Tsg101 Is Involved in the Sorting and Re-Distribution of Glucose Transporter-4 to the Sarcolemma Membrane of Cardiac Myocytes

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Ex Vivo Cardiac Ischemia/Reperfusion
2.3. Isolation of Neonatal Rat Cardiomyocytes and Transfection with Recombinant Adenoviruses
2.4. Measurement of Glucose Uptake
2.5. Measurement of ATP Content and Cell Viability
2.6. Western Blotting and Co-Immunoprecipitation (Co-IP) Analysis
2.7. Immunofluorescence Staining Analysis
2.8. Statistical Analysis
3. Results
3.1. Tsg101 and Sarcolemma Glut-4 Levels Are Upregulated in Ischemic Hearts
3.2. Cardiac-Specific Overexpression of Tsg101 Elevates Sarcolemma Levels of Glut-4 in Hearts
3.3. Overexpression of Tsg101 Enhances Glucose Uptake, ATP Generation, and Functional Recovery in Ischemic Hearts
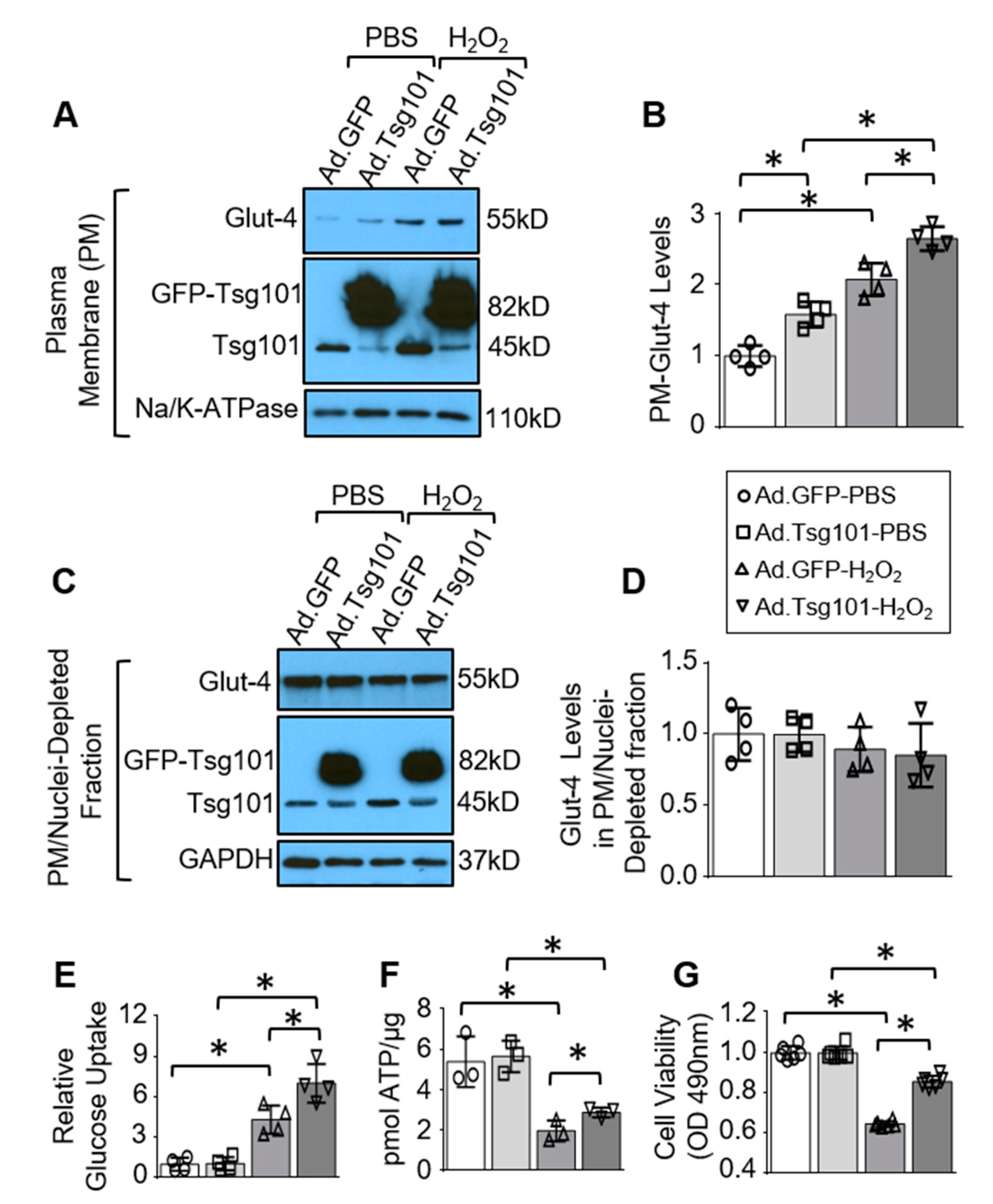
3.4. Elevation of Tsg101 in Cardiomyocytes Improves Glucose Uptake and Cardiomyocyte Survival upon Hypoxic Conditions
3.5. Knockdown of Tsg101 Diminishes Sarcolemma Levels of Glut-4 as Well as ATP Production and Function Recovery in Ischemic Hearts
3.6. Reduction of Tsg101 in Neonatal Myocytes Impedes Glucose Uptake, ATP Production and Promotes Cell Death upon Hypoxic Conditions
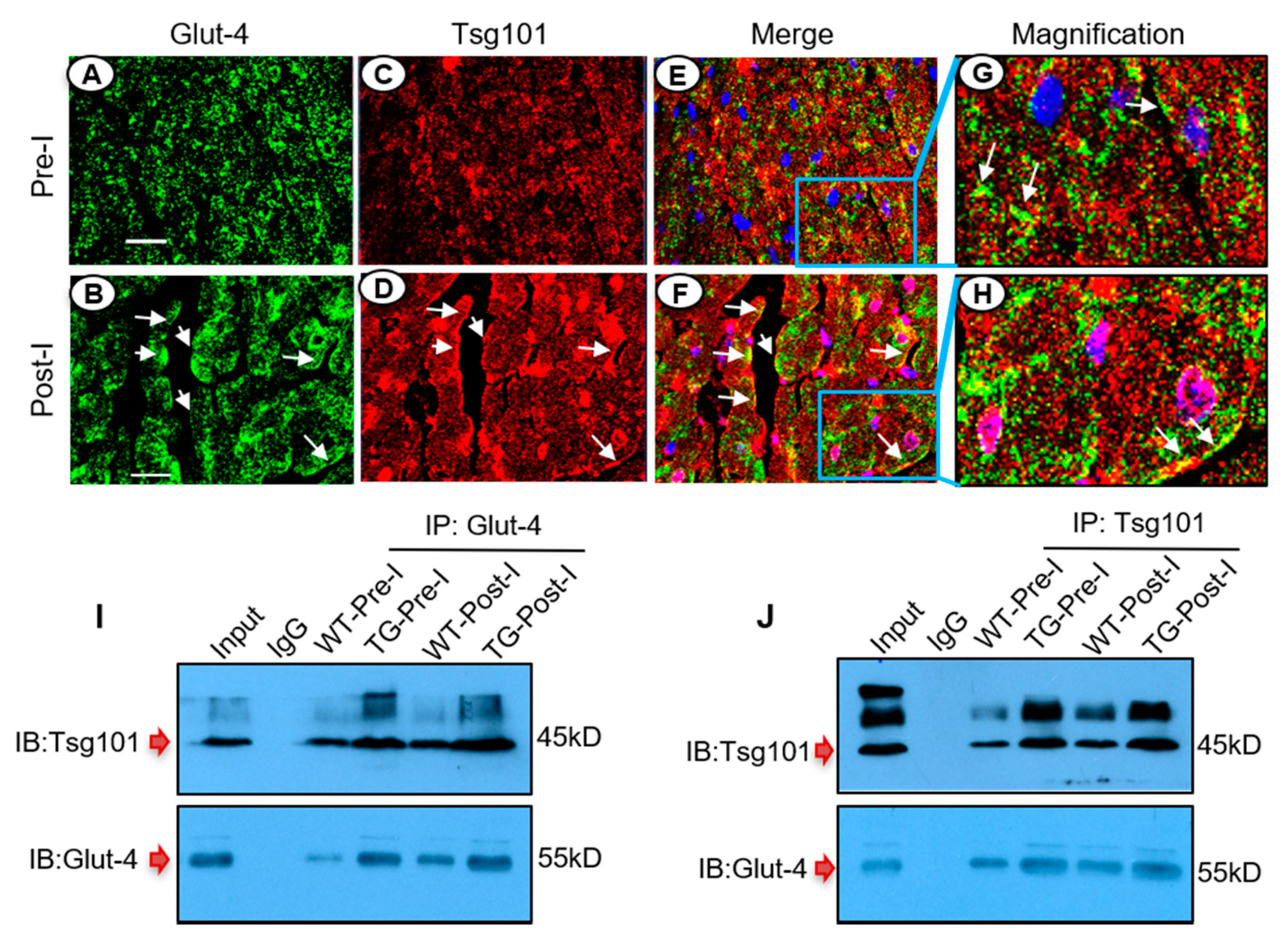
3.7. Tsg101 Interacts and Co-Localizes with Glut-4 in the Ischemic Heart
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wagner, R.; Piler, P.; Gabbasov, Z.A.; Maruyama, J.; Maruyama, K.; Nicovsky, J.; Kruzliak, P. Adjuvant Cardioprotection in Cardiac Surgery: Update. BioMed Res. Int. 2014, 2014, 1–12. [Google Scholar] [CrossRef]
- Patterson, B.; Fields, A.V.; Shannon, R.P. New insights into myocardial glucose metabolism: Surviving under stress. Curr. Opin. Clin. Nutr. Metab. Care 2009, 12, 424–430. [Google Scholar] [CrossRef]
- Frank, A.; Bonney, M.; Bonney, S.; Weitzel, L.; Koeppen, M.; Eckle, T. Myocardial ischemia reperfusion injury: From basic science to clinical bedside. Semin. Cardiothorac. Vasc. Anesth. 2012, 16, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.; Tian, R. Glucose Transporters in Cardiac Metabolism and Hypertrophy. Compr. Physiol. 2015, 6, 331–351. [Google Scholar] [CrossRef] [Green Version]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.L.; Jaswal, J.S.; Stanley, W.C. Myocardial Fatty Acid Metabolism in Health and Disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef]
