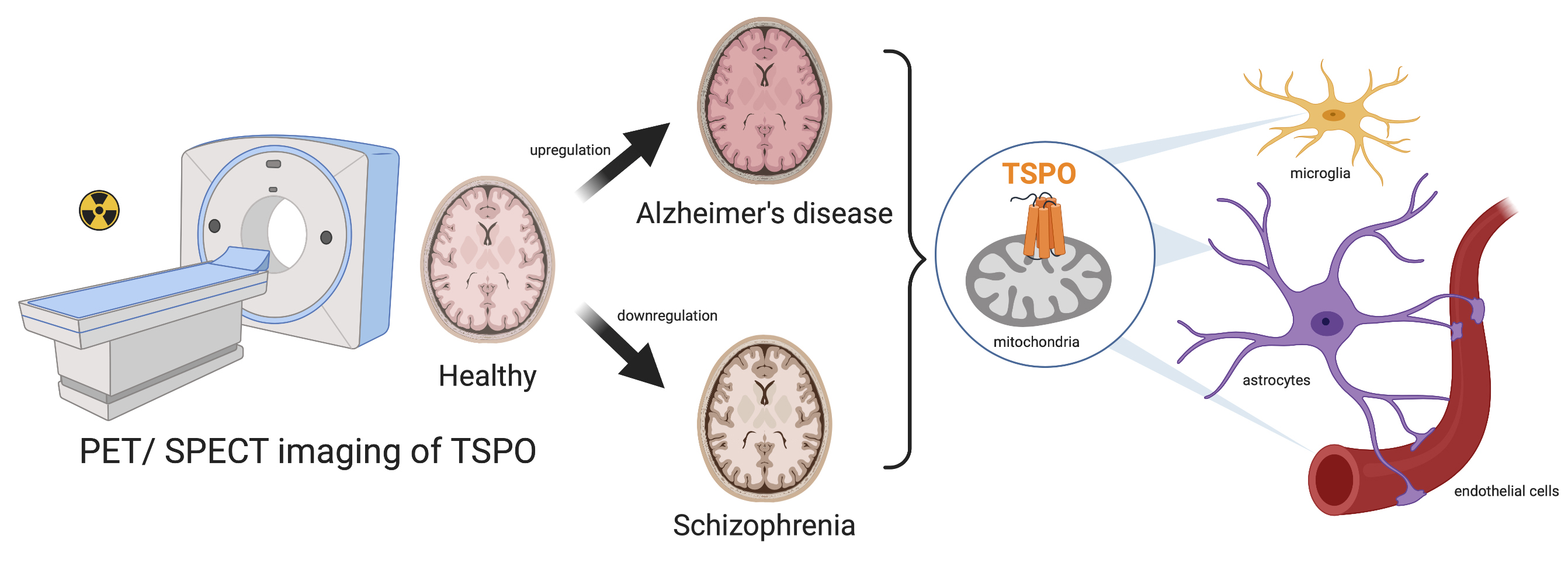
In Vivo TSPO Signal and Neuroinflammation in Alzheimer’s Disease

 , , ,
, , , 
Abstract
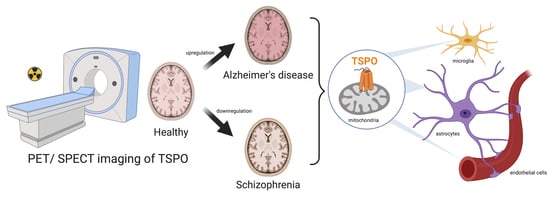
:
