Gene Expression Comparison between Sézary Syndrome and Lymphocytic-Variant Hypereosinophilic Syndrome Refines Biomarkers for Sézary Syndrome


Abstract
:1. Introduction
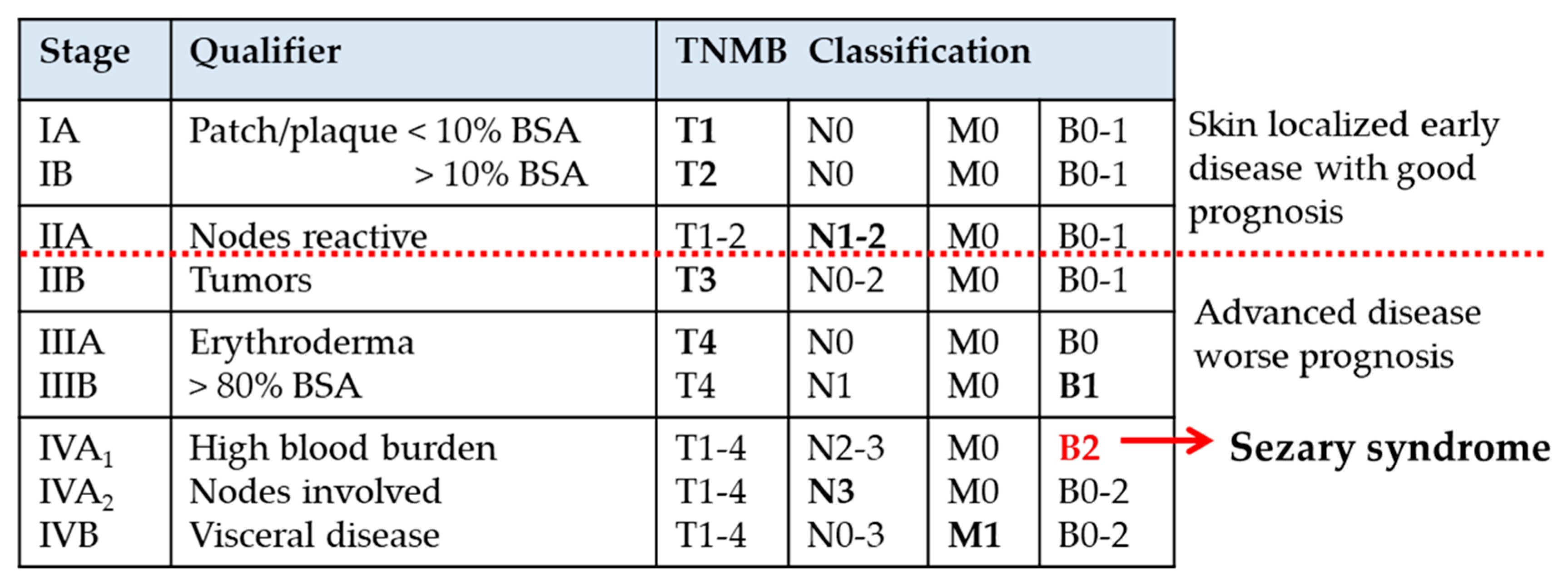
2. Clinical Features of SS and L-HES
3. Gene Expression in SS and L-HES
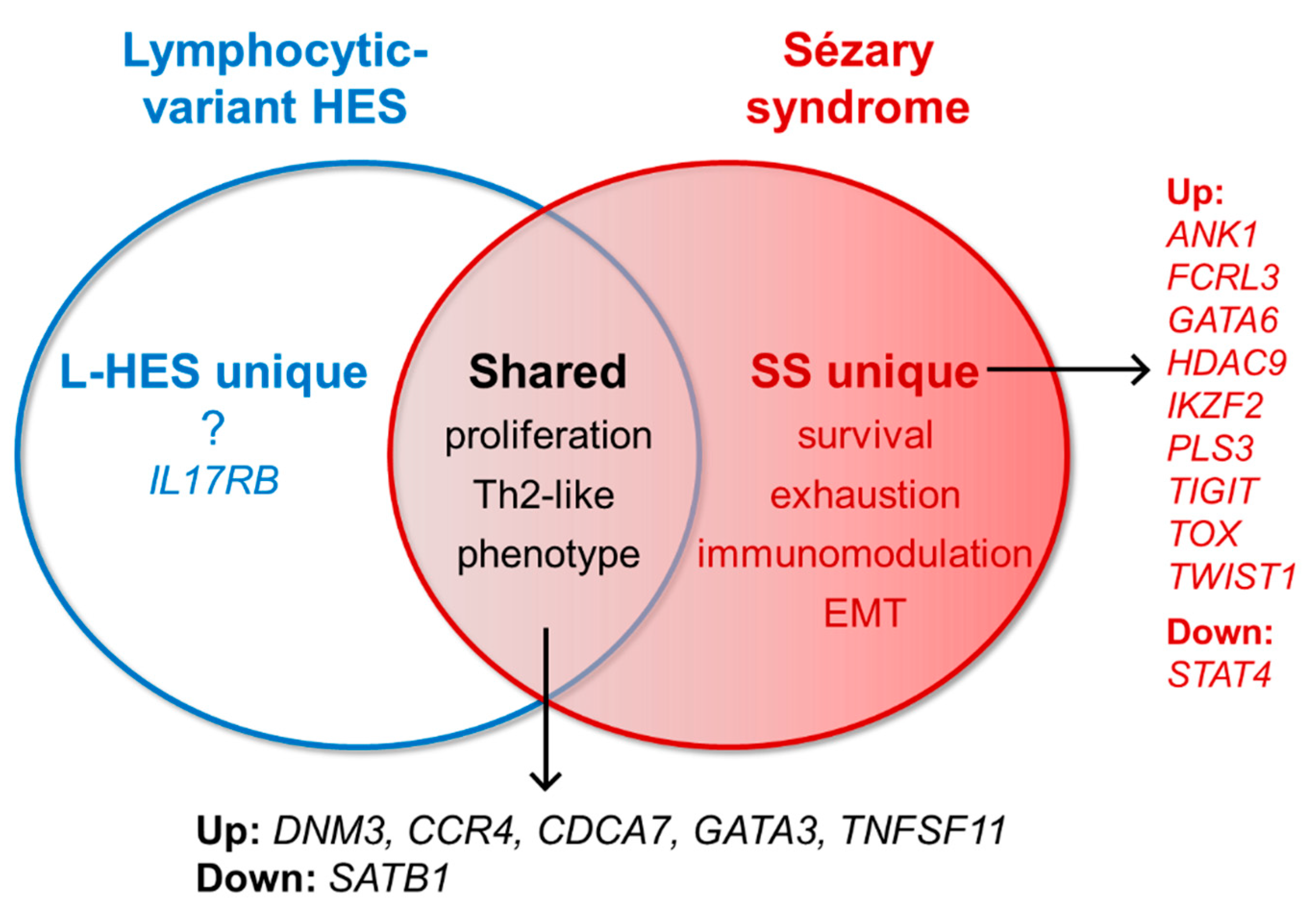
3.1. Gene Expression Shared by SS and L-HES
3.2. Gene Expression Unique to SS
3.2.1. Well-Established SS-Unique Biomarker Genes
3.2.2. SS-Unique Genes Associated with Regulatory and Exhaustion Phenotypes
3.2.3. New and Promising SS-Unique Biomarker Genes
4. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Hodak, E.; Amitay-Laish, I. Mycosis fungoides: A great imitator. Clin. Dermatol. 2019, 37, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Klemke, C.D.; Booken, N.; Weiss, C.; Nicolay, J.P.; Goerdt, S.; Felcht, M.; Geraud, C.; Kempf, W.; Assaf, C.; Ortonne, N.; et al. Histopathological and immunophenotypical criteria for the diagnosis of Sezary syndrome in differentiation from other erythrodermic skin diseases: A European Organisation for Research and Treatment of Cancer (EORTC) Cutaneous Lymphoma Task Force Study of 97 cases. Br. J. Dermatol. 2015, 173, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Mangold, A.R.; Thompson, A.K.; Davis, M.D.; Saulite, I.; Cozzio, A.; Guenova, E.; Hodak, E.; Amitay-Laish, I.; Pujol, R.M.; Pittelkow, M.R.; et al. Early clinical manifestations of Sezary syndrome: A multicenter retrospective cohort study. J. Am. Acad. Dermatol. 2017, 77, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Foo, S.H.; Shah, F.; Chaganti, S.; Stevens, A.; Scarisbrick, J.J. Unmasking mycosis fungoides/Sezary syndrome from preceding or co-existing benign inflammatory dermatoses requiring systemic therapies: Patients frequently present with advanced disease and have an aggressive clinical course. Br. J. Dermatol. 2016, 174, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Scarisbrick, J.J.; Quaglino, P.; Prince, H.M.; Papadavid, E.; Hodak, E.; Bagot, M.; Servitje, O.; Berti, E.; Ortiz-Romero, P.; Stadler, R.; et al. The PROCLIPI international registry of early-stage mycosis fungoides identifies substantial diagnostic delay in most patients. Br. J. Dermatol. 2019, 181, 350–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarisbrick, J.J.; Hodak, E.; Bagot, M.; Stranzenbach, R.; Stadler, R.; Ortiz-Romero, P.L.; Papadavid, E.; Evison, F.; Knobler, R.; Quaglino, P.; et al. Blood classification and blood response criteria in mycosis fungoides and Sezary syndrome using flow cytometry: Recommendations from the EORTC cutaneous lymphoma task force. Eur. J. Cancer 2018, 93, 47–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willemze, R.; Cerroni, L.; Kempf, W.; Berti, E.; Facchetti, F.; Swerdlow, S.H.; Jaffe, E.S. The 2018 update of the WHO-EORTC classification for primary cutaneous lymphomas. Blood 2019, 133, 1703–1714. [Google Scholar] [CrossRef]
- Boonk, S.E.; Zoutman, W.H.; Marie-Cardine, A.; van der Fits, L.; Out-Luiting, J.J.; Mitchell, T.J.; Tosi, I.; Morris, S.L.; Moriarty, B.; Booken, N.; et al. Evaluation of Immunophenotypic and Molecular Biomarkers for Sezary Syndrome Using Standard Operating Procedures: A Multicenter Study of 59 Patients. J. Investig. Dermatol. 2016, 136, 1364–1372. [Google Scholar] [CrossRef] [Green Version]
- Dulmage, B.; Geskin, L.; Guitart, J.; Akilov, O.E. The biomarker landscape in mycosis fungoides and Sezary syndrome. Exp. Dermatol. 2017, 26, 668–676. [Google Scholar] [CrossRef] [Green Version]
