Cancer Res. 2016, 22, 4391–4404. [Google Scholar] [CrossRef] [Green Version]
- Allen, W.L.; McLean, E.G.; Boyer, J.; McCulla, A.; Wilson, P.M.; Coyle, V.; Longley, D.B.; Casero, R.A.; Johnston, P.G. The role of spermidine/spermine N1-acetyltransferase in determining response to chemotherapeutic agents in colorectal cancer cells. Mol. Cancer Ther. 2007, 6, 128–137. [Google Scholar] [CrossRef] [Green Version]
- McCloskey, D.E.; Woster, P.M.; A Casero, R.; E Davidson, N. Effects of the polyamine analogues N1-ethyl-N11-((cyclopropyl)methyl)-4,8-diazaundecane and N1-ethylN-11-((cycloheptyl)methyl)-4,8-diazaundecane in human prostate cancer cells. Clin Cancer Res. 2000, 6, 17–23. [Google Scholar]
- Goyal, L.; Supko, J.G.; Berlin, J.; Blaszkowsky, L.S.; Carpenter, A.; Heuman, D.M.; Hilderbrand, S.L.; Stuart, K.E.; Cotler, S.; Senzer, N.N.; et al. Phase 1 study of N(1),N(11)-diethylnorspermine (DENSPM) in patients with advanced hepatocellular carcinoma. Cancer Chemother Pharmacol. 2013, 72, 1305–1314. [Google Scholar] [CrossRef]
- Wolff, A.C.; Armstrong, D.K.; Fetting, J.H.; Carducci, M.K.; Riley, C.D.; Bender, J.F.; Casero, R.A.; E Davidson, N. A Phase II study of the polyamine analog N1,N11-diethylnorspermine (DENSpm) daily for five days every 21 days in patients with previously treated metastatic breast cancer. Clin. Cancer Res. 2003, 9, 5922–5928. [Google Scholar] [PubMed]
- Hahm, H.A.; Ettinger, D.S.; Bowling, K.; Hoker, B.; Chen, T.L.; Zabelina, Y.; Casero, R.A. Phase I study of N(1),N(11)-diethylnorspermine in patients with non-small cell lung cancer. Clin. Cancer Res. 2002, 8, 684–690. [Google Scholar] [PubMed]



{kind=link}
{kind=link}
| Pathway | Target | Inhibitor | Phase | Trial ID | Target Cancer | Last Updated Dates, Status |
|---|---|---|---|---|---|---|
| Amino acid transporter | ASCT2 | MEDI7247 | Phase I | NCT03106428 | Hematological cancers | Completed |
| LAT1 | JPH203 | Phase II | UMIN000034080 | Advanced biliary tract cancer | Completed | |
| Macropinocytosis | PI3K | BKM120 | Phase II | NCT02301364 | Lymphoma | Completed |
| NCT01297491 | Lung cancer | Completed | ||||
| Phase III | NCT01610284 | Breast cancer | Completed | |||
| ZSTK474 | Phase I | NCT01280487 | Advanced solid tumors | Completed | ||
| Polyamines | ODC and polyamine transport | DFMO with AMXT1501 | Phase I | NCT03536728 | Advanced solid tumors | 13 May 2020 Recruiting |
| Serine-folate cycle | GART; SHMT1/2 | Lometrexol with paclitaxel | Phase I | NCT00024310 | Advanced solid tumors | 17 September 2013 |
| Lometrexol | Phase II | NCT00033722 | Stage IIIB and IV NSCLC | 6 January 2014 | ||
| Methionine cycle | MAT2A | AG-270 with docetaxel, nab-paclitaxel, and gemcitabine | Phase I | NCT03435250 | Advanced solid tumor or lymphoma with homozygous MTAP deletion | 10 July 2020 Recruiting |
| Glutathione | GCL | Buthionine sulfoximine (BSO) with Melphalan | Phase I | NCT03435250 | Neuroblastoma in pediatric patients | 1 February 2017 |
| BSO with Melphalan (followed by bone marrow or peripheral stem cell transplantation) | Phase I | NCT00002730 | Resistant or recurring neuroblastoma in pediatric patients | 30 August 2016 | ||
| Kynurenine | IDO1 | Epacadostat + chemoradiation | Phase I | NCT03516708 | Rectal cancer | 18 June 2020 Recruiting |
| Epacadostat+Pembrolizumab+CRS-207+/- Cyclophosphamide/GVAX | Phase II | NCT03006302 | Pancreas cancer | 12 February 2020 Recruiting | ||
| Epacadostat+Pembrolizumab | Phase II | NCT03291054 | Gastrointestinal cancer | 18 December 2019 Active | ||