1. Introduction
2. TSPO: An In Vivo Marker of Neuroinflammation
2.1. Evidence of TSPO Upregulation in Alzheimer’s Disease
2.1.1. TSPO Alterations Are Brain Region Specific
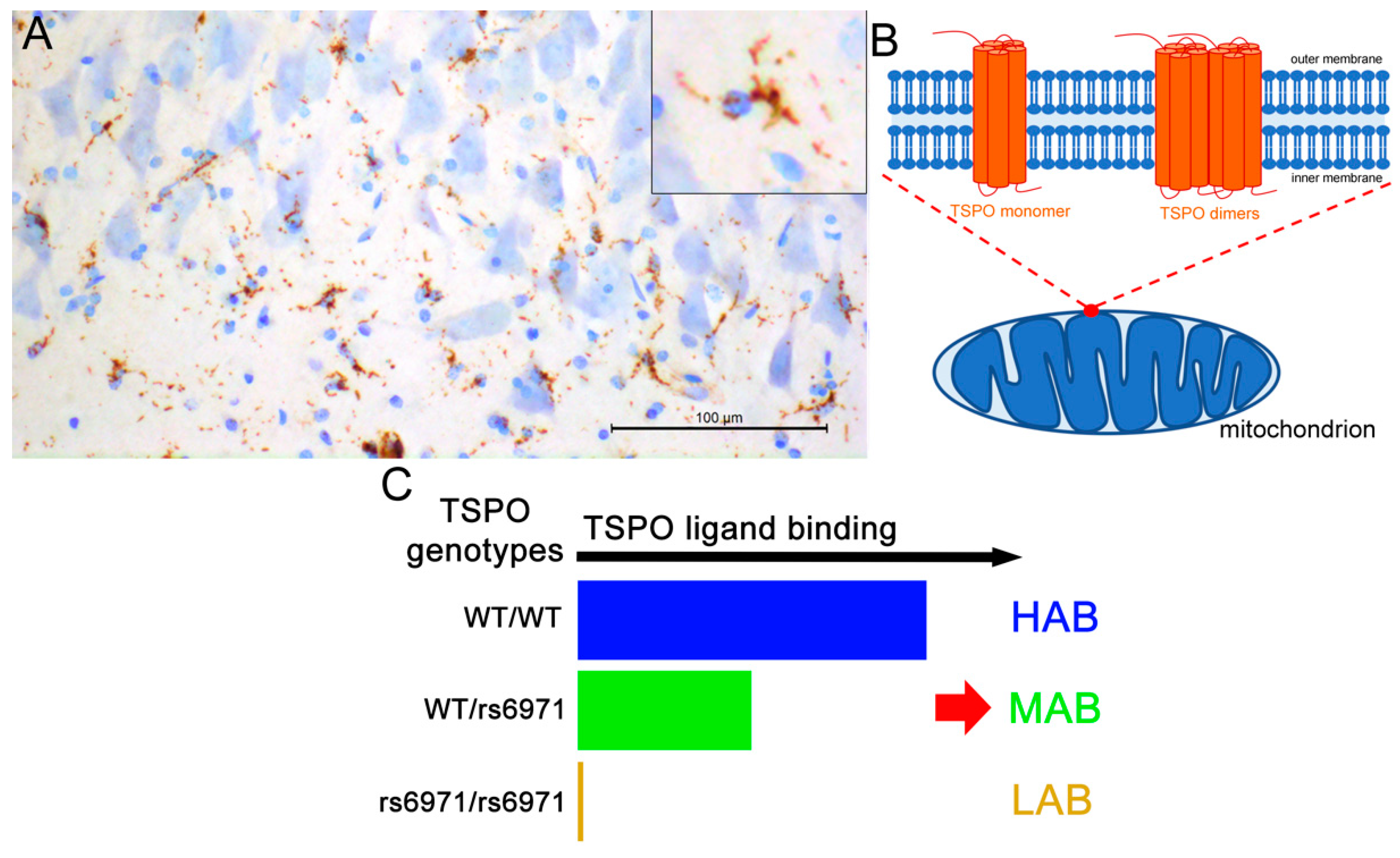
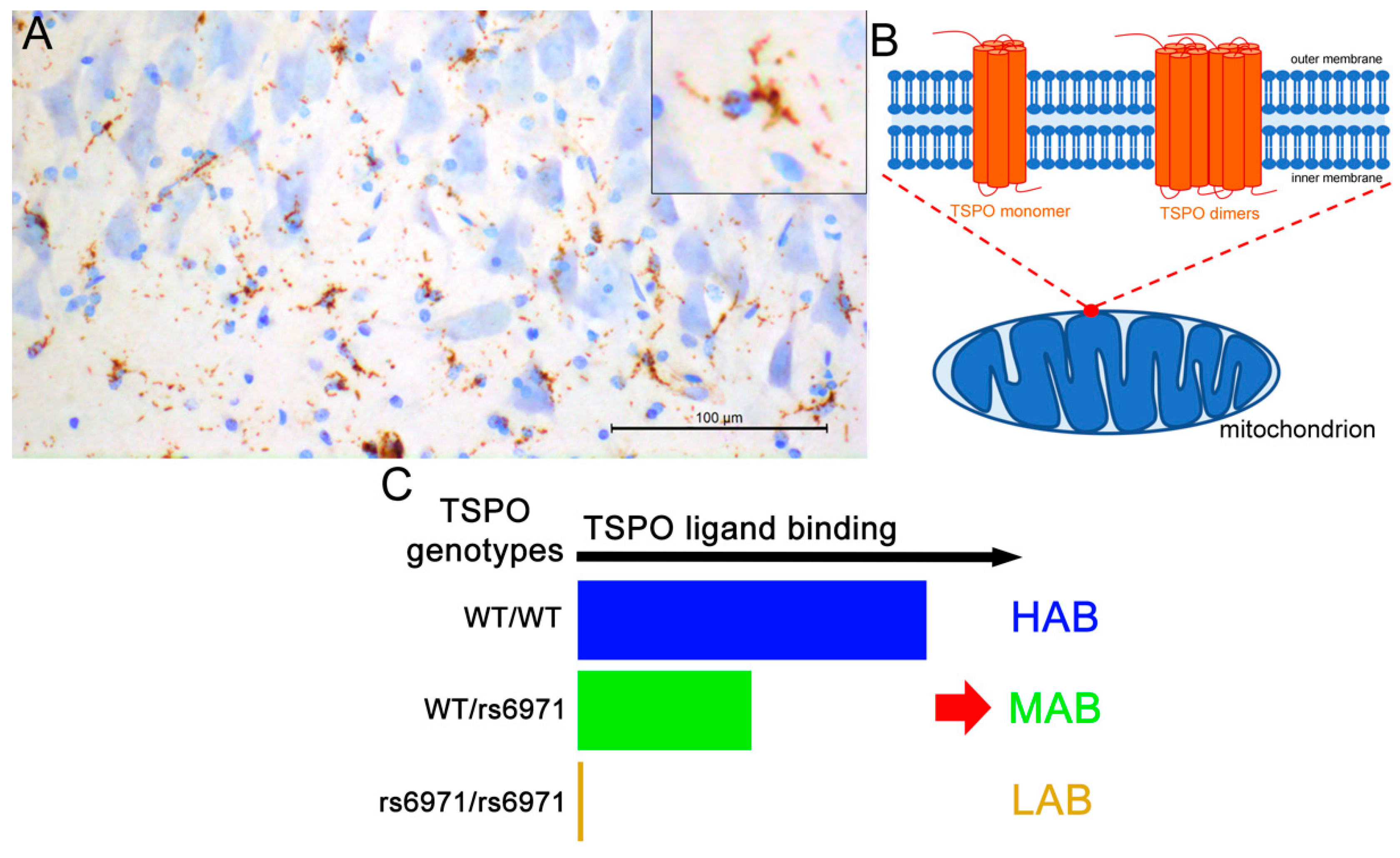
2.1.2. Impact of the Method of In Vivo TSPO Quantification
Methods of In Vivo TSPO Quantification
Impact of the Method of In Vivo TSPO Quantification
2.1.3. Other Influencing Factors
2.2. Evidence of a TSPO Upregulation in Preclinical Models of Alzheimer’s Disease
2.2.1. TSPO Is Increased in Tau, APP/PS1 and APP/PS1/Tau Mouse Models
2.2.2. TSPO Binding in Cerebellum and Choroid Plexus
2.3. Is TSPO Altered in Other Pathologies?
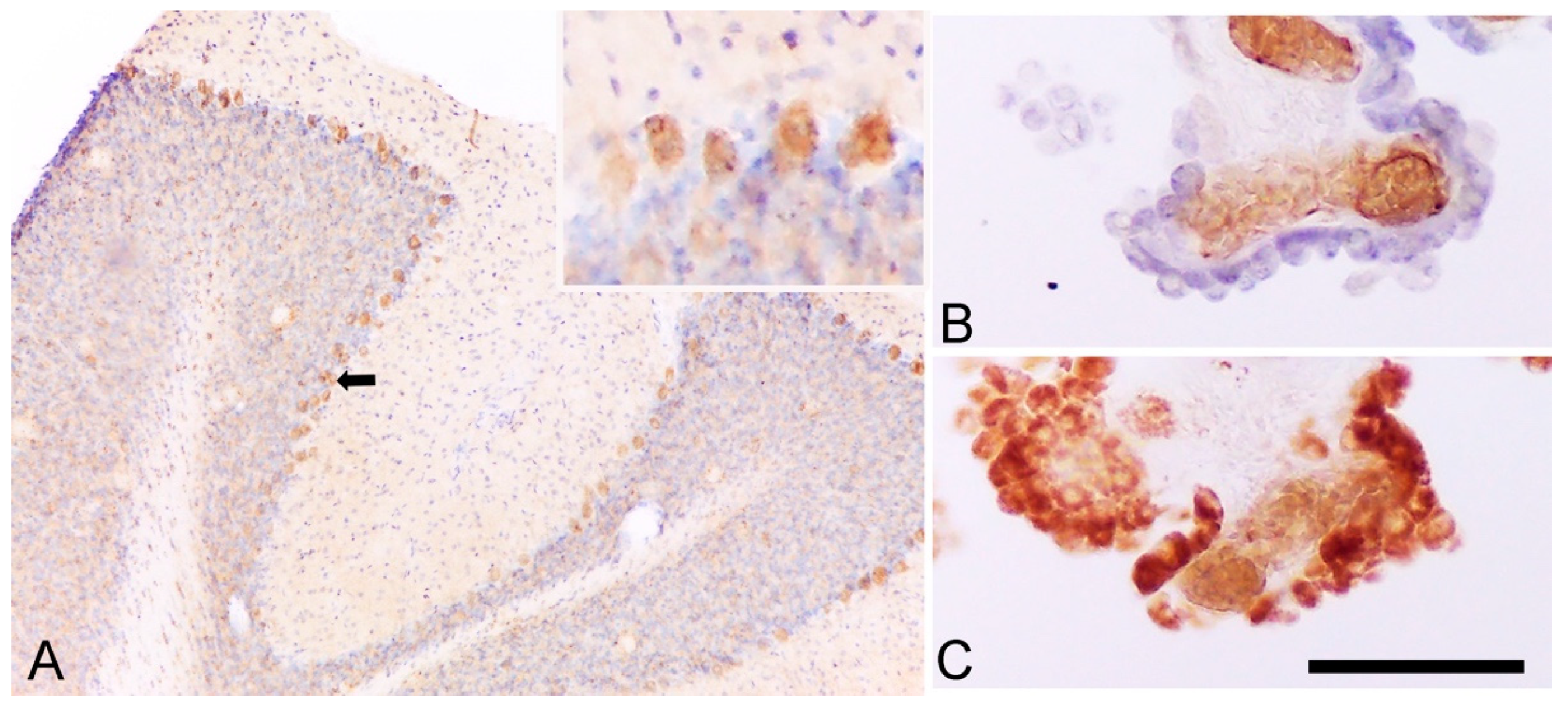
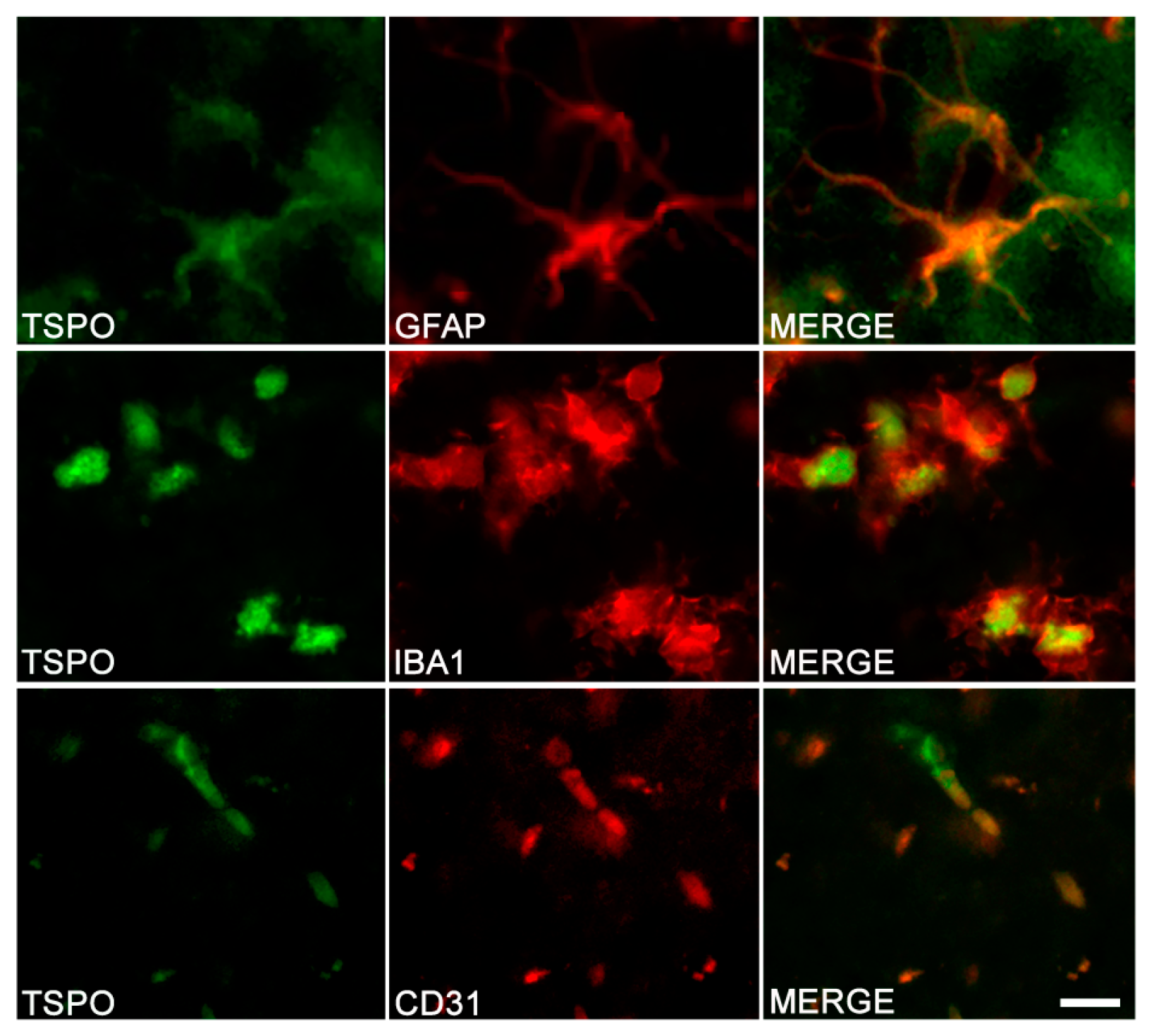
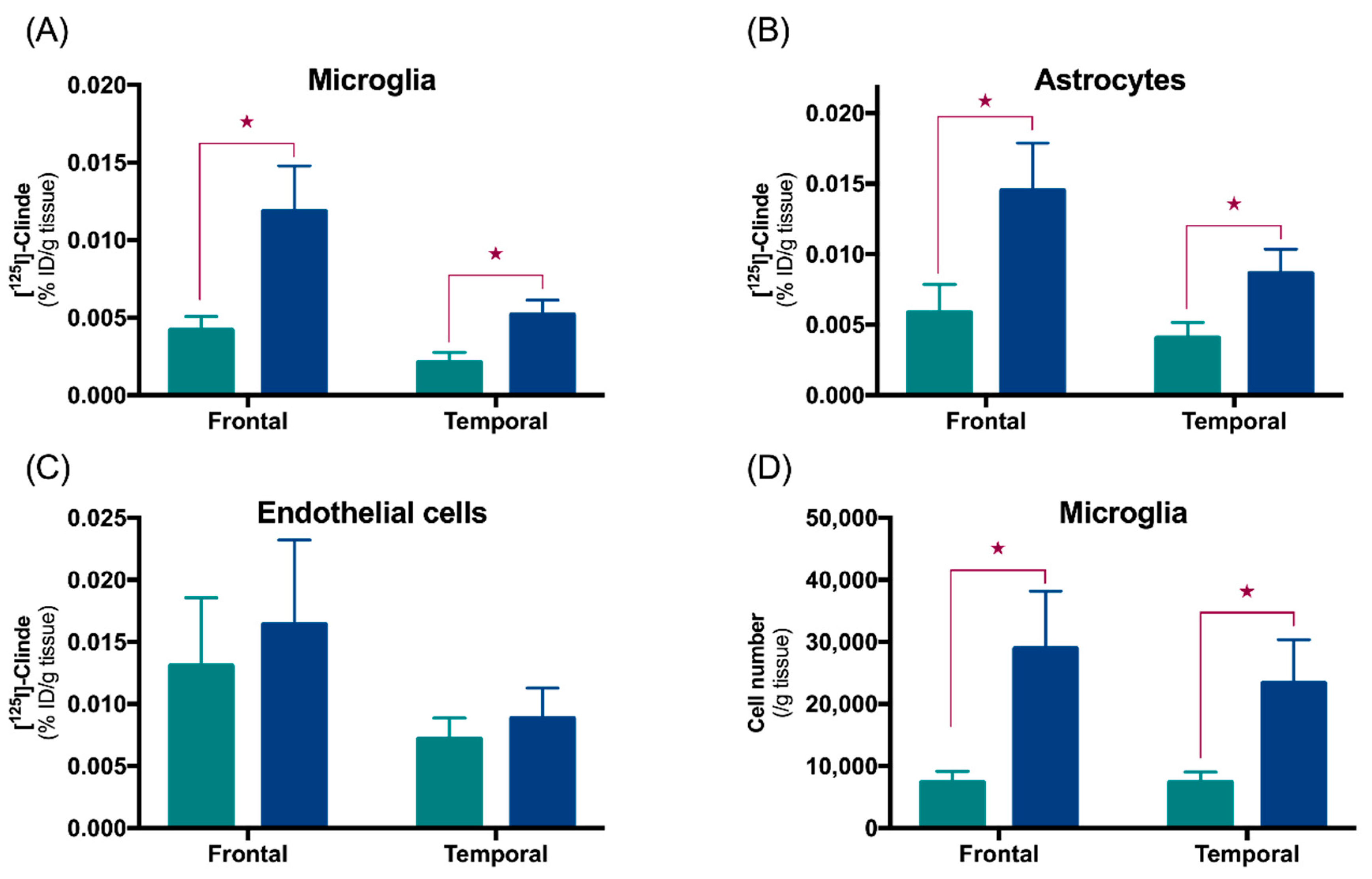
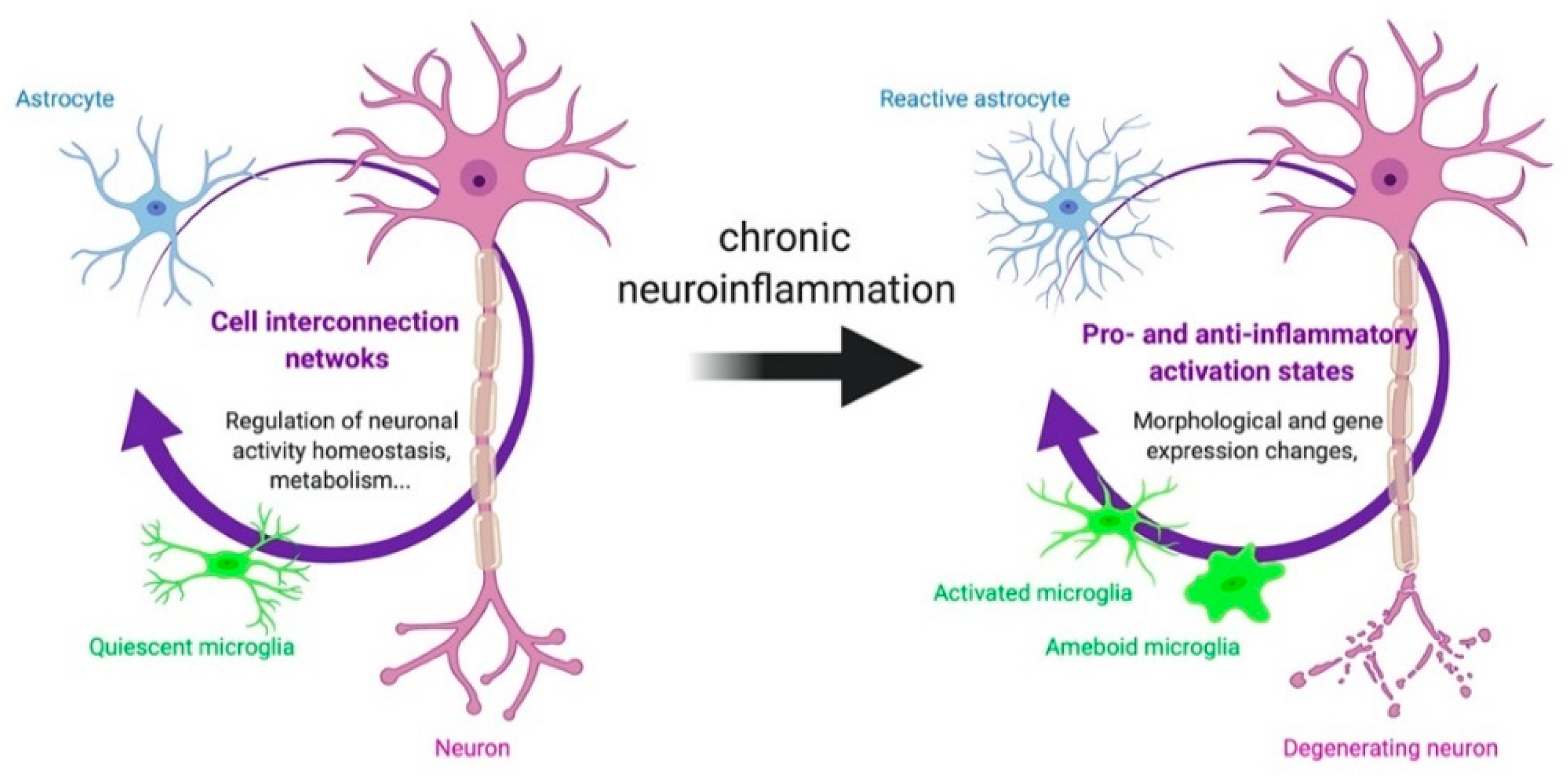
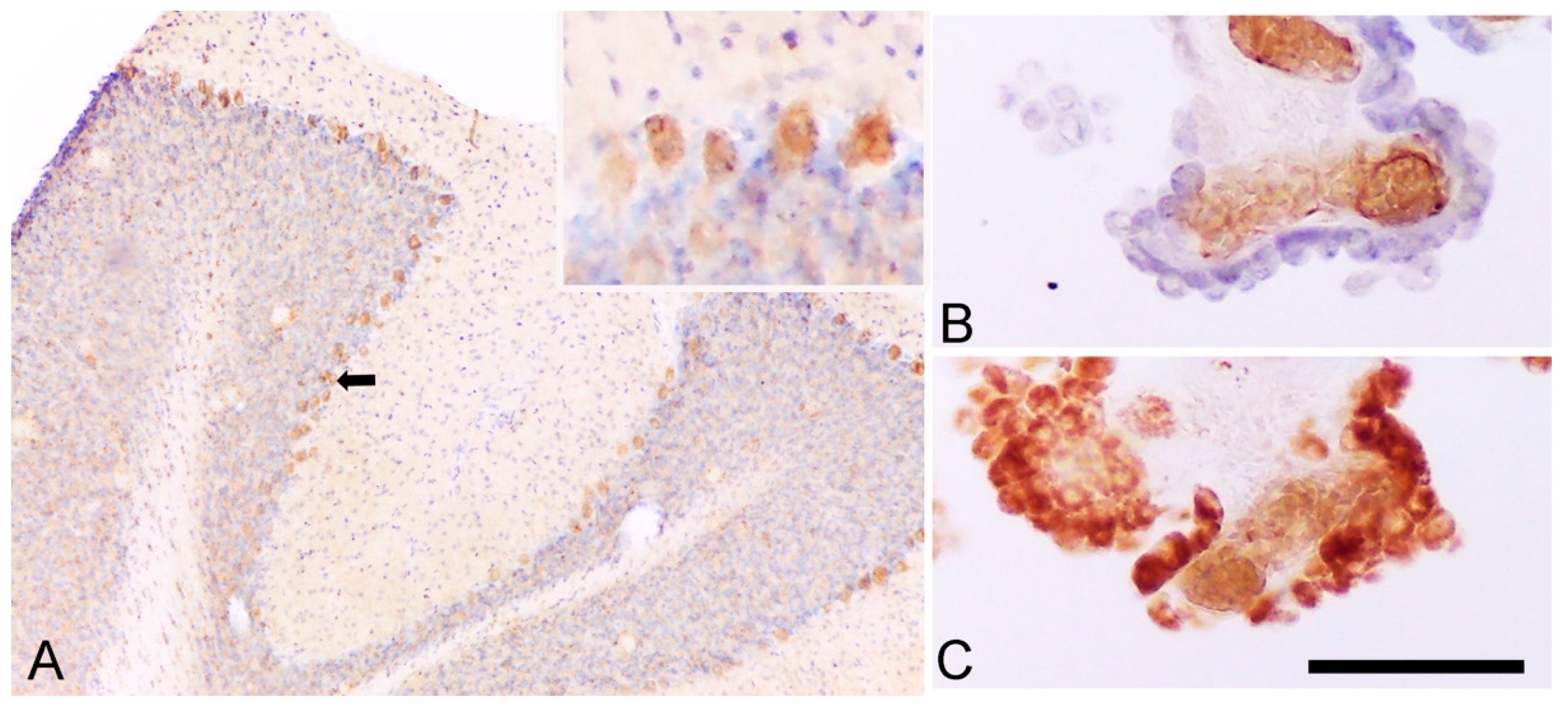
3. Cell Origin of TSPO Alterations
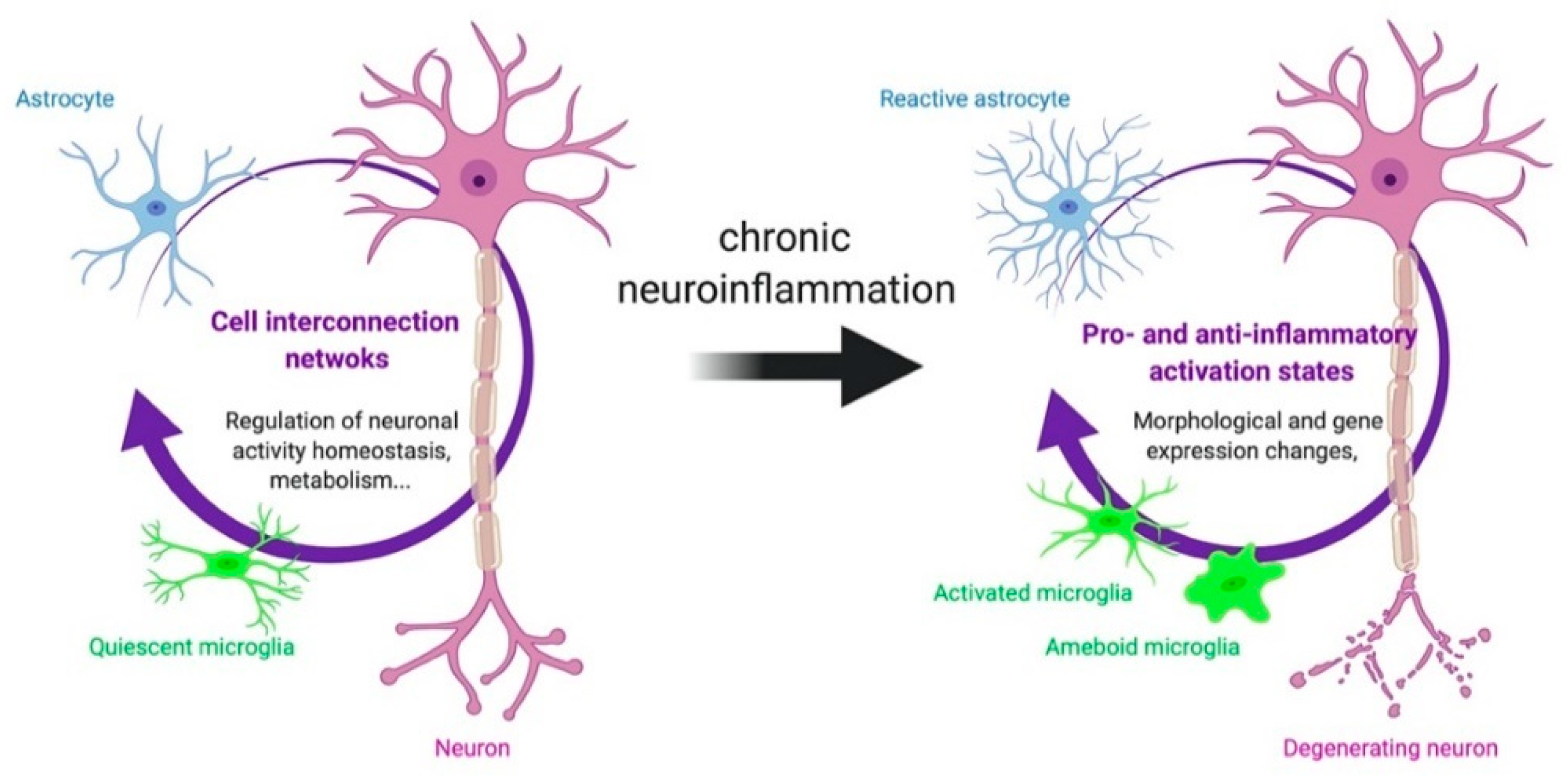
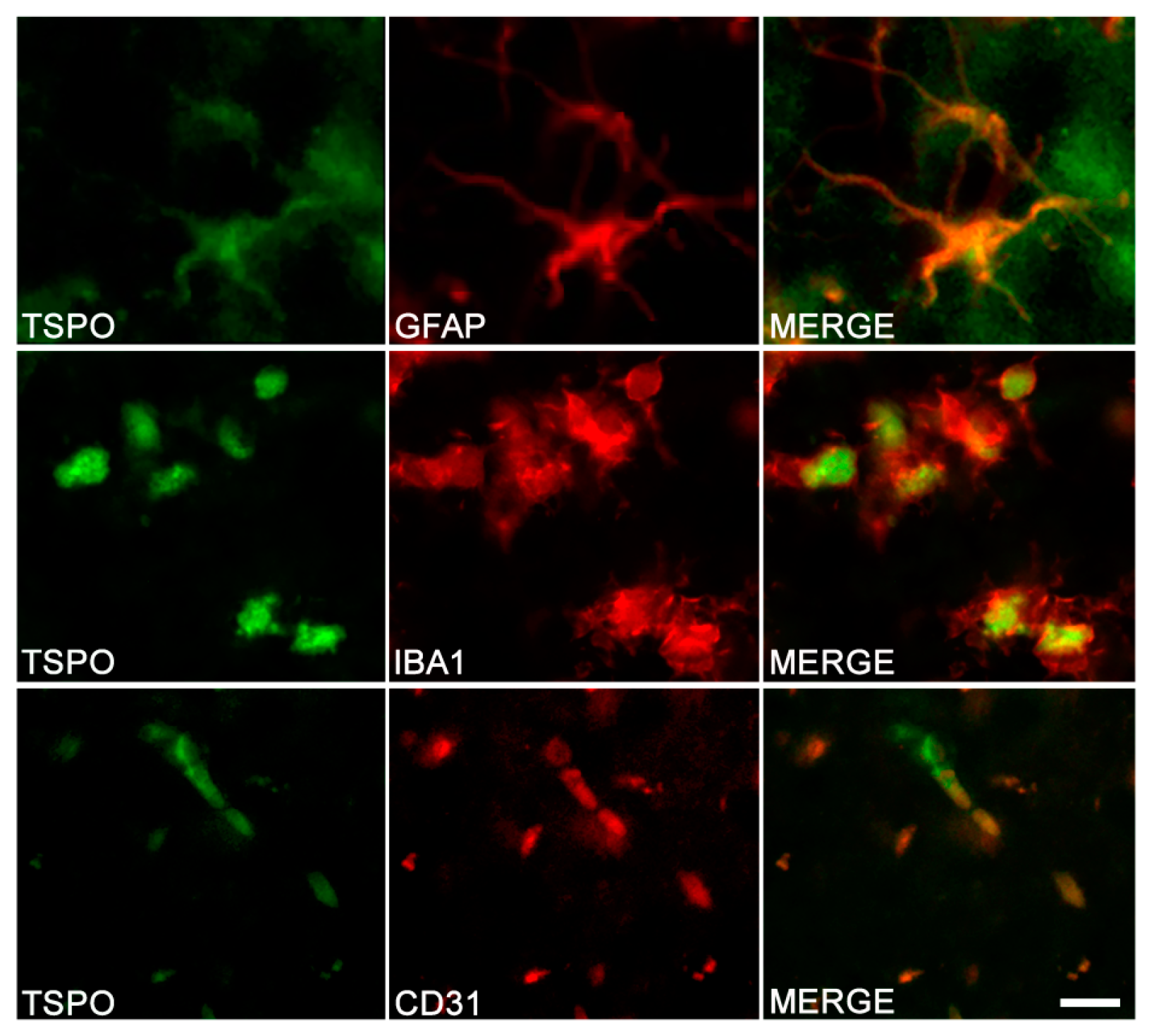
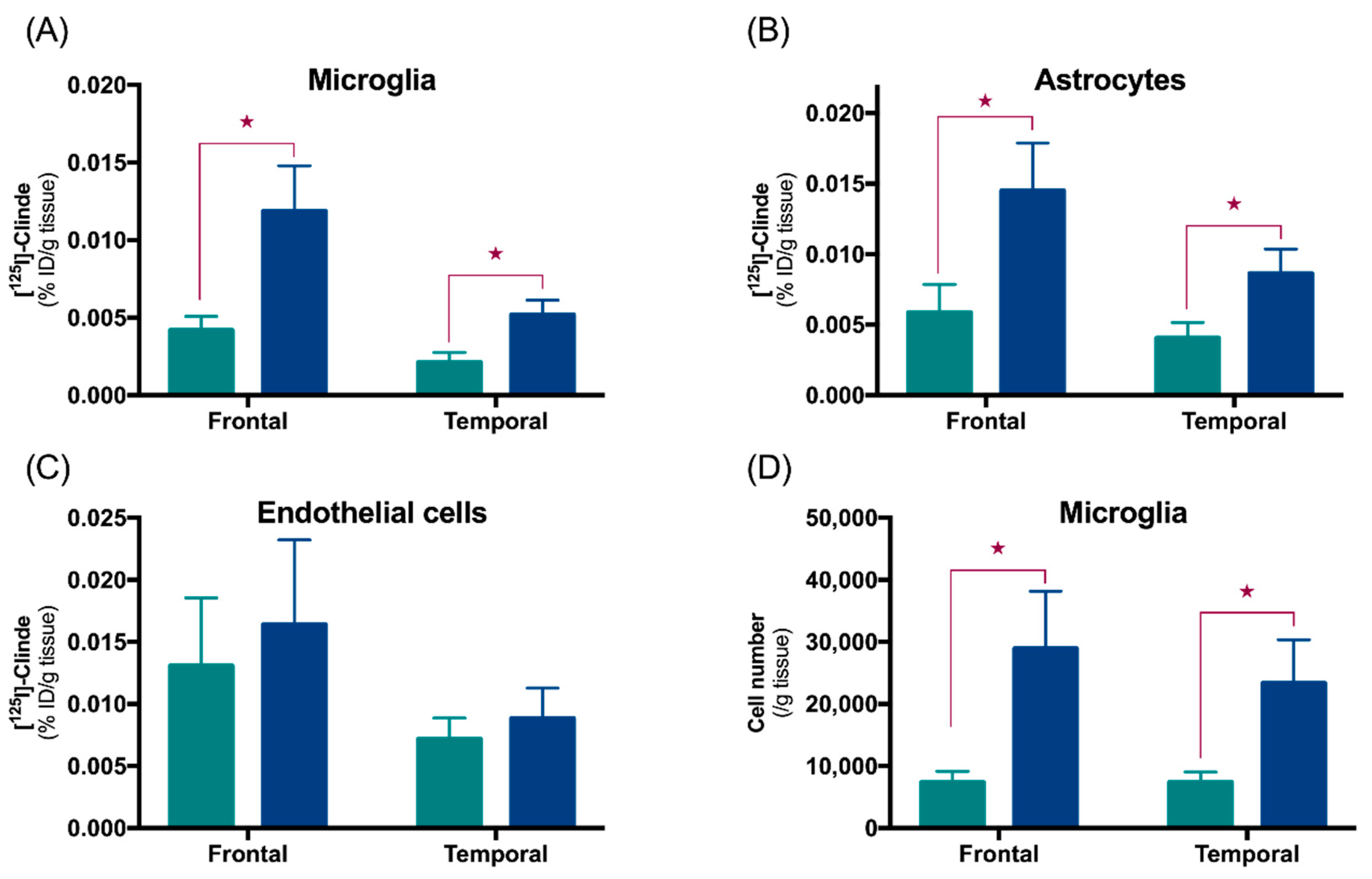
3.1. TSPO in Astrocytes and Microglia
3.2. Other Cells Types
3.3. Cells Expressing TSPO in Other Pathologies
4. Possible Biological Roles of TSPO
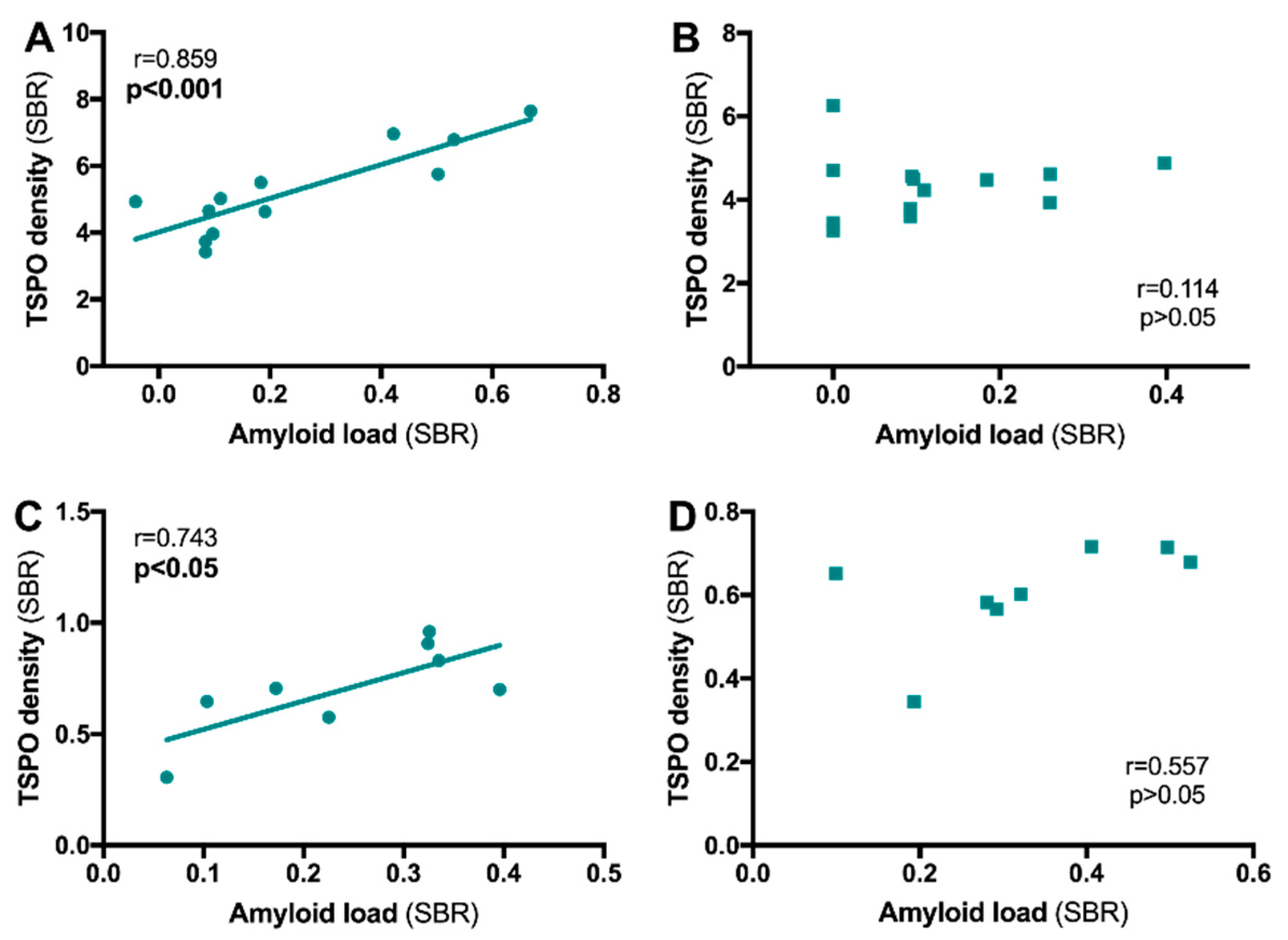
4.1. Relationship between TSPO and Alzheimer’s Disease
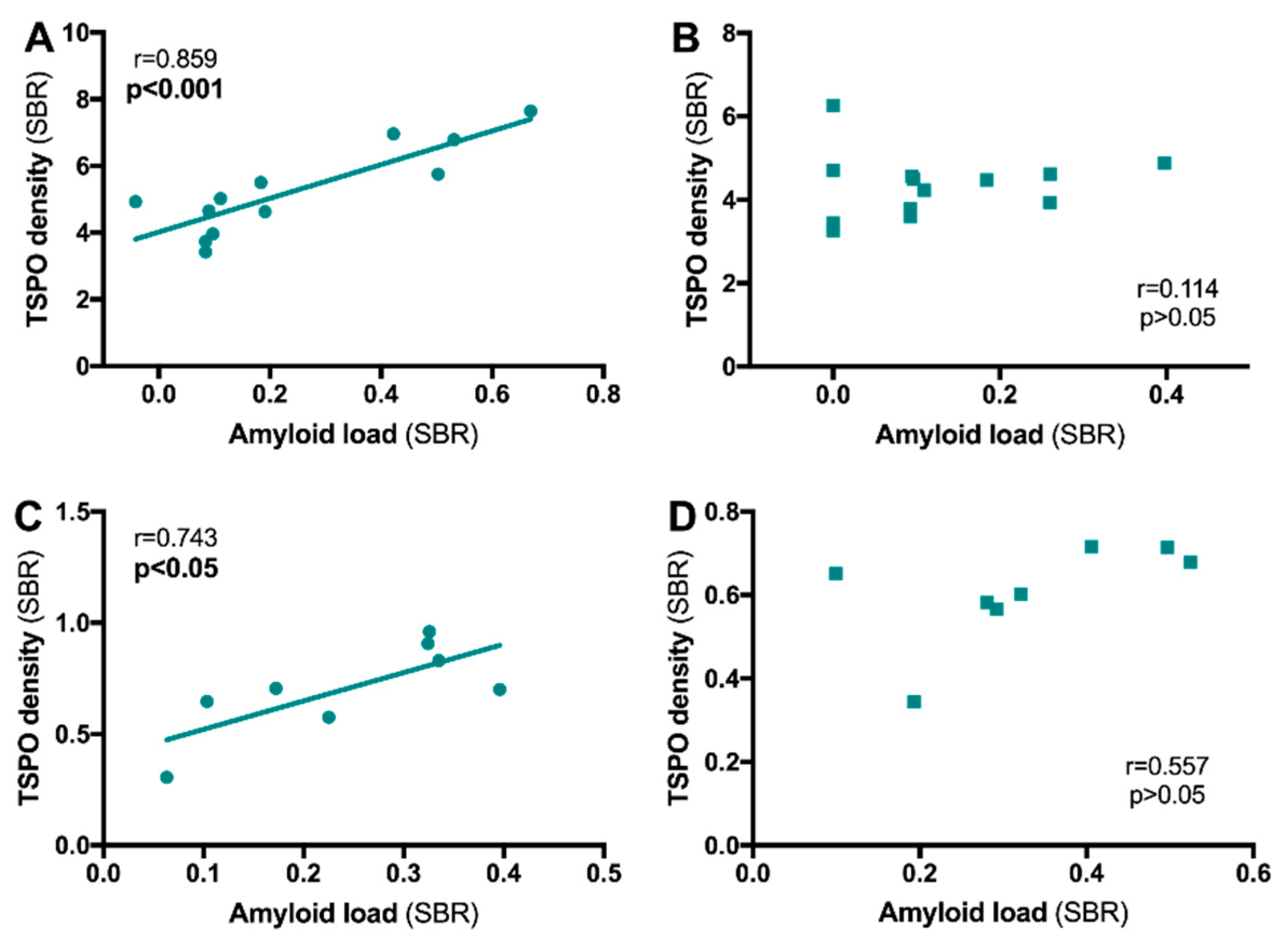
4.1.1. TSPO and Aβ Relationship
4.1.2. TSPO and Tau Relationship
4.1.3. TSPO and Clinical Symptoms of Alzheimer’s Disease
4.1.4. Is TSPO Protector or Aggravator?
4.2. Relationship between TSPO and Schizophrenia
5. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bauer, M.E.; Teixeira, A.L. Inflammation in psychiatric disorders: What comes first? Ann. NY Acad. Sci. 2019, 1437, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Guilarte, T.R. TSPO in diverse CNS pathologies and psychiatric disease: A critical review and a way forward. Pharmacol. Ther. 2018, 194, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Brosseron, F.; Traschutz, A.; Widmann, C.N.; Kummer, M.P.; Tacik, P.; Santarelli, F.; Jessen, F.; Heneka, M.T. Characterization and clinical use of inflammatory cerebrospinal fluid protein markers in Alzheimer’s disease. Alzheimer’s Res. Ther. 2018, 10, 25. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.S.P.; Liu, C.S.; Rau, A.; Lanctot, K.L.; Kohler, C.A.; Pakosh, M.; Carvalho, A.F.; Herrmann, N. Peripheral inflammatory markers in Alzheimer’s disease: A systematic review and meta-analysis of 175 studies. J. Neurol. Neurosurg. Psychiatry 2017, 88, 876–882. [Google Scholar] [CrossRef]
- Misiak, B.; Frydecka, D.; Stanczykiewicz, B.; Samochowiec, J. Editorial: Peripheral markers of immune response in major psychiatric disorders: Where are we now and where do we want to be? Front. Psychiatry 2019, 10, 5. [Google Scholar] [CrossRef]
- Fakhoury, M. Microglia and astrocytes in Alzheimer’s disease: Implications for therapy. Curr. Neuropharmacol. 2018, 16, 508–518. [Google Scholar] [CrossRef]
- Bayer, T.A.; Buslei, R.; Havas, L.; Falkai, P. Evidence for activation of microglia in patients with psychiatric illnesses. Neurosci. Lett. 1999, 271, 126–128. [Google Scholar] [CrossRef]
- Kolomeets, N.S.; Uranova, N. Ultrastructural abnormalities of astrocytes in the hippocampus in schizophrenia and duration of illness: A postortem morphometric study. World J. Biol. Psychiatry 2010, 11, 282–292. [Google Scholar] [CrossRef]
- Trepanier, M.O.; Hopperton, K.E.; Mizrahi, R.; Mechawar, N.; Bazinet, R.P. Postmortem evidence of cerebral inflammation in schizophrenia: A systematic review. Mol. Psychiatry 2016, 21, 1009–1026. [Google Scholar] [CrossRef]
- Tremblay, M.E.; Stevens, B.; Sierra, A.; Wake, H.; Bessis, A.; Nimmerjahn, A. The role of microglia in the healthy brain. J. Neurosci. 2011, 31, 16064–16069. [Google Scholar] [CrossRef]
- Yang, T.T.; Lin, C.; Hsu, C.T.; Wang, T.F.; Ke, F.Y.; Kuo, Y.M. Differential distribution and activation of microglia in the brain of male C57BL/6J mice. Brain Struct. Funct. 2013, 218, 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflammation 2014, 11, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, H.K.; Ji, K.; Min, K.; Joe, E.H. Brain inflammation and microglia: Facts and misconceptions. Exp. Neurobiol. 2013, 22, 59–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Maydell, D.; Jorfi, M. The interplay between microglial states and major risk factors in Alzheimer’s disease through the eyes of single-cell RNA-sequencing: Beyond black and white. J. Neurophysiol. 2019, 122, 1291–1296. [Google Scholar] [CrossRef]
- Werry, E.L.; King, V.A.; Barron, M.L.; Banister, S.D.; Sokias, R.; Kassiou, M. Derivatives of the pyrazolo[1,5-a]pyrimidine acetamide DPA-713 as translocator protein (TSPO) ligands and pro-apoptotic agents in human glioblastoma. Eur. J. Pharm. Sci. 2017, 96, 186–192. [Google Scholar] [CrossRef] [Green Version]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 2017, 169, 1276–1290. [Google Scholar] [CrossRef]
- Galatro, T.F.; Holtman, I.R.; Lerario, A.M.; Vainchtein, I.D.; Brouwer, N.; Sola, P.R.; Veras, M.M.; Pereira, T.F.; Leite, R.E.P.; Moller, T.; et al. Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat. Neurosci. 2017, 20, 1162–1171. [Google Scholar] [CrossRef]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Bottcher, C.; Amann, L.; Sagar; Scheiwe, C.; Nessler, S.; Kunz, P.; van Loo, G.; et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 2019, 566, 388–392. [Google Scholar] [CrossRef]
- Oberheim, N.A.; Goldman, S.A.; Nedergaard, M. Heterogeneity of astrocytic form and function. Methods Mol. Biol. 2012, 814, 23–45. [Google Scholar]
- Bushong, E.A.; Martone, M.E.; Jones, Y.Z.; Ellisman, M.H. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J. Neurosci. 2002, 22, 183–192. [Google Scholar] [CrossRef]
- Fiacco, T.A.; Agulhon, C.; McCarthy, K.D. Sorting out astrocyte physiology from pharmacology. Ann. Rev. Pharmacol. Toxicol. 2009, 49, 151–174. [Google Scholar] [CrossRef] [PubMed]
- Sul, J.Y.; Orosz, G.; Givens, R.S.; Haydon, P.G. Astrocytic connectivity in the hippocampus. Neuron Glia Biol. 2004, 1, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garwood, C.J.; Ratcliffe, L.E.; Simpson, J.E.; Heath, P.R.; Ince, P.G.; Wharton, S.B. Review: Astrocytes in Alzheimer’s disease and other age-associated dementias: A supporting player with a central role. Neuropathol. Appl. Neurobiol. 2017, 43, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.E.; Liddelow, S.A.; Chakraborty, C.; Munch, A.E.; Heiman, M.; Barres, B.A. Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. USA 2018, 115, E1896–E1905. [Google Scholar] [CrossRef] [Green Version]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef]
- Werry, E.L.; Bright, F.M.; Piguet, O.; Ittner, L.M.; Halliday, G.M.; Hodges, J.R.; Kiernan, M.C.; Loy, C.T.; Kril, J.J.; Kassiou, M. Recent developments in TSPO PET imaging as A biomarker of neuroinflammation in neurodegenerative disorders. Int. J. Mol. Sci. 2019, 20, 3161. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.; Torres, C. Astrocyte senescence: Evidence and significance. Aging Cell 2019, 18, e12937. [Google Scholar] [CrossRef] [Green Version]
- Chun, H.; Marriott, I.; Lee, C.J.; Cho, H. Elucidating the interactive roles of glia in Alzheimer’s disease using established and newly developed experimental models. Front. Neurol. 2018, 9, 797. [Google Scholar] [CrossRef] [Green Version]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Von Bernhardi, R.; Ramirez, G. Microglia-astrocyte interaction in Alzheimer’s disease: Friends or foes for the nervous system? Biol. Res. 2001, 34, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Giovannini, M.G.; Scali, C.; Prosperi, C.; Bellucci, A.; Vannucchi, M.G.; Rosi, S.; Pepeu, G.; Casamenti, F. Beta-amyloid-induced inflammation and cholinergic hypofunction in the rat brain in vivo: Involvement of the p38MAPK pathway. Neurobiol. Dis. 2002, 11, 257–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craft, J.M.; Watterson, D.M.; Van Eldik, L.J. Human amyloid beta-induced neuroinflammation is an early event in neurodegeneration. Glia 2006, 53, 484–490. [Google Scholar] [CrossRef]