- Tang, N.; Gibson, H.; Germeroth, T.; Porcu, P.; Lim, H.W.; Wong, H.K. T-plastin (PLS3) gene expression differentiates Sezary syndrome from mycosis fungoides and inflammatory skin diseases and can serve as a biomarker to monitor disease progression. Br. J. Dermatol. 2010, 162, 463–466. [Google Scholar] [CrossRef]
- Litvinov, I.V.; Netchiporouk, E.; Cordeiro, B.; Dore, M.A.; Moreau, L.; Pehr, K.; Gilbert, M.; Zhou, Y.; Sasseville, D.; Kupper, T.S. The Use of Transcriptional Profiling to Improve Personalized Diagnosis and Management of Cutaneous T-cell Lymphoma (CTCL). Clin. Cancer Res. 2015, 21, 2820–2829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walia, R.; Yeung, C.C.S. An Update on Molecular Biology of Cutaneous T Cell Lymphoma. Front. Oncol. 2019, 9, 1558. [Google Scholar] [CrossRef] [PubMed]
- Van Kester, M.S.; Borg, M.K.; Zoutman, W.H.; Out-Luiting, J.J.; Jansen, P.M.; Dreef, E.J.; Vermeer, M.H.; van Doorn, R.; Willemze, R.; Tensen, C.P. A meta-analysis of gene expression data identifies a molecular signature characteristic for tumor-stage mycosis fungoides. J. Investig. Dermatol. 2012, 132, 2050–2059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Doorn, R.; Dijkman, R.; Vermeer, M.H.; Out-Luiting, J.J.; van der Raaij-Helmer, E.M.; Willemze, R.; Tensen, C.P. Aberrant expression of the tyrosine kinase receptor EphA4 and the transcription factor twist in Sezary syndrome identified by gene expression analysis. Cancer Res. 2004, 64, 5578–5586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nebozhyn, M.; Loboda, A.; Kari, L.; Rook, A.H.; Vonderheid, E.C.; Lessin, S.; Berger, C.; Edelson, R.; Nichols, C.; Yousef, M.; et al. Quantitative PCR on 5 genes reliably identifies CTCL patients with 5% to 99% circulating tumor cells with 90% accuracy. Blood 2006, 107, 3189–3196. [Google Scholar] [CrossRef] [Green Version]
- Booken, N.; Gratchev, A.; Utikal, J.; Weiss, C.; Yu, X.; Qadoumi, M.; Schmuth, M.; Sepp, N.; Nashan, D.; Rass, K.; et al. Sezary syndrome is a unique cutaneous T-cell lymphoma as identified by an expanded gene signature including diagnostic marker molecules CDO1 and DNM3. Leukemia 2008, 22, 393–399. [Google Scholar] [CrossRef] [Green Version]
- Hahtola, S.; Tuomela, S.; Elo, L.; Hakkinen, T.; Karenko, L.; Nedoszytko, B.; Heikkila, H.; Saarialho-Kere, U.; Roszkiewicz, J.; Aittokallio, T.; et al. Th1 response and cytotoxicity genes are down-regulated in cutaneous T-cell lymphoma. Clin. Cancer Res. 2006, 12, 4812–4821. [Google Scholar] [CrossRef] [Green Version]
- Saulite, I.; Hoetzenecker, W.; Weidinger, S.; Cozzio, A.; Guenova, E.; Wehkamp, U. Sezary Syndrome and Atopic Dermatitis: Comparison of Immunological Aspects and Targets. BioMed Res. Int. 2016, 2016, 9717530. [Google Scholar] [CrossRef] [Green Version]
- Vieyra-Garcia, P.; Crouch, J.D.; O’Malley, J.T.; Seger, E.W.; Yang, C.H.; Teague, J.E.; Vromans, A.M.; Gehad, A.; Win, T.S.; Yu, Z.; et al. Benign T cells drive clinical skin inflammation in cutaneous T cell lymphoma. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Krejsgaard, T.; Lindahl, L.M.; Mongan, N.P.; Wasik, M.A.; Litvinov, I.V.; Iversen, L.; Langhoff, E.; Woetmann, A.; Odum, N. Malignant inflammation in cutaneous T-cell lymphoma-a hostile takeover. Semin. Immunopathol. 2017, 39, 269–282. [Google Scholar] [CrossRef]
- Dhingra, N.; Shemer, A.; da Rosa, J.C.; Rozenblit, M.; Fuentes-Duculan, J.; Gittler, J.K.; Finney, R.; Czarnowicki, T.; Zheng, X.; Xu, H.; et al. Molecular profiling of contact dermatitis skin identifies allergen-dependent differences in immune response. J. Allergy Clin. Immunol. 2014, 134, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Moerman-Herzog, A.M.; Acheampong, D.A.; Brooks, A.G.; Blair, S.M.; Hsu, P.C.; Wong, H.K. Transcriptome analysis of Sezary syndrome and lymphocytic-variant hypereosinophilic syndrome T cells reveals common and divergent genes. Oncotarget 2019, 10, 5052–5069. [Google Scholar] [CrossRef] [PubMed]