- Abel, E.D.; O’Shea, K.M.; Ramasamy, R. Insulin Resistance: Metabolic Mechanisms and Consequences in the Heart. Arter. Thromb. Vasc. Biol. 2012, 32, 2068–2076. [Google Scholar] [CrossRef] [Green Version]
- Cartee, G.D. Mechanisms for greater insulin-stimulated glucose uptake in normal and insulin-resistant skeletal muscle after acute exercise. Am. J. Physiol. Metab. 2015, 309, E949–E959. [Google Scholar] [CrossRef] [Green Version]
- Govers, R. Molecular mechanisms of GLUT4 regulation in adipocytes. Diabetes Metab. 2014, 40, 400–410. [Google Scholar] [CrossRef]
- Beauloye, C.; Bertrand, L.; Krause, U.; Marsin, A.-S.; Dresselaers, T.; Vanstapel, F.; Vanoverschelde, J.-L.; Hue, L. No-flow ischemia inhibits insulin signaling in heart by decreasing intracellular pH. Circ. Res. 2001, 88, 513–519. [Google Scholar] [CrossRef] [Green Version]
- Hue, L.; Beauloye, C.; Marsin, A.-S.; Bertrand, L.; Horman, S.; Rider, M.H. Insulin and ischemia stimulate glycolysis by acting on the same targets through different and opposing signaling pathways. J. Mol. Cell. Cardiol. 2002, 34, 1091–1097. [Google Scholar] [CrossRef] [PubMed]
- Fu, F.; Zhao, K.; Li, J.; Xu, J.; Zhang, Y.; Liu, C.; Yang, W.; Gao, C.; Li, J.; Zhang, H.; et al. Direct Evidence that Myocardial Insulin Resistance following Myocardial Ischemia Contributes to Post-Ischemic Heart Failure. Sci. Rep. 2015, 5, 17927. [Google Scholar] [CrossRef] [PubMed]
- Kloner, R.A.; Nesto, R.W. Glucose-Insulin-Potassium for Acute Myocardial Infarction: Continuing Controversy Over Cardioprotection. Circulation 2008, 117, 2523–2533. [Google Scholar] [CrossRef] [PubMed]
- Puskarich, M.A.; Runyon, M.S.; Trzeciak, S.; Kline, J.A.; Jones, A.E. Effect of Glucose-Insulin-Potassium Infusion on Mortality in Critical Care Settings: A Systematic Review and Meta-Analysis. J. Clin. Pharmacol. 2009, 49, 758–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabi, D.M.; Clement, F.M.; McAlister, F.A.; Majumdar, S.R.; Sauve, R.; Johnson, J.A.; Ghali, W.A. Effect of perioperative glucose-insulin-potassium infusions on mortality and atrial fibrillation after coronary artery bypass grafting: A systematic review and meta-analysis. Can. J. Cardiol. 2010, 26, e178–e184. [Google Scholar] [CrossRef] [Green Version]