- Medeiros, R.; LaFerla, F.M. Astrocytes: Conductors of the Alzheimer disease neuroinflammatory symphony. Exp. Neurol. 2013, 239, 133–138. [Google Scholar] [CrossRef]
- Capani, F.; Quarracino, C.; Caccuri, R.; Sica, R.E. Astrocytes as the main players in primary degenerative disorders of the human central nervous system. Front. Aging Neurosci. 2016, 8, 45. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, K.; Hayashi, M.; Nakano, H.; Shimazaki, M.; Sugimori, K.; Koshino, Y. Correlation between astrocyte apoptosis and Alzheimer changes in gray matter lesions in Alzheimer’s disease. J. Alzheimer’s Dis. 2004, 6, 623–632. [Google Scholar] [CrossRef]
- Baik, S.H.; Kang, S.; Son, S.M.; Mook-Jung, I. Microglia contributes to plaque growth by cell death due to uptake of amyloid beta in the brain of Alzheimer’s disease mouse model. Glia 2016, 64, 2274–2290. [Google Scholar] [CrossRef]
- Angelova, P.R.; Abramov, A.Y. Interaction of neurons and astrocytes underlies the mechanism of Abeta-induced neurotoxicity. Biochem. Soc. Trans. 2014, 42, 1286–1290. [Google Scholar] [CrossRef]
- Sollvander, S.; Nikitidou, E.; Brolin, R.; Soderberg, L.; Sehlin, D.; Lannfelt, L.; Erlandsson, A. Accumulation of amyloid-beta by astrocytes result in enlarged endosomes and microvesicle-induced apoptosis of neurons. Mol. Neurodegener. 2016, 11, 38. [Google Scholar] [CrossRef]
- De Strooper, B.; Karran, E. The cellular phase of Alzheimer’s disease. Cell 2016, 164, 603–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Kesteren, C.F.; Gremmels, H.; de Witte, L.D.; Hol, E.M.; Van Gool, A.R.; Falkai, P.G.; Kahn, R.S.; Sommer, I.E. Immune involvement in the pathogenesis of schizophrenia: A meta-analysis on postmortem brain studies. Transl. Psychiatry 2017, 7, e1075. [Google Scholar] [CrossRef] [PubMed]
- Woods, M.J.; Williams, D.C. Multiple forms and locations for the peripheral-type benzodiazepine receptor. Biochem. Pharmacol. 1996, 52, 1805–1814. [Google Scholar] [CrossRef]
- Taniguchi, T.; Wang, J.K.; Spector, S. [3H]Diazepam binding sites on rat heart and kidney. Biochem. Pharmacol. 1982, 31, 589–590. [Google Scholar] [CrossRef]
- Lacapere, J.J.; Delavoie, F.; Li, H.; Peranzi, G.; Maccario, J.; Papadopoulos, V.; Vidic, B. Structural and functional study of reconstituted peripheral benzodiazepine receptor. Biochem. Biophys. Res. Commun. 2001, 284, 536–541. [Google Scholar] [CrossRef]
- Murail, S.; Robert, J.C.; Coic, Y.M.; Neumann, J.M.; Ostuni, M.A.; Yao, Z.X.; Papadopoulos, V.; Jamin, N.; Lacapere, J.J. Secondary and tertiary structures of the transmembrane domains of the translocator protein TSPO determined by NMR. Stabilization of the TSPO tertiary fold upon ligand binding. Biochim. Biophys. Acta 2008, 1778, 1375–1381. [Google Scholar] [CrossRef] [Green Version]
- Delavoie, F.; Li, H.; Hardwick, M.; Robert, J.C.; Giatzakis, C.; Peranzi, G.; Yao, Z.X.; Maccario, J.; Lacapere, J.J.; Papadopoulos, V. In vivo and in vitro peripheral-type benzodiazepine receptor polymerization: Functional significance in drug ligand and cholesterol binding. Biochemistry 2003, 42, 4506–4519. [Google Scholar] [CrossRef]
- Klubo-Gwiezdzinska, J.; Jensen, K.; Bauer, A.; Patel, A.; Costello, J., Jr.; Burman, K.D.; Wartofsky, L.; Hardwick, M.J.; Vasko, V.V. The expression of translocator protein in human thyroid cancer and its role in the response of thyroid cancer cells to oxidative stress. J. Endocrinol. 2012, 214, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Jaipuria, G.; Leonov, A.; Giller, K.; Vasa, S.K.; Jaremko, L.; Jaremko, M.; Linser, R.; Becker, S.; Zweckstetter, M. Cholesterol-mediated allosteric regulation of the mitochondrial translocator protein structure. Nat. Commun. 2017, 8, 14893. [Google Scholar] [CrossRef] [Green Version]
- Selvaraj, V.; Stocco, D.M.; Clark, B.J. Current knowledge on the acute regulation of steroidogenesis. Biol. Reprod. 2018, 99, 13–26. [Google Scholar] [CrossRef] [Green Version]
- Zheng, P. Neuroactive steroid regulation of neurotransmitter release in the CNS: Action, mechanism and possible significance. Prog. Neurobiol. 2009, 89, 134–152. [Google Scholar] [CrossRef] [PubMed]
- Veenman, L.; Gavish, M. The role of 18 kDa mitochondrial translocator protein (TSPO) in programmed cell death, and effects of steroids on TSPO expression. Curr. Mol. Med. 2012, 12, 398–412. [Google Scholar] [PubMed]
- Sileikyte, J.; Petronilli, V.; Zulian, A.; Dabbeni-Sala, F.; Tognon, G.; Nikolov, P.; Bernardi, P.; Ricchelli, F. Regulation of the inner membrane mitochondrial permeability transition by the outer membrane translocator protein (peripheral benzodiazepine receptor). J. Biol. Chem. 2011, 286, 1046–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sileikyte, J.; Blachly-Dyson, E.; Sewell, R.; Carpi, A.; Menabo, R.; Di Lisa, F.; Ricchelli, F.; Bernardi, P.; Forte, M. Regulation of the mitochondrial permeability transition pore by the outer membrane does not involve the peripheral benzodiazepine receptor (Translocator Protein of 18 kDa (TSPO)). J. Biol. Chem. 2014, 289, 13769–13781. [Google Scholar] [CrossRef] [Green Version]
- Chelli, B.; Salvetti, A.; Da Pozzo, E.; Rechichi, M.; Spinetti, F.; Rossi, L.; Costa, B.; Lena, A.; Rainaldi, G.; Scatena, F.; et al. PK 11195 differentially affects cell survival in human wild-type and 18 kDa translocator protein-silenced ADF astrocytoma cells. J. Cell Biochem. 2008, 105, 712–723. [Google Scholar] [CrossRef]
- Lee, D.H.; Kang, S.K.; Lee, R.H.; Ryu, J.M.; Park, H.Y.; Choi, H.S.; Bae, Y.C.; Suh, K.T.; Kim, Y.K.; Jung, J.S. Effects of peripheral benzodiazepine receptor ligands on proliferation and differentiation of human mesenchymal stem cells. J. Cell Physiol. 2004, 198, 91–99. [Google Scholar] [CrossRef]
- Rechichi, M.; Salvetti, A.; Chelli, B.; Costa, B.; Da Pozzo, E.; Spinetti, F.; Lena, A.; Evangelista, M.; Rainaldi, G.; Martini, C.; et al. TSPO over-expression increases motility, transmigration and proliferation properties of C6 rat glioma cells. Biochim. Biophys. Acta 2008, 1782, 118–125. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, T.; Decaudin, D.; Susin, S.A.; Marchetti, P.; Larochette, N.; Resche-Rigon, M.; Kroemer, G. PK11195, a ligand of the mitochondrial benzodiazepine receptor, facilitates the induction of apoptosis and reverses Bcl-2-mediated cytoprotection. Exp. Cell Res. 1998, 241, 426–434. [Google Scholar] [CrossRef]
- Ching, A.S.; Kuhnast, B.; Damont, A.; Roeda, D.; Tavitian, B.; Dolle, F. Current paradigm of the 18-kDa translocator protein (TSPO) as a molecular target for PET imaging in neuroinflammation and neurodegenerative diseases. Insights Imag. 2012, 3, 111–119. [Google Scholar] [CrossRef] [Green Version]
- Owen, D.R.; Gunn, R.N.; Rabiner, E.A.; Bennacef, I.; Fujita, M.; Kreisl, W.C.; Innis, R.B.; Pike, V.W.; Reynolds, R.; Matthews, P.M.; et al. Mixed-affinity binding in humans with 18-kDa translocator protein ligands. J. Nucl. Med. 2011, 52, 24–32. [Google Scholar] [CrossRef] [Green Version]
- Janssen, B.; Vugts, D.J.; Funke, U.; Molenaar, G.T.; Kruijer, P.S.; van Berckel, B.N.; Lammertsma, A.A.; Windhorst, A.D. Imaging of neuroinflammation in Alzheimer’s disease, multiple sclerosis and stroke: Recent developments in positron emission tomography. Biochim. Biophys. Acta 2016, 1862, 425–441. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.A.; Lopresti, B.J.; Venneti, S.; Price, J.; Klunk, W.E.; DeKosky, S.T.; Mathis, C.A. Carbon 11-labeled Pittsburgh Compound B and carbon 11-labeled (R)-PK11195 positron emission tomographic imaging in Alzheimer disease. Arch. Neurol. 2009, 66, 60–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuitemaker, A.; Kropholler, M.A.; Boellaard, R.; van der Flier, W.M.; Kloet, R.W.; van der Doef, T.F.; Knol, D.L.; Windhorst, A.D.; Luurtsema, G.; Barkhof, F.; et al. Microglial activation in Alzheimer’s disease: An (R)-[(1)(1)C]PK11195 positron emission tomography study. Neurobiol. Aging 2013, 34, 128–136. [Google Scholar] [CrossRef] [Green Version]