- Roufosse, F.; Cogan, E.; Goldman, M. Lymphocytic Variant Hypereosinophilic Syndromes. Immunol. Allergy Clin. N. Am. 2007, 27, 389–413. [Google Scholar] [CrossRef]
- Roufosse, F.; Schandene, L.; Sibille, C.; Willard-Gallo, K.; Kennes, B.; Efira, A.; Goldman, M.; Cogan, E. Clonal Th2 lymphocytes in patients with the idiopathic hypereosinophilic syndrome. Br. J. Haematol. 2000, 109, 540–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefevre, G.; Copin, M.C.; Roumier, C.; Aubert, H.; Avenel-Audran, M.; Grardel, N.; Poulain, S.; Staumont-Salle, D.; Seneschal, J.; Salles, G.; et al. CD3-CD4+ lymphoid variant of hypereosinophilic syndrome: Nodal and extranodal histopathological and immunophenotypic features of a peripheral indolent clonal T-cell lymphoproliferative disorder. Haematologica 2015, 100, 1086–1095. [Google Scholar] [CrossRef]
- Ogbogu, P.U.; Bochner, B.S.; Butterfield, J.H.; Gleich, G.J.; Huss-Marp, J.; Kahn, J.E.; Leiferman, K.M.; Nutman, T.B.; Pfab, F.; Ring, J.; et al. Hypereosinophilic syndrome: A multicenter, retrospective analysis of clinical characteristics and response to therapy. J. Allergy Clin. Immunol. 2009, 124, 1319–1325. [Google Scholar] [CrossRef] [Green Version]
- Leiferman, K.M.; Gleich, G.J.; Peters, M.S. Dermatologic manifestations of the hypereosinophilic syndromes. Immunol. Allergy Clin. N. Am. 2007, 27, 415–441. [Google Scholar] [CrossRef]
- Simon, H.U.; Plotz, S.G.; Dummer, R.; Blaser, K. Abnormal clones of T cells producing interleukin-5 in idiopathic eosinophilia. N. Engl. J. Med. 1999, 341, 1112–1120. [Google Scholar] [CrossRef]
- Roufosse, F.; Garaud, S.; de Leval, L. Lymphoproliferative disorders associated with hypereosinophilia. Semin. Hematol. 2012, 49, 138–148. [Google Scholar] [CrossRef]
- Ravoet, M.; Sibille, C.; Gu, C.; Libin, M.; Haibe-Kains, B.; Sotiriou, C.; Goldman, M.; Roufosse, F.; Willard-Gallo, K. Molecular profiling of CD3-CD4+ T cells from patients with the lymphocytic variant of hypereosinophilic syndrome reveals targeting of growth control pathways. Blood 2009, 114, 2969–2983. [Google Scholar] [CrossRef] [Green Version]
- Piccolo, V.; Russo, T.; Agozzino, M.; Vitiello, P.; Caccavale, S.; Alfano, R.; Argenziano, G. Dermoscopy of Cutaneous Lymphoproliferative Disorders: Where Are We Now? Dermatology 2018, 234, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Cerroni, L. Lymphoproliferative lesions of the skin. J. Clin. Pathol. 2006, 59, 813–826. [Google Scholar] [CrossRef] [PubMed]
- Olsen, E.A.; Whittaker, S.; Kim, Y.H.; Duvic, M.; Prince, H.M.; Lessin, S.R.; Wood, G.S.; Willemze, R.; Demierre, M.F.; Pimpinelli, N.; et al. Clinical end points and response criteria in mycosis fungoides and Sezary syndrome: A consensus statement of the International Society for Cutaneous Lymphomas, the United States Cutaneous Lymphoma Consortium, and the Cutaneous Lymphoma Task Force of the European Organisation for Research and Treatment of Cancer. J. Clin. Oncol. 2011, 29, 2598–2607. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.J.; Clark, R.A.; Watanabe, R.; Kupper, T.S. Sezary syndrome and mycosis fungoides arise from distinct T-cell subsets: A biologic rationale for their distinct clinical behaviors. Blood 2010, 116, 767–771. [Google Scholar] [CrossRef] [PubMed]