- Klip, A.; McGraw, T.E.; James, D.E. Thirty sweet years of GLUT4. J. Biol. Chem. 2019, 294, 11369–11381. [Google Scholar] [CrossRef] [Green Version]
- Heather, L.C.; Pates, K.M.; Atherton, H.J.; Cole, M.A.; Ball, D.R.; Evans, R.D.; Glatz, J.F.; Luiken, J.J.; Griffin, J.L.; Clarke, K. Differential Translocation of the Fatty Acid Transporter, FAT/CD36, and the Glucose Transporter, GLUT4, Coordinates Changes in Cardiac Substrate Metabolism During Ischemia and Reperfusion. Circ. Heart Fail. 2013, 6, 1058–1066. [Google Scholar] [CrossRef] [Green Version]
- Tian, R.; Abel, E.D. Responses of GLUT4-deficient hearts to ischemia underscore the importance of glycolysis. Circulation 2001, 103, 2961–2966. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Gilchrist, R.; Borley, N.; Chng, H.W.; Morgan, M.R.; Marshall, J.F.; Camplejohn, R.S.; Muir, G.H.; Hart, I.R. Reduction of TSG101 protein has a negative impact on tumor cell growth. Int. J. Cancer 2004, 109, 541–547. [Google Scholar] [CrossRef]
- Moberg, K.H.; Schelble, S.; Burdick, S.K.; Hariharan, I.K. Mutations in erupted, the Drosophila Ortholog of Mammalian Tumor Susceptibility Gene 101, Elicit Non-Cell-Autonomous Overgrowth. Dev. Cell 2005, 9, 699–710. [Google Scholar] [CrossRef] [Green Version]
- Nabhan, J.F.; Hu, R.; Oh, R.S.; Cohen, S.N.; Lü, Q. Formation and release of arrestin domain-containing protein 1-mediated microvesicles (ARMMs) at plasma membrane by recruitment of TSG101 protein. Proc. Natl. Acad. Sci. USA 2012, 109, 4146–4151. [Google Scholar] [CrossRef] [Green Version]
- Shtanko, O.; Nikitina, R.A.; Altuntas, C.Z.; Chepurnov, A.A.; Davey, R.A. Crimean-Congo Hemorrhagic Fever Virus Entry into Host Cells Occurs through the Multivesicular Body and Requires ESCRT Regulators. PLoS Pathog. 2014, 10, e1004390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, A.; He, X.; Zhang, L.; Yang, L.; Woodman, P.; Li, W. Biogenesis of Lysosome-related Organelles Complex-1 Subunit 1 (BLOS1) Interacts with Sorting Nexin 2 and the Endosomal Sorting Complex Required for Transport-I (ESCRT-I) Component TSG101 to Mediate the Sorting of Epidermal Growth Factor Receptor into Endosomal Compartments. J. Biol. Chem. 2014, 289, 29180–29194. [Google Scholar] [CrossRef] [Green Version]