- Gulyas, B.; Vas, A.; Toth, M.; Takano, A.; Varrone, A.; Cselenyi, Z.; Schain, M.; Mattsson, P.; Halldin, C. Age and disease related changes in the translocator protein (TSPO) system in the human brain: Positron emission tomography measurements with [11C]vinpocetine. Neuroimage 2011, 56, 1111–1121. [Google Scholar] [CrossRef]
- Varrone, A.; Mattsson, P.; Forsberg, A.; Takano, A.; Nag, S.; Gulyas, B.; Borg, J.; Boellaard, R.; Al-Tawil, N.; Eriksdotter, M.; et al. In vivo imaging of the 18-kDa translocator protein (TSPO) with [18F]FEDAA1106 and PET does not show increased binding in Alzheimer’s disease patients. Eur. J. Nucl. Med. Mol. Imag. 2013, 40, 921–931. [Google Scholar] [CrossRef]
- Golla, S.S.; Boellaard, R.; Oikonen, V.; Hoffmann, A.; van Berckel, B.N.; Windhorst, A.D.; Virta, J.; Haaparanta-Solin, M.; Luoto, P.; Savisto, N.; et al. Quantification of [18F]DPA-714 binding in the human brain: Initial studies in healthy controls and Alzheimer’s disease patients. J. Cereb. Blood Flow Metab. 2015, 35, 766–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Versijpt, J.J.; Dumont, F.; Van Laere, K.J.; Decoo, D.; Santens, P.; Audenaert, K.; Achten, E.; Slegers, G.; Dierckx, R.A.; Korf, J. Assessment of neuroinflammation and microglial activation in Alzheimer’s disease with radiolabelled PK11195 and single photon emission computed tomography. A pilot study. Eur. Neurol. 2003, 50, 39–47. [Google Scholar] [CrossRef]
- Cagnin, A.; Brooks, D.J.; Kennedy, A.M.; Gunn, R.N.; Myers, R.; Turkheimer, F.E.; Jones, T.; Banati, R.B. In-vivo measurement of activated microglia in dementia. Lancet 2001, 358, 461–467. [Google Scholar] [CrossRef]
- Edison, P.; Archer, H.A.; Gerhard, A.; Hinz, R.; Pavese, N.; Turkheimer, F.E.; Hammers, A.; Tai, Y.F.; Fox, N.; Kennedy, A.; et al. Microglia, amyloid, and cognition in Alzheimer’s disease: An [11C](R)PK11195-PET and [11C]PIB-PET study. Neurobiol. Dis. 2008, 32, 412–419. [Google Scholar] [CrossRef]
- Fan, Z.; Aman, Y.; Ahmed, I.; Chetelat, G.; Landeau, B.; Ray Chaudhuri, K.; Brooks, D.J.; Edison, P. Influence of microglial activation on neuronal function in Alzheimer’s and Parkinson’s disease dementia. Alzheimer’s Dement. 2015, 11, 608–621. [Google Scholar] [CrossRef]
- Fan, Z.; Okello, A.A.; Brooks, D.J.; Edison, P. Longitudinal influence of microglial activation and amyloid on neuronal function in Alzheimer’s disease. Brain 2015, 138, 3685–3698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parbo, P.; Ismail, R.; Hansen, K.V.; Amidi, A.; Marup, F.H.; Gottrup, H.; Braendgaard, H.; Eriksson, B.O.; Eskildsen, S.F.; Lund, T.E.; et al. Brain inflammation accompanies amyloid in the majority of mild cognitive impairment cases due to Alzheimer’s disease. Brain 2017, 140, 2002–2011. [Google Scholar] [CrossRef] [PubMed]
- Varrone, A.; Oikonen, V.; Forsberg, A.; Joutsa, J.; Takano, A.; Solin, O.; Haaparanta-Solin, M.; Nag, S.; Nakao, R.; Al-Tawil, N.; et al. Positron emission tomography imaging of the 18-kDa translocator protein (TSPO) with [18F]FEMPA in Alzheimer’s disease patients and control subjects. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 438–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreisl, W.C.; Lyoo, C.H.; McGwier, M.; Snow, J.; Jenko, K.J.; Kimura, N.; Corona, W.; Morse, C.L.; Zoghbi, S.S.; Pike, V.W.; et al. In vivo radioligand binding to translocator protein correlates with severity of Alzheimer’s disease. Brain 2013, 136, 2228–2238. [Google Scholar] [CrossRef] [Green Version]
- Lyoo, C.H.; Ikawa, M.; Liow, J.S.; Zoghbi, S.S.; Morse, C.L.; Pike, V.W.; Fujita, M.; Innis, R.B.; Kreisl, W.C. Cerebellum can serve as a pseudo-reference region in Alzheimer disease to detect neuroinflammation measured with PET radioligand binding to translocator protein. J. Nucl. Med. 2015, 56, 701–706. [Google Scholar] [CrossRef] [Green Version]
- Kreisl, W.C.; Lyoo, C.H.; Liow, J.S.; Wei, M.; Snow, J.; Page, E.; Jenko, K.J.; Morse, C.L.; Zoghbi, S.S.; Pike, V.W.; et al. (11)C-PBR28 binding to translocator protein increases with progression of Alzheimer’s disease. Neurobiol. Aging 2016, 44, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Hamelin, L.; Lagarde, J.; Dorothee, G.; Leroy, C.; Labit, M.; Comley, R.A.; de Souza, L.C.; Corne, H.; Dauphinot, L.; Bertoux, M.; et al. Early and protective microglial activation in Alzheimer’s disease: A prospective study using 18F-DPA-714 PET imaging. Brain 2016, 139, 1252–1264. [Google Scholar] [CrossRef] [Green Version]
- Hamelin, L.; Lagarde, J.; Dorothee, G.; Potier, M.C.; Corlier, F.; Kuhnast, B.; Caille, F.; Dubois, B.; Fillon, L.; Chupin, M.; et al. Distinct dynamic profiles of microglial activation are associated with progression of Alzheimer’s disease. Brain 2018, 141, 1855–1870. [Google Scholar] [CrossRef]
- Tsartsalis, S.; Moulin-Sallanon, M.; Dumas, N.; Tournier, B.B.; Ghezzi, C.; Charnay, Y.; Ginovart, N.; Millet, P. Quantification of GABAA receptors in the rat brain with [(123)I]Iomazenil SPECT from factor analysis-denoised images. Nucl. Med. Biol. 2014, 41, 186–195. [Google Scholar] [CrossRef] [Green Version]
- Millet, P.; Graf, C.; Buck, A.; Walder, B.; Ibanez, V. Evaluation of the reference tissue models for PET and SPECT benzodiazepine binding parameters. Neuroimage 2002, 17, 928–942. [Google Scholar] [CrossRef]
- Millet, P.; Moulin-Sallanon, M.; Tournier, B.B.; Dumas, N.; Charnay, Y.; Ibanez, V.; Ginovart, N. Quantification of dopamine D(2/3) receptors in rat brain using factor analysis corrected [18F] Fallypride images. Neuroimage 2012, 62, 1455–1468. [Google Scholar] [CrossRef]
- Shrestha, S.; Hirvonen, J.; Hines, C.S.; Henter, I.D.; Svenningsson, P.; Pike, V.W.; Innis, R.B. Serotonin-1A receptors in major depression quantified using PET: Controversies, confounds, and recommendations. Neuroimage 2012, 59, 3243–3251. [Google Scholar] [CrossRef] [Green Version]
- Rupprecht, R.; Papadopoulos, V.; Rammes, G.; Baghai, T.C.; Fan, J.; Akula, N.; Groyer, G.; Adams, D.; Schumacher, M. Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat. Rev. Drug Discov. 2010, 9, 971–988. [Google Scholar] [CrossRef]
- Rizzo, G.; Veronese, M.; Tonietto, M.; Zanotti-Fregonara, P.; Turkheimer, F.E.; Bertoldo, A. Kinetic modeling without accounting for the vascular component impairs the quantification of [(11)C]PBR28 brain PET data. J. Cereb. Blood Flow Metab. 2014, 34, 1060–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veronese, M.; Reis Marques, T.; Bloomfield, P.S.; Rizzo, G.; Singh, N.; Jones, D.; Agushi, E.; Mosses, D.; Bertoldo, A.; Howes, O.; et al. Kinetic modelling of [(11)C]PBR28 for 18 kDa translocator protein PET data: A validation study of vascular modelling in the brain using XBD173 and tissue analysis. J. Cereb. Blood Flow Metab. 2017, 38, 1227–1242. [Google Scholar] [CrossRef]
- Salinas, C.A.; Searle, G.E.; Gunn, R.N. The simplified reference tissue model: Model assumption violations and their impact on binding potential. J. Cereb. Blood Flow Metab. 2015, 35, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Thie, J.A. Understanding the standardized uptake value, its methods, and implications for usage. J. Nucl. Med. 2004, 45, 1431–1434. [Google Scholar] [PubMed]
- Innis, R.B.; Cunningham, V.J.; Delforge, J.; Fujita, M.; Gjedde, A.; Gunn, R.N.; Holden, J.; Houle, S.; Huang, S.C.; Ichise, M.; et al. Consensus nomenclature for in vivo imaging of reversibly binding radioligands. J. Cereb. Blood Flow Metab. 2007, 27, 1533–1539. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Muzik, O.; Shandal, V.; Chugani, D.; Chakraborty, P.; Chugani, H.T. Evaluation of age-related changes in translocator protein (TSPO) in human brain using (11)C-[R]-PK11195 PET. J. Neuroinflammation 2012, 9, 232. [Google Scholar] [CrossRef] [Green Version]