- Gotlib, J. World Health Organization-defined eosinophilic disorders: 2015 update on diagnosis, risk stratification, and management. Am. J. Hematol. 2015, 90, 1077–1089. [Google Scholar] [CrossRef]
- Izykowska, K.; Przybylski, G.K.; Gand, C.; Braun, F.C.; Grabarczyk, P.; Kuss, A.W.; Olek-Hrab, K.; Bastidas Torres, A.N.; Vermeer, M.H.; Zoutman, W.H.; et al. Genetic rearrangements result in altered gene expression and novel fusion transcripts in Sezary syndrome. Oncotarget 2017, 8, 39627–39639. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Goh, G.; Walradt, T.; Hong, B.S.; Bunick, C.G.; Chen, K.; Bjornson, R.D.; Maman, Y.; Wang, T.; Tordoff, J.; et al. Genomic landscape of cutaneous T cell lymphoma. Nat. Genet. 2015, 47, 1011–1019. [Google Scholar] [CrossRef]
- da Silva Almeida, A.C.; Abate, F.; Khiabanian, H.; Martinez-Escala, E.; Guitart, J.; Tensen, C.P.; Vermeer, M.H.; Rabadan, R.; Ferrando, A.; Palomero, T. The mutational landscape of cutaneous T cell lymphoma and Sezary syndrome. Nat. Genet. 2015, 47, 1465–1470. [Google Scholar] [CrossRef] [Green Version]
- Van Doorn, R.; Slieker, R.C.; Boonk, S.E.; Zoutman, W.H.; Goeman, J.J.; Bagot, M.; Michel, L.; Tensen, C.P.; Willemze, R.; Heijmans, B.T.; et al. Epigenomic Analysis of Sezary Syndrome Defines Patterns of Aberrant DNA Methylation and Identifies Diagnostic Markers. J. Investig. Dermatol. 2016, 136, 1876–1884. [Google Scholar] [CrossRef] [Green Version]
- Qu, K.; Zaba, L.C.; Satpathy, A.T.; Giresi, P.G.; Li, R.; Jin, Y.; Armstrong, R.; Jin, C.; Schmitt, N.; Rahbar, Z.; et al. Chromatin Accessibility Landscape of Cutaneous T Cell Lymphoma and Dynamic Response to HDAC Inhibitors. Cancer Cell 2017, 32, 27–41. [Google Scholar] [CrossRef] [Green Version]
- Caprini, E.; Cristofoletti, C.; Arcelli, D.; Fadda, P.; Citterich, M.H.; Sampogna, F.; Magrelli, A.; Censi, F.; Torreri, P.; Frontani, M.; et al. Identification of key regions and genes important in the pathogenesis of sezary syndrome by combining genomic and expression microarrays. Cancer Res. 2009, 69, 8438–8446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roufosse, F.; Schandene, L.; Sibille, C.; Kennes, B.; Efira, A.; Cogan, E.; Goldman, M. T-cell receptor-independent activation of clonal Th2 cells associated with chronic hypereosinophilia. Blood 1999, 94, 994–1002. [Google Scholar] [CrossRef] [PubMed]
- Roelens, M.; Delord, M.; Ram-Wolff, C.; Marie-Cardine, A.; Alberdi, A.; Maki, G.; Homyrda, L.; Bensussan, A.; Bagot, M.; Toubert, A.; et al. Circulating and skin-derived Sezary cells: Clonal but with phenotypic plasticity. Blood 2017, 130, 1468–1471. [Google Scholar] [CrossRef] [PubMed]
- Kahn, J.E.; Groh, M.; Lefevre, G. (A Critical Appraisal of) Classification of Hypereosinophilic Disorders. Front. Med. 2017, 4, 216. [Google Scholar] [CrossRef] [Green Version]
- Pardanani, A.; Brockman, S.R.; Paternoster, S.F.; Flynn, H.C.; Ketterling, R.P.; Lasho, T.L.; Ho, C.L.; Li, C.Y.; Dewald, G.W.; Tefferi, A. FIP1L1-PDGFRA fusion: Prevalence and clinicopathologic correlates in 89 consecutive patients with moderate to severe eosinophilia. Blood 2004, 104, 3038–3045. [Google Scholar] [CrossRef]
- Tancrede-Bohin, E.; Ionescu, M.A.; de La Salmoniere, P.; Dupuy, A.; Rivet, J.; Rybojad, M.; Dubertret, L.; Bachelez, H.; Lebbe, C.; Morel, P. Prognostic value of blood eosinophilia in primary cutaneous T-cell lymphomas. Arch. Dermatol. 2004, 140, 1057–1061. [Google Scholar] [CrossRef] [Green Version]
- Netchiporouk, E.; Litvinov, I.V.; Moreau, L.; Gilbert, M.; Sasseville, D.; Duvic, M. Deregulation in STAT signaling is important for cutaneous T-cell lymphoma (CTCL) pathogenesis and cancer progression. Cell Cycle 2014, 13, 3331–3335. [Google Scholar] [CrossRef]