| Pembrolizumab +/- Epacadostat | Phase II | NCT03322540 | Lung cancer | 5 February 2020 Active | ||
| Pembrolizumab+ platimun based therapy (pemetrexed, carboplatin, cisplatin or paclitaxel) +/- Epacadostat | Phase II | NCT03322566 | Metastatic NSCLC | 29 January 2020 Active | ||
| Pembrolizumab +/- Epacadostat | Phase III | NCT02752074 | Melanoma | Completed | ||
| Pembrolizumab +/- Epacadostat | Phase III | NCT03361865 | Urothelial cancer | Active | ||
| Pembrolizumab +/- Epacadostat | Phase III | NCT03358472 | Head and neck cancer | Active | ||
| BMS986205 with Atezolizumab | Phase I | NCT02471846 | Advanced solid tumors | Completed | ||
| BMS986205 with Nivolumab | Phase I/II | NCT03695250 | Liver cancer | 25 February 2020 Recruiting | ||
| BMS986205 with Nivolumab+ Ipilimumab | Phase I/II | NCT02658890 | Lung cancer and Melanoma | 9 June 2020 Recruiting | ||
| BMS986205 with Nivolumab+Gemcitabine+Cisplatin | Phase III | NCT03661320 | Bladder cancer | 29 June 2020 Recruiting | ||
| BMS986205 with Nivolumab | Phase III | NCT03329846 | Melanoma | 16 June 2020 Active | ||
| IDO1/TDO | HTI-1090/SHR-9146 with SHR-1210 and Apatinib | Phase I | NCT03491631 | Advanced solid tumors | 6 June 2018 Unknown | |
| Pyrimidine synthesis | DHODH | Leflunomide | Phase I/II | NCT03709446 | Metastatic Triple Negative Breast cancer | 17 March 2020 Recruiting |
| Mitoxantrone and Prednisone +/- Leflunomide | Phase II/III | NCT00004071 | Prostate Cancer | 11 September 2012 | ||
| Leflunomide | Phase II | NCT04463615 | Recurrent and refractory lymphoproliferative disorders | 9 July 2020 Not yet recruiting | ||
| Leflunomide | Phase II | NCT00003775 | Brain and central nervous system tumor | 13 September 2012 | ||
| Leflunomide | Phase I/II | NCT02509052 | Recurrent and refractory plasma cell myeloma | 6 September 2019 Active |
| Pathway | Inhibitor | FDA Appoved for | Original Target | Metabolic Target |
|---|---|---|---|---|
| Amno acid transporter | Sulfasalazine | Anti-inflammatory | Unclear | xCT |
| Sorafenib | Cancer (Liver; kidney; thyroid) | RTK | xCT | |
| Macropinocytosis | Imipramine | Anti-depressant | Tricyclic antidepressants | Membrane ruffle formation |
| BYL719 | Cancer (HR+/HER2- advanced breast cancer) | PI3K | Actin polymerization for membran ruffle formation | |
| Folate cycle | Pemextred | Cancer (Non-small cell lung cancer; pleural mesothelioma) | DHFR; TS; AICART;GART | DHFR; TS; AICART;GART |
| Methotrexate | Cancer (Leukemia; breast cancer; lymphoma; osteosarcoma; primary central nervous system lymphoma) | DHFR | DHFR | |
| Glutathione | Ethacrynic Acid (EA) | Diuretic agent | Na-K-Cl cotransporter in the thick ascending loop of Henle and the macula densa | GSTP1-1 |
| Nucleotide synthesis | 5-Flurouracil (5-FU) | Anti-neoplastic agent; uracil analog | TS | TS |
| Leflunomide | Immunosuppressive agent | DHODH | DHODH | |
| Mycophenolic Acid (MPA) | Immunosuppressive agent to prevent organ rejection | IMPDH | IMPDH | |
| Polyamine | Difluoromethylornithine(DFMO) | Trypansoma brucei infection | ODC | ODC |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muhammad, N.; Lee, H.M.; Kim, J. Oncology Therapeutics Targeting the Metabolism of Amino Acids. Cells 2020, 9, 1904. https://doi.org/10.3390/cells9081904
Muhammad N, Lee HM, Kim J. Oncology Therapeutics Targeting the Metabolism of Amino Acids. Cells. 2020; 9(8):1904. https://doi.org/10.3390/cells9081904
Chicago/Turabian StyleMuhammad, Nefertiti, Hyun Min Lee, and Jiyeon Kim. 2020. "Oncology Therapeutics Targeting the Metabolism of Amino Acids" Cells 9, no. 8: 1904. https://doi.org/10.3390/cells9081904