- Pike, C.J. Sex and the development of Alzheimer’s disease. J. Neurosci. Res. 2017, 95, 671–680. [Google Scholar] [CrossRef] [Green Version]
- Bradburn, S.; Murgatroyd, C.; Ray, N. Neuroinflammation in mild cognitive impairment and Alzheimer’s disease: A meta-analysis. Age. Res. Rev. 2019, 50, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ji, B.; Maeda, J.; Sawada, M.; Ono, M.; Okauchi, T.; Inaji, M.; Zhang, M.R.; Suzuki, K.; Ando, K.; Staufenbiel, M.; et al. Imaging of peripheral benzodiazepine receptor expression as biomarkers of detrimental versus beneficial glial responses in mouse models of Alzheimer’s and other CNS pathologies. J. Neurosci. 2008, 28, 12255–12267. [Google Scholar] [CrossRef] [PubMed]
- Maeda, J.; Zhang, M.R.; Okauchi, T.; Ji, B.; Ono, M.; Hattori, S.; Kumata, K.; Iwata, N.; Saido, T.C.; Trojanowski, J.Q.; et al. In vivo positron emission tomographic imaging of glial responses to amyloid-beta and tau pathologies in mouse models of Alzheimer’s disease and related disorders. J. Neurosci. 2011, 31, 4720–4730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, M.L.; Belichenko, N.P.; Nguyen, T.V.; Andrews, L.E.; Ding, Z.; Liu, H.; Bodapati, D.; Arksey, N.; Shen, B.; Cheng, Z.; et al. PET imaging of translocator protein (18 kDa) in a mouse model of Alzheimer’s disease using N-(2,5-dimethoxybenzyl)-2-18F-fluoro-N-(2-phenoxyphenyl)acetamide. J. Nucl. Med. 2015, 56, 311–316. [Google Scholar] [CrossRef] [Green Version]
- Sacher, C.; Blume, T.; Beyer, L.; Peters, F.; Eckenweber, F.; Sgobio, C.; Deussing, M.; Albert, N.L.; Unterrainer, M.; Lindner, S.; et al. Longitudinal PET monitoring of amyloidosis and microglial activation in a second generation amyloid-beta mouse model. J. Nucl. Med. 2019, 60, 1787–1793. [Google Scholar] [CrossRef]
- Liu, B.; Le, K.X.; Park, M.A.; Wang, S.; Belanger, A.P.; Dubey, S.; Frost, J.L.; Holton, P.; Reiser, V.; Jones, P.A.; et al. In vivo detection of age- and disease-related increases in neuroinflammation by 18F-GE180 TSPO microPET imaging in wild-type and Alzheimer’s transgenic mice. J. Neurosci. 2015, 35, 15716–15730. [Google Scholar] [CrossRef] [Green Version]
- Mirzaei, N.; Tang, S.P.; Ashworth, S.; Coello, C.; Plisson, C.; Passchier, J.; Selvaraj, V.; Tyacke, R.J.; Nutt, D.J.; Sastre, M. In vivo imaging of microglial activation by positron emission tomography with [(11)C]PBR28 in the 5XFAD model of Alzheimer’s disease. Glia 2016, 64, 993–1006. [Google Scholar]
- Rapic, S.; Backes, H.; Viel, T.; Kummer, M.P.; Monfared, P.; Neumaier, B.; Vollmar, S.; Hoehn, M.; Van der Linden, A.; Heneka, M.T.; et al. Imaging microglial activation and glucose consumption in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2013, 34, 351–354. [Google Scholar] [CrossRef] [Green Version]
- Serriere, S.; Tauber, C.; Vercouillie, J.; Mothes, C.; Pruckner, C.; Guilloteau, D.; Kassiou, M.; Domene, A.; Garreau, L.; Page, G.; et al. Amyloid load and translocator protein 18 kDa in APPswePS1-dE9 mice: A longitudinal study. Neurobiol. Aging 2015, 36, 1639–1652. [Google Scholar] [CrossRef] [Green Version]
- Venneti, S.; Lopresti, B.J.; Wang, G.; Hamilton, R.L.; Mathis, C.A.; Klunk, W.E.; Apte, U.M.; Wiley, C.A. PK11195 labels activated microglia in Alzheimer’s disease and in vivo in a mouse model using PET. Neurobiol. Aging 2009, 30, 1217–1226. [Google Scholar] [CrossRef] [Green Version]
- Tournier, B.B.; Tsartsalis, S.; Rigaud, D.; Fossey, C.; Cailly, T.; Fabis, F.; Pham, T.; Gregoire, M.C.; Kovari, E.; Moulin-Sallanon, M.; et al. TSPO and amyloid deposits in sub-regions of the hippocampus in the 3xTgAD mouse model of Alzheimer’s disease. Neurobiol. Dis. 2019, 121, 95–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betlazar, C.; Harrison-Brown, M.; Middleton, R.J.; Banati, R.; Liu, G.J. Cellular sources and regional variations in the expression of the neuroinflammatory marker translocator protein (TSPO) in the normal brain. Int. J. Mol. Sci. 2018, 19, 2707. [Google Scholar] [CrossRef] [Green Version]
- Chaney, A.; Bauer, M.; Bochicchio, D.; Smigova, A.; Kassiou, M.; Davies, K.E.; Williams, S.R.; Boutin, H. Longitudinal investigation of neuroinflammation and metabolite profiles in the APPswe xPS1Deltae9 transgenic mouse model of Alzheimer’s disease. J. Neurochem. 2018, 144, 318–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setiawan, E.; Wilson, A.A.; Mizrahi, R.; Rusjan, P.M.; Miler, L.; Rajkowska, G.; Suridjan, I.; Kennedy, J.L.; Rekkas, P.V.; Houle, S.; et al. Role of translocator protein density, a marker of neuroinflammation, in the brain during major depressive episodes. JAMA Psychiatry 2015, 72, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Bloomfield, P.S.; Selvaraj, S.; Veronese, M.; Rizzo, G.; Bertoldo, A.; Owen, D.R.; Bloomfield, M.A.; Bonoldi, I.; Kalk, N.; Turkheimer, F.; et al. Microglial activity in people at ultra high risk of psychosis and in schizophrenia: An [(11)C]PBR28 PET brain imaging study. Am. J. Psychiatry 2016, 173, 44–52. [Google Scholar] [CrossRef] [Green Version]
- Collste, K.; Plaven-Sigray, P.; Fatouros-Bergman, H.; Victorsson, P.; Schain, M.; Forsberg, A.; Amini, N.; Aeinehband, S.; Karolinska Schizophrenia Project (KaSP) consortium; Erhardt, S.; et al. Lower levels of the glial cell marker TSPO in drug-naive first-episode psychosis patients as measured using PET and [11C]PBR28. Mol. Psychiatry 2017, 22, 850–856. [Google Scholar] [CrossRef] [Green Version]
- Coughlin, J.M.; Wang, Y.; Ambinder, E.B.; Ward, R.E.; Minn, I.; Vranesic, M.; Kim, P.K.; Ford, C.N.; Higgs, C.; Hayes, L.N.; et al. In vivo markers of inflammatory response in recent-onset schizophrenia: A combined study using [(11)C]DPA-713 PET and analysis of CSF and plasma. Transl. Psychiatry 2016, 6, e777. [Google Scholar] [CrossRef]
- Hafizi, S.; Tseng, H.H.; Rao, N.; Selvanathan, T.; Kenk, M.; Bazinet, R.P.; Suridjan, I.; Wilson, A.A.; Meyer, J.H.; Remington, G.; et al. Imaging microglial activation in untreated first-episode psychosis: A PET study with [18F]FEPPA. Am. J. Psychiatry 2017, 174, 118–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, S.E.; Hinz, R.; Drake, R.J.; Gregory, C.J.; Conen, S.; Matthews, J.C.; Anton-Rodriguez, J.M.; Gerhard, A.; Talbot, P.S. In vivo imaging of brain microglial activity in antipsychotic-free and medicated schizophrenia: A [11C](R)-PK11195 positron emission tomography study. Mol. Psychiatry 2016, 21, 1672–1679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenk, M.; Selvanathan, T.; Rao, N.; Suridjan, I.; Rusjan, P.; Remington, G.; Meyer, J.H.; Wilson, A.A.; Houle, S.; Mizrahi, R. Imaging neuroinflammation in gray and white matter in schizophrenia: An in-vivo PET study with [18F]-FEPPA. Schizophr. Bull. 2015, 41, 85–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Notter, T.; Coughlin, J.M.; Gschwind, T.; Weber-Stadlbauer, U.; Wang, Y.; Kassiou, M.; Vernon, A.C.; Benke, D.; Pomper, M.G.; Sawa, A.; et al. Translational evaluation of translocator protein as a marker of neuroinflammation in schizophrenia. Mol. Psychiatry 2017, 23, 323–334. [Google Scholar] [CrossRef]