- Fredholm, S.; Gjerdrum, L.M.; Willerslev-Olsen, A.; Petersen, D.L.; Nielsen, I.O.; Kauczok, C.S.; Wobser, M.; Ralfkiaer, U.; Bonefeld, C.M.; Wasik, M.A.; et al. STAT3 activation and infiltration of eosinophil granulocytes in mycosis fungoides. Anticancer Res. 2014, 34, 5277–5286. [Google Scholar]
- Waldmann, T.A.; Chen, J. Disorders of the JAK/STAT Pathway in T Cell Lymphoma Pathogenesis: Implications for Immunotherapy. Annu. Rev. Immunol. 2017, 35, 533–550. [Google Scholar] [CrossRef]
- Damsky, W.; King, B.A. JAK inhibitors in dermatology: The promise of a new drug class. J. Am. Acad. Dermatol. 2017, 76, 736–744. [Google Scholar] [CrossRef]
- Stritesky, G.L.; Muthukrishnan, R.; Sehra, S.; Goswami, R.; Pham, D.; Travers, J.; Nguyen, E.T.; Levy, D.E.; Kaplan, M.H. The transcription factor STAT3 is required for T helper 2 cell development. Immunity 2011, 34, 39–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, M.; Nissen, M.H.; Gerwien, J.; Zocca, M.B.; Rasmussen, H.M.; Nakajima, K.; Ropke, C.; Geisler, C.; Kaltoft, K.; Odum, N. Spontaneous interleukin-5 production in cutaneous T-cell lymphoma lines is mediated by constitutively activated Stat3. Blood 2002, 99, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.; Wang, C.; Walradt, T.; Hong, B.S.; Tanner, J.R.; Levinsohn, J.L.; Goh, G.; Subtil, A.; Lessin, S.R.; Heymann, W.R.; et al. Identification of a gain-of-function STAT3 mutation (p.Y640F) in lymphocytic variant hypereosinophilic syndrome. Blood 2016, 127, 948–951. [Google Scholar] [CrossRef] [PubMed]
- Suchin, K.R.; Cassin, M.; Gottleib, S.L.; Sood, S.; Cucchiara, A.J.; Vonderheid, E.C.; Rook, A.H. Increased interleukin 5 production in eosinophilic Sezary syndrome: Regulation by interferon alfa and interleukin 12. J. Am. Acad. Dermatol. 2001, 44, 28–32. [Google Scholar] [CrossRef]
- Olsen, E.; Vonderheid, E.; Pimpinelli, N.; Willemze, R.; Kim, Y.; Knobler, R.; Zackheim, H.; Duvic, M.; Estrach, T.; Lamberg, S.; et al. Revisions to the staging and classification of mycosis fungoides and Sezary syndrome: A proposal of the International Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force of the European Organization of Research and Treatment of Cancer (EORTC). Blood 2007, 110, 1713–1722. [Google Scholar] [CrossRef] [Green Version]
- Roufosse, F. Peripheral T-cell lymphoma developing after diagnosis of lymphocytic variant hypereosinophilic syndrome: Misdiagnosed lymphoma or natural disease progression? Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2014, 118, 506–510. [Google Scholar] [CrossRef]
- Whittaker, S.; Hoppe, R.; Prince, H.M. How I treat mycosis fungoides and Sezary syndrome. Blood 2016, 127, 3142–3153. [Google Scholar] [CrossRef] [Green Version]
- Olsen, E.A.; Rook, A.H.; Zic, J.; Kim, Y.; Porcu, P.; Querfeld, C.; Wood, G.; Demierre, M.F.; Pittelkow, M.; Wilson, L.D.; et al. Sezary syndrome: Immunopathogenesis, literature review of therapeutic options, and recommendations for therapy by the United States Cutaneous Lymphoma Consortium (USCLC). J. Am. Acad. Dermatol. 2011, 64, 352–404. [Google Scholar] [CrossRef]
- Klion, A.D. How I treat hypereosinophilic syndromes. Blood 2015, 126, 1069–1077. [Google Scholar] [CrossRef] [Green Version]
- Khoury, P.; Bochner, B.S. Consultation for Elevated Blood Eosinophils: Clinical Presentations, High Value Diagnostic Tests, and Treatment Options. J. Allergy Clin. Immunol. Pract. 2018, 6, 1446–1453. [Google Scholar] [CrossRef]