- Rush, J.S.; Ceresa, B.P. RAB7 and TSG101 are required for the constitutive recycling of unliganded EGFRs via distinct mechanisms. Mol. Cell. Endocrinol. 2013, 381, 188–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Essandoh, K.; Deng, S.; Wang, X.; Jiang, M.; Mu, X.; Peng, J.; Li, Y.; Peng, T.; Wagner, K.-U.; Rubinstein, J.; et al. Tsg101 positively regulates physiologic-like cardiac hypertrophy through FIP3-mediated endosomal recycling of IGF-1R. FASEB J. 2019, 33, 7451–7466. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Essandoh, K.; Wang, X.; Li, Y.; Huang, W.; Chen, J.; Peng, J.; Jiang, D.-S.; Mu, X.; Wang, C.; et al. Tsg101 positively regulates P62-Keap1-Nrf2 pathway to protect hearts against oxidative damage. Redox Biol. 2020, 32, 101453. [Google Scholar] [CrossRef] [PubMed]
- Essandoh, K.; Wang, X.; Huang, W.; Deng, S.; Gardner, G.; Mu, X.; Li, Y.; Kranias, E.G.; Wang, Y.; Fan, G.-C. Tumor susceptibility gene 101 ameliorates endotoxin-induced cardiac dysfunction by enhancing Parkin-mediated mitophagy. J. Biol. Chem. 2019, 294, 18057–18068. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.-U.; Krempler, A.; Qi, Y.; Park, K.; Henry, M.D.; Triplett, A.A.; Riedlinger, G.; Rucker, E.B., III; Hennighausen, L. Tsg101 Is Essential for Cell Growth, Proliferation, and Cell Survival of Embryonic and Adult Tissues. Mol. Cell. Biol. 2003, 23, 150–162. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.; Nguyen, N.; DeGrado, T.R.; Schwaiger, M.; Brosius, F.C. Ischemia induces translocation of the insulin-responsive glucose transporter GLUT4 to the plasma membrane of cardiac myocytes. Circulation 1994, 89, 793–798. [Google Scholar] [CrossRef] [Green Version]
- Zhou, T.; Chuang, C.-C.; Zuo, L. Molecular Characterization of Reactive Oxygen Species in Myocardial Ischemia-Reperfusion Injury. BioMed Res. Int. 2015, 2015, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef] [Green Version]