- Selvaraj, S.; Bloomfield, P.S.; Cao, B.; Veronese, M.; Turkheimer, F.; Howes, O.D. Brain TSPO imaging and gray matter volume in schizophrenia patients and in people at ultra high risk of psychosis: An [11C]PBR28 study. Schizophr. Res. 2017, 195, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Van der Doef, T.F.; de Witte, L.D.; Sutterland, A.L.; Jobse, E.; Yaqub, M.; Boellaard, R.; de Haan, L.; Eriksson, J.; Lammertsma, A.A.; Kahn, R.S.; et al. In vivo (R)-[(11)C]PK11195 PET imaging of 18kDa translocator protein in recent onset psychosis. NPJ Schizophr. 2016, 2, 16031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Doef, T.F.; Doorduin, J.; van Berckel, B.N.M.; Cervenka, S. Assessing brain immune activation in psychiatric disorders: Clinical and preclinical PET imaging studies of the 18-kDa translocator protein. Clin. Transl. Imaging 2015, 3, 449–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Picker, L.J.; Morrens, M.; Chance, S.A.; Boche, D. Microglia and brain plasticity in acute psychosis and schizophrenia illness course: A meta-review. Front. Psychiatry 2017, 8, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plaven-Sigray, P.; Matheson, G.J.; Collste, K.; Ashok, A.H.; Coughlin, J.M.; Howes, O.D.; Mizrahi, R.; Pomper, M.G.; Rusjan, P.; Veronese, M.; et al. Positron emission tomography studies of the glial cell marker translocator protein in patients with psychosis: A meta-analysis using individual participant data. Biol. Psychiatry 2018, 84, 433–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verkhratsky, A.; Zorec, R.; Parpura, V. Stratification of astrocytes in healthy and diseased brain. Brain Pathol. 2017, 27, 629–644. [Google Scholar] [CrossRef] [PubMed]
- Haarman, B.C.; Riemersma-Van der Lek, R.F.; de Groot, J.C.; Ruhe, H.G.; Klein, H.C.; Zandstra, T.E.; Burger, H.; Schoevers, R.A.; de Vries, E.F.; Drexhage, H.A.; et al. Neuroinflammation in bipolar disorder—A [(11)C]-(R)-PK11195 positron emission tomography study. Brain Behav. Immun. 2014, 40, 219–225. [Google Scholar] [CrossRef]
- Cosenza-Nashat, M.; Zhao, M.L.; Suh, H.S.; Morgan, J.; Natividad, R.; Morgello, S.; Lee, S.C. Expression of the translocator protein of 18 kDa by microglia, macrophages and astrocytes based on immunohistochemical localization in abnormal human brain. Neuropathol. Appl. Neurobiol. 2009, 35, 306–328. [Google Scholar] [CrossRef] [Green Version]
- Gui, Y.; Marks, J.D.; Das, S.; Hyman, B.T.; Serrano-Pozo, A. Characterization of the 18 kDa translocator protein (TSPO) expression in post-mortem normal and Alzheimer’s disease brains. Brain Pathol. 2019, 30, 151–164. [Google Scholar] [CrossRef] [Green Version]
- Venneti, S.; Wang, G.; Nguyen, J.; Wiley, C.A. The positron emission tomography ligand DAA1106 binds with high affinity to activated microglia in human neurological disorders. J. Neuropathol. Exp. Neurol. 2008, 67, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Tournier, B.B.; Tsartsalis, S.; Ceyzeriat, K.; Fraser, B.H.; Gregoire, M.C.; Kovari, E.; Millet, P. Astrocytic TSPO upregulation appears before microglial TSPO in Alzheimer’s disease. J. Alzheimer’s Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.R.; Narayan, N.; Wells, L.; Healy, L.; Smyth, E.; Rabiner, E.A.; Galloway, D.; Williams, J.B.; Lehr, J.; Mandhair, H.; et al. Pro-inflammatory activation of primary microglia and macrophages increases 18 kDa translocator protein expression in rodents but not humans. J. Cereb. Blood Flow Metab. 2017, 37, 2679–2690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckers, L.; Ory, D.; Geric, I.; Declercq, L.; Koole, M.; Kassiou, M.; Bormans, G.; Baes, M. Increased expression of translocator protein (TSPO) marks pro-inflammatory microglia but does not predict neurodegeneration. Mol. Imag. Biol. 2018, 20, 94–102. [Google Scholar] [CrossRef] [Green Version]
- Narayan, N.; Mandhair, H.; Smyth, E.; Dakin, S.G.; Kiriakidis, S.; Wells, L.; Owen, D.; Sabokbar, A.; Taylor, P. The macrophage marker translocator protein (TSPO) is down-regulated on pro-inflammatory ‘M1’ human macrophages. PLoS ONE 2017, 12, e0185767. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Gordon, M.N.; Holcomb, L.A.; Jantzen, P.T.; DiCarlo, G.; Wilcock, D.; Boyett, K.W.; Connor, K.; Melachrino, J.; O’Callaghan, J.P.; Morgan, D. Time course of the development of Alzheimer-like pathology in the doubly transgenic PS1+APP mouse. Exp. Neurol. 2002, 173, 183–195. [Google Scholar] [CrossRef] [Green Version]
- Carter, S.F.; Scholl, M.; Almkvist, O.; Wall, A.; Engler, H.; Langstrom, B.; Nordberg, A. Evidence for astrocytosis in prodromal Alzheimer disease provided by 11C-deuterium-L-deprenyl: A multitracer PET paradigm combining 11C-Pittsburgh compound B and 18F-FDG. J. Nucl. Med. 2012, 53, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Tournier, B.B.; Tsartsalis, S.; Ceyzeriat, K.; Medina, Z.; Fraser, B.H.; Gregoire, M.C.; Kovari, E.; Millet, P. Fluorescence-activated cell sorting to reveal the cell origin of radioligand binding. J. Cereb. Blood Flow Metab. 2020, 40, 1242–1255. [Google Scholar] [CrossRef]
- Lopez-Picon, F.R.; Snellman, A.; Eskola, O.; Helin, S.; Solin, O.; Haaparanta-Solin, M.; Rinne, J.O. Neuroinflammation appears early and then plateaus in a mouse model of Alzheimer’s disease shown by PET imaging. J. Nucl. Med. 2017, 59, 509–515. [Google Scholar] [CrossRef] [Green Version]
- Thal, D.R.; Rub, U.; Schultz, C.; Sassin, I.; Ghebremedhin, E.; Del Tredici, K.; Braak, E.; Braak, H. Sequence of Abeta-protein deposition in the human medial temporal lobe. J. Neuropathol. Exp. Neurol. 2000, 59, 733–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Del Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, S.; Wu, M.D.; Shaftel, S.S.; Kyrkanides, S.; LaFerla, F.M.; Olschowka, J.A.; O’Banion, M.K. Sustained interleukin-1beta overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer’s mouse model. J. Neurosci. 2013, 33, 5053–5064. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.T.; Gao, J.; Cao, S.; Sandhu, N.; Cui, J.Z.; Chou, C.L.; Fang, E.; Matsubara, J.A. Inflammatory mediators induced by amyloid-beta in the retina and RPE in vivo: Implications for inflammasome activation in age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 2013, 54, 2225–2237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di, J.; Cohen, L.S.; Corbo, C.P.; Phillips, G.R.; El Idrissi, A.; Alonso, A.D. Abnormal tau induces cognitive impairment through two different mechanisms: Synaptic dysfunction and neuronal loss. Sci. Rep. 2016, 6, 20833. [Google Scholar] [CrossRef] [PubMed]
- Maphis, N.; Xu, G.; Kokiko-Cochran, O.N.; Cardona, A.E.; Ransohoff, R.M.; Lamb, B.T.; Bhaskar, K. Loss of tau rescues inflammation-mediated neurodegeneration. Front. Neurosci. 2015, 9, 196. [Google Scholar] [CrossRef] [PubMed]
- Currais, A.; Quehenberger, O.; Armando, M.A.; Daugherty, D.; Maher, P.; Schubert, D. Amyloid proteotoxicity initiates an inflammatory response blocked by cannabinoids. NPJ Aging Mech. Dis. 2016, 2, 16012. [Google Scholar] [CrossRef]
- Parbo, P.; Ismail, R.; Sommerauer, M.; Stokholm, M.G.; Hansen, A.K.; Hansen, K.V.; Amidi, A.; Schaldemose, J.L.; Gottrup, H.; Braendgaard, H.; et al. Does inflammation precede tau aggregation in early Alzheimer’s disease? A PET study. Neurobiol. Dis. 2018, 117, 211–216. [Google Scholar] [CrossRef] [Green Version]
- Focke, C.; Blume, T.; Zott, B.; Yuan Shi, Y.; Deussing, M.; Peters, F.; Schmidt, C.; Kleinberger, G.; Lindner, S.; Gildehaus, F.J.; et al. Early and longitudinal microglial activation but not amyloid accumulation predict cognitive outcome in PS2APP mice. J. Nucl. Med. 2018, 60, 548–554. [Google Scholar] [CrossRef]
- Ma, L.; Zhang, H.; Liu, N.; Wang, P.Q.; Guo, W.Z.; Fu, Q.; Jiao, L.B.; Ma, Y.Q.; Mi, W.D. TSPO ligand PK11195 alleviates neuroinflammation and beta-amyloid generation induced by systemic LPS administration. Brain Res. Bull. 2016, 121, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Veiga, S.; Carrero, P.; Pernia, O.; Azcoitia, I.; Garcia-Segura, L.M. Translocator protein 18 kDa is involved in the regulation of reactive gliosis. Glia 2007, 55, 1426–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.; Ifuku, M.; Noda, M.; Guilarte, T.R. Translocator protein (18 kDa)/peripheral benzodiazepine receptor specific ligands induce microglia functions consistent with an activated state. Glia 2011, 59, 219–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlstetter, M.; Nothdurfter, C.; Aslanidis, A.; Moeller, K.; Horn, F.; Scholz, R.; Neumann, H.; Weber, B.H.; Rupprecht, R.; Langmann, T. Translocator protein (18 kDa) (TSPO) is expressed in reactive retinal microglia and modulates microglial inflammation and phagocytosis. J. Neuroinflammation 2014, 11, 3. [Google Scholar] [CrossRef] [Green Version]