- Butt, N.M.; Lambert, J.; Ali, S.; Beer, P.A.; Cross, N.C.; Duncombe, A.; Ewing, J.; Harrison, C.N.; Knapper, S.; McLornan, D.; et al. Guideline for the investigation and management of eosinophilia. Br. J. Haematol. 2017, 176, 553–572. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Rook, A.H. Treatment of Sézary syndrome. In UpToDate; Kuzel, T.W., Zic, J.A., Eds.; Wolters Kluwer: Waltham, MA, USA, 2020; Available online: https://www.uptodate.com/contents/treatment-of-sezary-syndrome#H9585818 (accessed on 27 August 2020).
- Agar, N.S.; Wedgeworth, E.; Crichton, S.; Mitchell, T.J.; Cox, M.; Ferreira, S.; Robson, A.; Calonje, E.; Stefanato, C.M.; Wain, E.M.; et al. Survival outcomes and prognostic factors in mycosis fungoides/Sezary syndrome: Validation of the revised International Society for Cutaneous Lymphomas/European Organisation for Research and Treatment of Cancer staging proposal. J. Clin. Oncol. 2010, 28, 4730–4739. [Google Scholar] [CrossRef] [PubMed]
- Buus, T.B.; Willerslev-Olsen, A.; Fredholm, S.; Blumel, E.; Nastasi, C.; Gluud, M.; Hu, T.; Lindahl, L.M.; Iversen, L.; Fogh, H.; et al. Single-cell heterogeneity in Sezary syndrome. Blood Adv. 2018, 2, 2115–2126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokolowska-Wojdylo, M.; Wenzel, J.; Gaffal, E.; Lenz, J.; Speuser, P.; Erdmann, S.; Abuzahra, F.; Bowman, E.; Roszkiewicz, J.; Bieber, T.; et al. Circulating clonal CLA(+) and CD4(+) T cells in Sezary syndrome express the skin-homing chemokine receptors CCR4 and CCR10 as well as the lymph node-homing chemokine receptor CCR7. Br. J. Dermatol. 2005, 152, 258–264. [Google Scholar] [CrossRef]
- Bernengo, M.G.; Novelli, M.; Quaglino, P.; Lisa, F.; De Matteis, A.; Savoia, P.; Cappello, N.; Fierro, M.T. The relevance of the CD4+ CD26- subset in the identification of circulating Sezary cells. Br. J. Dermatol. 2001, 144, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; La Perle, K.; Kwiatkowski, S.; Sullivan, L.A.; Sams, G.H.; Johns, J.; Curphey, D.P.; Wen, J.; McConnell, K.; Qi, J.; et al. Mechanism, Consequences, and Therapeutic Targeting of Abnormal IL15 Signaling in Cutaneous T-cell Lymphoma. Cancer Discov. 2016, 6, 986–1005. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Ni, X.; Covington, K.R.; Yang, B.Y.; Shiu, J.; Zhang, X.; Xi, L.; Meng, Q.; Langridge, T.; Drummond, J.; et al. Genomic profiling of Sezary syndrome identifies alterations of key T cell signaling and differentiation genes. Nat. Genet. 2015, 47, 1426–1434. [Google Scholar] [CrossRef] [Green Version]
- Woollard, W.J.; Pullabhatla, V.; Lorenc, A.; Patel, V.M.; Butler, R.M.; Bayega, A.; Begum, N.; Bakr, F.; Dedhia, K.; Fisher, J.; et al. Candidate driver genes involved in genome maintenance and DNA repair in Sezary syndrome. Blood 2016, 127, 3387–3397. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.K.; Mishra, A.; Hake, T.; Porcu, P. Evolving insights in the pathogenesis and therapy of cutaneous T-cell lymphoma (mycosis fungoides and Sezary syndrome). Br. J. Haematol. 2011, 155, 150–166. [Google Scholar] [CrossRef] [Green Version]
- Kari, L.; Loboda, A.; Nebozhyn, M.; Rook, A.H.; Vonderheid, E.C.; Nichols, C.; Virok, D.; Chang, C.; Horng, W.H.; Johnston, J.; et al. Classification and prediction of survival in patients with the leukemic phase of cutaneous T cell lymphoma. J. Exp. Med. 2003, 197, 1477–1488. [Google Scholar] [CrossRef]