- Ku, H.-C.; Chen, W.P.; Su, M.-J. DPP4 Deficiency Exerts Protective Effect against H2O2 Induced Oxidative Stress in Isolated Cardiomyocytes. PLoS ONE 2013, 8, e54518. [Google Scholar] [CrossRef] [PubMed]
- Fillmore, N.; Mori, J.; Lopaschuk, G.D. Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. Br. J. Pharmacol. 2014, 171, 2080–2090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, Y.; Thomas, J.; Sevilla, L.; Muñoz, P.; Becker, C.; Holman, G.; Kozka, I.J.; Palacín, M.; Testar, X.; Kammermeier, H.; et al. Insulin-induced recruitment of glucose transporter 4 (GLUT4) and GLUT1 in isolated rat cardiac myocytes. Evidence of the existence of different intracellular GLUT4 vesicle populations. J. Biol. Chem. 1997, 272, 7085–7092. [Google Scholar] [CrossRef] [Green Version]
- Kessler, A.; Tomás, E.; Immler, D.; Meyer, H.E.; Zorzano, A.; Eckel, J. Rab11 is associated with GLUT4-containing vesicles and redistributes in response to insulin. Diabetologia 2000, 43, 1518–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlig, M.; Passlack, W.; Eckel, J. Functional role of Rab11 in GLUT4 trafficking in cardiomyocytes. Mol. Cell. Endocrinol. 2005, 235, 1–9. [Google Scholar] [CrossRef]
- Schwenk, R.W.; Eckel, J. A novel method to monitor insulin-stimulated GTP-loading of Rab11a in cardiomyocytes. Cell. Signal. 2007, 19, 825–830. [Google Scholar] [CrossRef]
- Ferraiuolo, R.-M.; Manthey, K.C.; Stanton, M.J.; Triplett, A.A.; Wagner, K.-U. The Multifaceted Roles of the Tumor Susceptibility Gene 101 (TSG101) in Normal Development and Disease. Cancers 2020, 12, 450. [Google Scholar] [CrossRef] [Green Version]
- Koumanov, F.; Pereira, V.J.; Whitley, P.R.; Holman, G.D. GLUT4 Traffic through an ESCRT-III-Dependent Sorting Compartment in Adipocytes. PLoS ONE 2012, 7, e44141. [Google Scholar] [CrossRef] [Green Version]
- Ismaili, N.; Blind, R.; Garabedian, M.J. Stabilization of the Unliganded Glucocorticoid Receptor by TSG101. J. Biol. Chem. 2005, 280, 11120–11126. [Google Scholar] [CrossRef] [Green Version]
- Kantamneni, S.; Holman, D.; Wilkinson, K.A.; Corrêa, S.A.L.; Feligioni, M.; Ogden, S.; Fraser, W.; Nishimune, A.; Henley, J.M. GISP binding to TSG101 increases GABA receptor stability by down-regulating ESCRT-mediated lysosomal degradation. J. Neurochem. 2008, 107, 86–95. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Hu, X.; Selvakumar, P.; Russell, R.R.; Cushman, S.W.; Holman, G.D.; Young, L.H. Role of the nitric oxide pathway in AMPK-mediated glucose uptake and GLUT4 translocation in heart muscle. Am. J. Physiol. Metab. 2004, 287, E834–E841. [Google Scholar] [CrossRef] [PubMed]