- Mills, C.; Makwana, M.; Wallace, A.; Benn, S.; Schmidt, H.; Tegeder, I.; Costigan, M.; Brown, R.H., Jr.; Raivich, G.; Woolf, C.J. Ro5-4864 promotes neonatal motor neuron survival and nerve regeneration in adult rats. Eur. J. Neurosci. 2008, 27, 937–946. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, L.; Zhang, X.; Xue, R.; Li, L.; Zhao, W.; Fu, Q.; Mi, W.; Li, Y. Lentiviral-mediated overexpression of the 18 kDa translocator protein (TSPO) in the hippocampal dentate gyrus ameliorates LPS-induced cognitive impairment in mice. Front. Pharmacol. 2016, 7, 384. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Ma, L.; Yin, Y.L.; Dong, L.Q.; Cheng, G.G.; Ma, Y.Q.; Li, Y.F.; Xu, B.N. Over-expression of TSPO in the hippocampal CA1 area alleviates cognitive dysfunction caused by lipopolysaccharide in mice. Brain Res. 2016, 1646, 402–409. [Google Scholar] [CrossRef]
- Kohler, O.; Benros, M.E.; Nordentoft, M.; Farkouh, M.E.; Iyengar, R.L.; Mors, O.; Krogh, J. Effect of anti-inflammatory treatment on depression, depressive symptoms, and adverse effects: A systematic review and meta-analysis of randomized clinical trials. JAMA Psychiatry 2014, 71, 1381–1391. [Google Scholar] [CrossRef]
- Oya, K.; Kishi, T.; Iwata, N. Efficacy and tolerability of minocycline augmentation therapy in schizophrenia: A systematic review and meta-analysis of randomized controlled trials. Hum. Psychopharmacol. 2014, 29, 483–491. [Google Scholar] [CrossRef]
- Sommer, I.E.; de Witte, L.; Begemann, M.; Kahn, R.S. Nonsteroidal anti-inflammatory drugs in schizophrenia: Ready for practice or a good start? A meta-analysis. J. Clin. Psychiatry 2012, 73, 414–419. [Google Scholar] [CrossRef]
- Capuron, L.; Gumnick, J.F.; Musselman, D.L.; Lawson, D.H.; Reemsnyder, A.; Nemeroff, C.B.; Miller, A.H. Neurobehavioral effects of interferon-alpha in cancer patients: Phenomenology and paroxetine responsiveness of symptom dimensions. Neuropsychopharmacology 2002, 26, 643–652. [Google Scholar] [CrossRef]
- Capuron, L.; Pagnoni, G.; Drake, D.F.; Woolwine, B.J.; Spivey, J.R.; Crowe, R.J.; Votaw, J.R.; Goodman, M.M.; Miller, A.H. Dopaminergic mechanisms of reduced basal ganglia responses to hedonic reward during interferon alfa administration. Arch. Gen. Psychiatry 2012, 69, 1044–1053. [Google Scholar] [CrossRef]
- Majer, M.; Welberg, L.A.; Capuron, L.; Pagnoni, G.; Raison, C.L.; Miller, A.H. IFN-alpha-induced motor slowing is associated with increased depression and fatigue in patients with chronic hepatitis C. Brain Behav. Immun. 2008, 22, 870–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenberger, N.I.; Berkman, E.T.; Inagaki, T.K.; Rameson, L.T.; Mashal, N.M.; Irwin, M.R. Inflammation-induced anhedonia: Endotoxin reduces ventral striatum responses to reward. Biol. Psychiatry 2010, 68, 748–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisht, K.; Sharma, K.P.; Lecours, C.; Sanchez, M.G.; El Hajj, H.; Milior, G.; Olmos-Alonso, A.; Gomez-Nicola, D.; Luheshi, G.; Vallieres, L.; et al. Dark microglia: A new phenotype predominantly associated with pathological states. Glia 2016, 64, 826–839. [Google Scholar] [CrossRef] [Green Version]
- Friedman, B.A.; Srinivasan, K.; Ayalon, G.; Meilandt, W.J.; Lin, H.; Huntley, M.A.; Cao, Y.; Lee, S.H.; Haddick, P.C.G.; Ngu, H.; et al. Diverse brain myeloid expression profiles reveal distinct microglial activation states and aspects of Alzheimer’s disease not evident in mouse models. Cell Rep. 2018, 22, 832–847. [Google Scholar] [CrossRef] [Green Version]
- Caballero, B.; Veenman, L.; Bode, J.; Leschiner, S.; Gavish, M. Concentration-dependent bimodal effect of specific 18 kDa translocator protein (TSPO) ligands on cell death processes induced by ammonium chloride: Potential implications for neuropathological effects due to hyperammonemia. CNS Neurol. Disord. Drug Targets 2014, 13, 574–592. [Google Scholar] [CrossRef]
- Barron, A.M.; Garcia-Segura, L.M.; Caruso, D.; Jayaraman, A.; Lee, J.W.; Melcangi, R.C.; Pike, C.J. Ligand for translocator protein reverses pathology in a mouse model of Alzheimer’s disease. J. Neurosci. 2013, 33, 8891–8897. [Google Scholar] [CrossRef] [Green Version]
- Christensen, A.; Pike, C.J. TSPO ligand PK11195 improves Alzheimer-related outcomes in aged female 3xTg-AD mice. Neurosci. Lett. 2018, 683, 7–12. [Google Scholar] [CrossRef]
- Ceyzériat, K.; Zilli, T.; Millet, P.; Frisoni, G.B.; Garibotto, V.; Tournier, B.B. Learning from the past: A review of clinical trials targeting amyloid, tau and neuroinflammation in Alzheimer’s disease. Curr. Alzheimer Res. 2020, 17, 112–125. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Radiotracer | Population | m/f (%males) | Age Range (mean) | MMSE Score Range (mean) | TSPO Methodology: Scan Acquisition and Analysis | Main Results | Reference |
|---|---|---|---|---|---|---|---|
| Studies showing no main effect of AD on TSPO | |||||||
| 11C-PK11195 * 11C-PiB | 6 modAD-A+ 6 MCI (4 AD-A+) 5 HC (2 HC-A+) | 4/2 (67) 4/2 (67) 3/2 (60) | 65–94 (76) 61–81 (72) 65–79 (72) | 13–28 (19.3) 27–30 (28.7) 28–30 (29.4) | 0–90min BPSRTM with RefCE 10–60min images SUR with Refsubcortical white matter | BPSRTM and SUR: AD = MCI = HC A+ = A− | [62] |
| 11C-PK11195 * No amyloid detection | 20 AD 13 proAD 21 HC | 11/8 (58) 7/3 (70) 13/8 (62) | (69) (72) (68) | (23) (26) (29) | 0–60.5 min. BPSRTM with RefCluster Analysis VWA-SPM | BPSRTM: AD = prodromal = HC VWA-SPM: AD > HC:Occ No correlations between PK11195 and neuropsychological tests. No difference between prodromal AD patients who remained clinically stable and those who progressed clinically to dementia. | [63] |
| 11C-Vinpocetine No amyloid detection | 6 AD 6 young HC 6 old HC | 3/3 (50) 6/0 (100) 6/0 (100) | 67–82 (73) 54–78 (67) 25–44 (35) | (dnf) | 0–66 min SUVR with RefCE BPLogan with RefCE | SUVR Old HC > Young HC: pF, medT, latT, Occ, Th, St AD = Old HC BPLogan AD = HC: Th AD = Old HC > Young HC: whole-brain, grey matter | [64] |
| 18F-FEDAA1106 No amyloid detection | 9 AD 7 HC | 6/3 (67) 5/2 (71) | 64–76 (69) 63–73 (68) | 21–30 (25) 28–30 (29) | 0–60 min and 80–140 min. VT BPND VT Logan | AD: 8 AChE inhibitor + 1 AchE inhibitor- VT, BPND, VT Logan AD = HC | [65] |
| 18F-DAP-714 No amyloid detection | 9 AD 6 HC | 4/4 (50) 1/5 (17) | 60–80 (73.7) 60–73 (64.5) | 20–29 (24.5) 28–30 (28.8) | 0–90 min and 120–150 min VT BPSRTM with RefCE | Age effect: AD > HC VT, BPSRTM AD = HC | [66] |
| Studies showing TSPO overexpression in AD | |||||||
| 123I-PK11195 * No amyloid detection | 10 AD 9 HC | 4/6 (40) 6/3 (67) | 55–87 (77) 53–76 (67) | 9–25 (19) | 60–80 min SUR with RefCE | HC: no age effect. AD > HC: loF, pF, latF, rMT, bG Diverse inverse correlations between regional PK1195 and neuropsychological tests. | [67] |
| 11C-PK11195 * No amyloid detection | 8 AD 15 HC | 4/4 (50) 7/8 (47) | 58–68 (65) 32–80 (75) | 6–24 (17.25) | 0–60 min. BPSRTM with RefCluster Analysis | HC: age effect in Th AD > HC: iTG, mTG, fG, lPG, lAm, lpCin, iPG, Pu, rPall AD trend to > HC: rCE (p=0.06) | [68] |
| 11C-PK11195 * No amyloid detection | 13 AD 10 HC | 8/5 (62) 6/4 (60) | 54–73 (65) 54–71 (64) | 15–26 (21) (30) | 0–60 min. BPSRTM with RefCluster Analysis VWA-SPM | BPSRTM AD = HC: Th, Hipp AD > HC: aCin, pCin, St, F, T, P, Occ, WC, Am, PG, mTG, CE (+22%) VWA-SPM: AD > HC: iTG, mTG, mFG, rF, latOcc, loF, lpostCG, rpostT, infF, roF, preCG, supFG. Negative correlations between regional PK11195 and MMSE scores | [69] |
| 11C-PK11195 * 11C-PiB | 10 AD-A+ 10 MCI (5 MCI-A+) 8 HC | 4/6 (40) 5/5 (50) 4/4 (50) | 51–74 (66) 55–77 (67) 58–71 (65) | (20.5) (28.2) (30) | (dnf) min BPSRTM with RefCluster Analysis VWA-SPM | BPSRTM AD > HC: aCin, F, T, P, Occ, mTG, Am, Hipp MCI > HC: aCin, F, T, P, Occ, mTG, Am, Hipp, pCin VWA-SPM AD > HC: lHipp, rPG, lpostT, lpreCG, lpostCG, lsupTG, liTG, lmTG, latOcc. MCI > HC: liTG, lmTG, rsupFG, rsupPG, lIns, lPu, rmedOG, lmFG, raOG, rStG, rlatOcc In different regions (AD only): Positive correlations between PK1195 and MMSE scores; Positive correlations between PK1195 and PiB | [70] |