- Wang, Y.; Su, M.; Zhou, L.L.; Tu, P.; Zhang, X.; Jiang, X.; Zhou, Y. Deficiency of SATB1 expression in Sezary cells causes apoptosis resistance by regulating FasL/CD95L transcription. Blood 2011, 117, 3826–3835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fanok, M.H.; Sun, A.; Fogli, L.K.; Narendran, V.; Eckstein, M.; Kannan, K.; Dolgalev, I.; Lazaris, C.; Heguy, A.; Laird, M.E.; et al. Role of Dysregulated Cytokine Signaling and Bacterial Triggers in the Pathogenesis of Cutaneous T-Cell Lymphoma. J. Investig. Dermatol. 2018, 138, 1116–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borcherding, N.; Voigt, A.P.; Liu, V.; Link, B.K.; Zhang, W.; Jabbari, A. Single-Cell Profiling of Cutaneous T-Cell Lymphoma Reveals Underlying Heterogeneity Associated with Disease Progression. Clin. Cancer Res. 2019, 25, 2996–3005. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.S.; Ungewickell, A.; Bhaduri, A.; Qu, K.; Webster, D.E.; Armstrong, R.; Weng, W.K.; Aros, C.J.; Mah, A.; Chen, R.O.; et al. Transcriptome sequencing in Sezary syndrome identifies Sezary cell and mycosis fungoides-associated lncRNAs and novel transcripts. Blood 2012, 120, 3288–3297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pomerantz, R.G.; Mirvish, E.D.; Erdos, G.; Falo, L.D., Jr.; Geskin, L.J. Novel approach to gene expression profiling in Sezary syndrome. Br. J. Dermatol. 2010, 163, 1090–1094. [Google Scholar] [CrossRef] [Green Version]
- Michel, L.; Jean-Louis, F.; Begue, E.; Bensussan, A.; Bagot, M. Use of PLS3, Twist, CD158k/KIR3DL2, and NKp46 gene expression combination for reliable Sezary syndrome diagnosis. Blood 2013, 121, 1477–1478. [Google Scholar] [CrossRef] [Green Version]
- Wysocka, M.; Kossenkov, A.V.; Benoit, B.M.; Troxel, A.B.; Singer, E.; Schaffer, A.; Kim, B.; Dentchev, T.; Nagata, S.; Ise, T.; et al. CD164 and FCRL3 are highly expressed on CD4+CD26- T cells in Sezary syndrome patients. J. Investig. Dermatol. 2014, 134, 229–236. [Google Scholar] [CrossRef] [Green Version]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdottir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef]
- Gibson, H.M.; Mishra, A.; Chan, D.V.; Hake, T.S.; Porcu, P.; Wong, H.K. Impaired proteasome function activates GATA3 in T cells and upregulates CTLA-4: Relevance for Sezary syndrome. J. Investig. Dermatol. 2013, 133, 249–257. [Google Scholar] [CrossRef] [Green Version]
- Geskin, L.J.; Viragova, S.; Stolz, D.B.; Fuschiotti, P. Interleukin-13 is overexpressed in cutaneous T-cell lymphoma cells and regulates their proliferation. Blood 2015, 125, 2798–2805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredholm, S.; Willerslev-Olsen, A.; Met, O.; Kubat, L.; Gluud, M.; Mathiasen, S.L.; Friese, C.; Blumel, E.; Petersen, D.L.; Hu, T.; et al. SATB1 in Malignant T Cells. J. Investig. Dermatol. 2018, 138, 1805–1815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshie, O.; Matsushima, K. CCR4 and its ligands: From bench to bedside. Int. Immunol. 2015, 27, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristofoletti, C.; Bresin, A.; Picozza, M.; Picchio, M.C.; Monzo, F.; Helmer Citterich, M.; Passarelli, F.; Frezzolini, A.; Scala, E.; Monopoli, A.; et al. Blood and skin-derived Sezary cells: Differences in proliferation-index, activation of PI3K/AKT/mTORC1 pathway and its prognostic relevance. Leukemia 2019, 33, 1231–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duvic, M.; Pinter-Brown, L.C.; Foss, F.M.; Sokol, L.; Jorgensen, J.L.; Challagundla, P.; Dwyer, K.M.; Zhang, X.; Kurman, M.R.; Ballerini, R.; et al. Phase 1/2 study of mogamulizumab, a defucosylated anti-CCR4 antibody, in previously treated patients with cutaneous T-cell lymphoma. Blood 2015, 125, 1883–1889. [Google Scholar] [CrossRef]