- Quan, N.; Sun, W.; Wang, L.; Chen, X.; Bogan, J.; Zhou, X.; Cates, C.; Liu, Q.; Zheng, Y.; Li, J. Sestrin2 prevents age-related intolerance to ischemia and reperfusion injury by modulating substrate metabolism. FASEB J. 2017, 31, 4153–4167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malide, D.; Davies-Hill, T.M.; Levine, M.; Simpson, I.A. Distinct localization of GLUT-1, -3, and -5 in human monocyte-derived macrophages: Effects of cell activation. Am. J. Physiol. Content 1998, 274, E516–E526. [Google Scholar] [CrossRef] [PubMed]
- Davey, K.A.B.; Garlick, P.B.; Warley, A.; Southworth, R. Immunogold labeling study of the distribution of GLUT-1 and GLUT-4 in cardiac tissue following stimulation by insulin or ischemia. Am. J. Physiol. Circ. Physiol. 2007, 292, H2009–H2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Essandoh, K.; Deng, S.; Wang, X.; Li, Y.; Li, Q.; Mu, X.; Peng, T.; Fan, G.-C. Tsg101 Is Involved in the Sorting and Re-Distribution of Glucose Transporter-4 to the Sarcolemma Membrane of Cardiac Myocytes. Cells 2020, 9, 1936. https://doi.org/10.3390/cells9091936
Essandoh K, Deng S, Wang X, Li Y, Li Q, Mu X, Peng T, Fan G-C. Tsg101 Is Involved in the Sorting and Re-Distribution of Glucose Transporter-4 to the Sarcolemma Membrane of Cardiac Myocytes. Cells. 2020; 9(9):1936. https://doi.org/10.3390/cells9091936
Chicago/Turabian StyleEssandoh, Kobina, Shan Deng, Xiaohong Wang, Yutian Li, Qianqian Li, Xingjiang Mu, Tianqing Peng, and Guo-Chang Fan. 2020. "Tsg101 Is Involved in the Sorting and Re-Distribution of Glucose Transporter-4 to the Sarcolemma Membrane of Cardiac Myocytes" Cells 9, no. 9: 1936. https://doi.org/10.3390/cells9091936
APA StyleEssandoh, K., Deng, S., Wang, X., Li, Y., Li, Q., Mu, X., Peng, T., & Fan, G.-C. (2020). Tsg101 Is Involved in the Sorting and Re-Distribution of Glucose Transporter-4 to the Sarcolemma Membrane of Cardiac Myocytes. Cells, 9(9), 1936. https://doi.org/10.3390/cells9091936