| 11C-PK11195 * 11C-PiB | 8 AD (7 AD-A+) 8 HC out of 14 | 3/5 (37) 7/9 (44) ** | 51–74 (66) 54–75 (65) ** | (21) | 0–60 min BPSRTM with RefCluster Analysis VWA-SPM | BPSRTM and VWA-SPM AD > HC: F, T, P, Occ, Hipp, St Positive correlations between PK1195 and PiB | [71] |
| 11C-PK11195 * 11C-PiB | 26 MCI-A+ 16 MCI-A− 10 HC out of 15 | 17/9 (65) 7/9 (44) 6/) (40) ** | 62–83 (73) 50–79 (66) 58–80 (68) ** | 23–30 (27) 23–30 (28) 25–30 (29) ** | 0–60min BPSRTM with RefCluster Analysis | MCI-A+ > HC: F, latT, P MCI-A+ > MCI-A−: Hipp MCI-A− = HC In different regions: positive correlations PK1195 and PiB | [72] |
| 18F-FEMPA No amyloid detection | 5 HAB-AD 3 MAB-AD 2 ?-AD 4 HAB-HC 3 MAB-HC | 2/3 (40) 2/1 (67) 1/1 (50) 1/3 (25) 2/1 (67) | 67–73 (71.2) 55–67 (61) 56–74 (65) 66–71 (69) 55–58 (56) | 23–28 (25.6) 22–28 (25) 23–29 (26) 28–30 (29) 29–30 (29.7) | 0–90 min and 120–150 min. VT VT Logan | VT Logan HAB/MAB-AD > HAB/MAB-HC: medT HAB-AD > HAB-HC: medT, latT, pCin, Cau, Pu, Th, CE (+19%) No correlations between FEMPA and MMSE scores | [73] |
| 11C-PBR28 No amyloid detection | 9 HAB-AD 10 MAB-AD 4 HAB-MCI 6 MAB-MCI 5 HAB-HC 8 MAB-HC | 11/8 6/4 9/4 | (63.1) (72.6) (62.9) | (20.3) (27.5) (29.8) | 0–90 min VT | Age effect: MCI > AD = HC AD: 15 AchE inhibitor +, 4 AchE inhibitor- MCI: 4 AchE inhibitor +, 6 AchE inhibitor- HAB > MAB (controls and patients combined) in whole brain. AD > HC: pF, iP, supT, iTG, mTG, Prec, pCin, Occ, Hipp, Ent AD > MCI: pF, iP, supT, iTG, mTG, Occ HAB-AD > HAB-HC: iP HAB-AD > HAB-MCI: iP MAB-AD > MAB-HC: iP Correlation between regional PBR28 (AD and MCI) and neuropsychological tests, grey matter volume and age of symptom onset. | [74] |
| 11C-PBR28 11C-PiB | 11 HAB-AD-A+ 14 MAB-AD-A+ 5 HAB-MCI-A+ 6 MAB-MCI-A+ 7 HAB-HC 14 MAB-HC | AD: 11/14 MCI: 7/4 HC: 15/6 | AD: (63) MCI: (72) HC: (55) | 0–90 min. VT SUVR with RefCE (60–90 min) DVR (VT target/VT CE) | Some subjects were previously included in Reference [74]. VT uncorrected for plasma free fraction of radioligand AD=MCI=HC VT corrected for plasma free fraction of radioligand AD > HC: iP, mTG, iTG, Ent AD > MCI: mTG, iTG, Ent SUVR with genotype correction: AD > HC: mTG, iTG, iP, Ent, PG MAB > HAB: in all diagnostic groups. DVR AD > HC: mTG, iTG, iP, Prec, Hipp, Ent, PG AD > MCI: mTG, iTG, iP, Ent, PG, Occ Positive correlations between PBR28 (combined mTG-iTG) and CDR | [75] | |
| 11C-PBR28 No amyloid detection | 5 HAB-MCI/AD 9 MAB-MCI/AD 3 HAB-HC 5 MAB-HC | MCI/AD: (65) HC: (62) | MCI/AD: 14–30 (22) HC: >29 (30) | 0–90 min VT (n = 17). DVR (n = 17) SUVR with RefCE (60–90 min) 2nd PBR28 scan sessions: 1.2–5.7 years after. | AD: 10 AchE inhibitor +, 4 AchE inhibitor- at baseline, and 8 AchE inhibitor +, 6 AchE inhibitor- at the 2nd examination. Time to the 2nd PBR28 imaging: AD (2.5 years) < HC (4 years). Follow-up CDR: 5 AD stable and 9 with increased CDR. Magnitude of SUVR increase AD > HC: iTG, mTG, iP, Prec, Occ, Hipp, Ent. Magnitude of DVR increase AD > HC: iTG, mTG, iP, Prec, Occ, Ent. Annual increased in PBR binding: AD with increased CDR > stable AD Correlation between increase in PBR28 (pF, Prec, supP, iP) and increase in CDR score. | [76] | |
| 18F-DPA-714 11C-PiB | 12 HAB-AD 12 MAB-AD 2 LAB-AD 17 HAB-proAD 17 MAB-proAD 4 LAB-proAD 11 HAB-HC-A− 9 MAB-HC-A− 4 LAB-HC- A− 2 HAB-HC-A+ 4 MAB-HC-A+ 2 LAB-HC-A+ | AD: 8/22 (27) proAD: 16/22 (42) HC-A−: 6/18 (25) HC-A+: 4/4 (50) | AD: (68.3) proAD: (67.8) HC-A−: (68.2) HC- A+: (74.3) | AD: (15.8) proAD: (24) HC-A−: (29.5) HC-A+: (29.1) | 0–90 min. SUVR with RefCE (60–90 min) VWA-SPM Clinical follow up: 2 years | SUVR LAB-AD=LAB-proAD=LAB-HC HAB-AD > HAB-HC: pCin, T, Prec HAB-proAD > HAB-HC: GCI, medCin, pCin, P, T, Prec MAB-AD > MAB-HC: P, T, Prec MAB-proAD > MAB-HC: GCI, P, T, Prec VWA-SPM HAB/MAD AD > HC: P, T HAB/MAD proAD > HC: F, P, T At baseline: Slow decliners (stable CDR) > Fast decliners (≥0.5 CDR) No correlation between age and DPA in the whole population. Positive correlations between DPA (global cortical binding) and MMSE scores, grey matter volume. In different regions: positive correlations between DPA and PiB | [77] |
| 18F-DPA-714 No amyloid detetion | 33proAD-19AD: 22 HAB-AD 30 MAB-AD 9 HAB-HC 8 MAB-HC | (dnf) | proAD/AD: (67) HC: (69) | proAD/AD: (21) HC: (29) | 0–90 min. SUVR with RefCE (60–90 min) Clinical follow up: 2 years defining slow and fast CDR and MMSE decliners. Slow: stable CDR or ΔMMSE = −1; Fast: increase in CDR or ΔMMSE = −8 | SUVR proAD = AD HAB/MAD AD > HC: GCI, medCin, pCin, F, P, T, Prec, Occ At baseline: proAD slow decliners (CDR, n = 11) > Fast decliners (n = 22) proAD slow decliners (ΔMMSE, n = 15) > Fast decliners (n = 18) AD slow decliners (CDR, n = 6) > Fast decliners (n = 13) AD slow decliners (ΔMMSE, n = 8) > Fast decliners (n = 8) Positive correlations between DPA (global cortical binding) and MMSE scores | [78] |
| Radiotracer | Population | m/f (%males) | Age Range (mean) | Main Information | TSPO Methodology: Scan Acquisition and Analysis | Main Results | Reference |
|---|---|---|---|---|---|---|---|
| 11C-PBR28 | 8 HAB-FEP 8 MAB-FEP 9 HAB-HC 7 MAB-HC | FEP: 11/5 HC: 7/9 | FEP: (28) HC: (26) | Nicotine 2 in FEP 0 in HC FEP: 5 with benzodiazepine treatment | 0–91 min VT | FEP < HC: F, T, Hipp | [106] |
| 11C-DPA-713 | 8 HAB-SCZ 4 MAB-SCZ 2 LAB-SCZ 9 HAB-HC 5 MAB-HC 2 LAB-HC | SCZ: 11/3 (79) HC: 9/7 (56) | SCZ: (24) HC: (25) | Nicotine: 2 in HC 3 in SCZ SCZ: 2 unmedicated (last month) 1 with two antipsychotic 11 with one antipsyxchotic [range: 0–1119 chlorpromazine equivalent] | 0–90 min VT Logan | LAB were excluded. HAB-SCZ = HAB-HC MAB-SCZ = MAB-HC | [107] |
| 18F-FEPPA | 14 HAB-FEP 5 MAB-FEP 14 HAB-HC 6 MAB-HC | FEP: 12/19 (63) HC: 9/11 (45) | FEP: (28) HC: (28) | FEP: 14 antipsychotic naïve, 5 unmedicated (last 4 weeks) | 0–125 min VT | HAB-FEP = HAB-HC MAB-FEP = MAB-HC | [108] |
| 18F-FEPPA | 10 HAB-SCZ 6 MAB-SCZ 1 LAB-SCZ 19 HAB-HC 8 MAB-HC 0 LAB-HC | SCZ. 10/6 HC: 10/17 | SCZ: (43) HC: (44) | SCZ: 16 with antipsychotics and others (antidepressants…) | 0–125 min VT | HAB-SCZ = HAB-HC MAB-SCZ = MAB-HC | [110] |
| 11C-PK11195 | 16 SCZ 16 HC | 11/5 11/5 | (33) (33) | Nicotine 11 in SCZ 0 in HC SCZ: 8 antipsychotic-free | 0–60 min BPSRTM with RefCE | Nicotine status SCZ > HC BPSRTM SCZ antipsychotic-free = HC SCZ > SCZ antipsychotic-free (83% of increase but p = 0.097) | [109] |
| 11C-PK11195 | 19 SCZ 17 HC | 16/3 14/3 | (24) (47) | Nicotine 13 in SCZ 5 in HC SCZ: 4 antipsychotic-free, 16 with antipsychotic | 60 min BPSRTM with RefCluster Analysis | SCZ = HC | [113] |
| 11C-PBR28 | 7 HAB-UHR 7 MAB-UHR 10 HAB-HC 4 MAB-HC 13 HAB-SCZ 1 MAB-SCZ 14 HAB-HC | UHR: 7/7 (50) HC: 4/10 SCZ: 3/12 3/12 | UHR: (24) HC: (28) SCZ: (47) (46) | 2 UHR with citalopram by the past | 0–90 min. DVR (VT target/VT whole brain) | UHR > HC: Gm, F, T UHR excluding 2 subjects with citalopram > HC: F HAB-UHR > HAB-HC: Gm MAB-UHR > MAB-HC: Gm SCZ > HC: F, T UHR: Positive correlations between the mean PBR28 DVR and CAARMS | [105] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tournier, B.B.; Tsartsalis, S.; Ceyzériat, K.; Garibotto, V.; Millet, P. In Vivo TSPO Signal and Neuroinflammation in Alzheimer’s Disease. Cells 2020, 9, 1941. https://doi.org/10.3390/cells9091941
Tournier BB, Tsartsalis S, Ceyzériat K, Garibotto V, Millet P. In Vivo TSPO Signal and Neuroinflammation in Alzheimer’s Disease. Cells. 2020; 9(9):1941. https://doi.org/10.3390/cells9091941
Chicago/Turabian StyleTournier, Benjamin B., Stergios Tsartsalis, Kelly Ceyzériat, Valentina Garibotto, and Philippe Millet. 2020. "In Vivo TSPO Signal and Neuroinflammation in Alzheimer’s Disease" Cells 9, no. 9: 1941. https://doi.org/10.3390/cells9091941
APA StyleTournier, B. B., Tsartsalis, S., Ceyzériat, K., Garibotto, V., & Millet, P. (2020). In Vivo TSPO Signal and Neuroinflammation in Alzheimer’s Disease. Cells, 9(9), 1941. https://doi.org/10.3390/cells9091941