- Kitadate, A.; Ikeda, S.; Abe, F.; Takahashi, N.; Shimizu, N.; Matsue, K.; Tagawa, H. Histone deacetylase inhibitors downregulate CCR4 expression and decrease mogamulizumab efficacy in CCR4-positive mature T-cell lymphomas. Haematologica 2018, 103, 126–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, S.K.; Newton, S.B.; Bach, T.L.; Budgin, J.B.; Benoit, B.M.; Lin, J.H.; Yoon, J.S.; Wysocka, M.; Abrams, C.S.; Rook, A.H. Bexarotene blunts malignant T-cell chemotaxis in Sezary syndrome: Reduction of chemokine receptor 4-positive lymphocytes and decreased chemotaxis to thymus and activation-regulated chemokine. Am. J. Hematol. 2007, 82, 792–797. [Google Scholar] [CrossRef]
- Grzanka, D.; Gagat, M.; Izdebska, M.; Marszalek, A. Expression of special AT-rich sequence-binding protein 1 is an independent prognostic factor in cutaneous T-cell lymphoma. Oncol. Rep. 2015, 33, 250–266. [Google Scholar] [CrossRef] [Green Version]
- Renema, N.; Navet, B.; Heymann, M.F.; Lezot, F.; Heymann, D. RANK-RANKL signalling in cancer. Biosci. Rep. 2016, 36. [Google Scholar] [CrossRef] [Green Version]
- Boonk, S.E.; Zoutman, W.H.; Putter, H.; Ram-Wolff, C.; Felcht, M.; Klemke, C.D.; Ranki, A.; Quaglino, P.; Whittaker, S.; Bagot, M.; et al. Increased Expression of PLS3 Correlates with Better Outcome in Sezary Syndrome. J. Investig. Dermatol. 2017, 137, 754–757. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Chen, C.; Guo, W.; Zheng, S.; Sun, Z.; Geng, X. DNM3 Attenuates Hepatocellular Carcinoma Growth by Activating P53. Med. Sci. Monit. 2016, 22, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Osthus, R.C.; Karim, B.; Prescott, J.E.; Smith, B.D.; McDevitt, M.; Huso, D.L.; Dang, C.V. The Myc target gene JPO1/CDCA7 is frequently overexpressed in human tumors and has limited transforming activity in vivo. Cancer Res. 2005, 65, 5620–5627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez, P.R.; Martin-Cortazar, C.; Kourani, O.; Chiodo, Y.; Cordoba, R.; Dominguez-Franjo, M.P.; Redondo, J.M.; Iglesias, T.; Campanero, M.R. CDCA7 is a critical mediator of lymphomagenesis that selectively regulates anchorage-independent growth. Haematologica 2018, 103, 1669–1678. [Google Scholar] [CrossRef] [PubMed]
- Martin-Cortazar, C.; Chiodo, Y.; Jimenez, P.R.; Bernabe, M.; Cayuela, M.L.; Iglesias, T.; Campanero, M.R. CDCA7 finely tunes cytoskeleton dynamics to promote lymphoma migration and invasion. Haematologica 2019. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Ho, C.S.; Ponzielli, R.; Barsyte-Lovejoy, D.; Bouffet, E.; Picard, D.; Hawkins, C.E.; Penn, L.Z. Identification of a novel c-Myc protein interactor, JPO2, with transforming activity in medulloblastoma cells. Cancer Res. 2005, 65, 5607–5619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Begue, E.; Jean-Louis, F.; Bagot, M.; Jauliac, S.; Cayuela, J.M.; Laroche, L.; Parquet, N.; Bachelez, H.; Bensussan, A.; Courtois, G.; et al. Inducible expression and pathophysiologic functions of T-plastin in cutaneous T-cell lymphoma. Blood 2012, 120, 143–154. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.L.; Ferreira, S.; McKenzie, R.C.; Tosi, I.; Caesar, J.A.; Bagot, M.; Whittaker, S.J.; Mitchell, T.J. Regulation of T-plastin expression by promoter hypomethylation in primary cutaneous T-cell lymphoma. J. Investig. Dermatol. 2012, 132, 2042–2049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henn, A.; Michel, L.; Fite, C.; Deschamps, L.; Ortonne, N.; Ingen-Housz-Oro, S.; Marinho, E.; Beylot-Barry, M.; Bagot, M.; Laroche, L.; et al. Sezary syndrome without erythroderma. J. Am. Acad. Dermatol. 2015, 72, 1003–1009 e1001. [Google Scholar] [CrossRef]
- Su, M.W.; Dorocicz, I.; Dragowska, W.H.; Ho, V.; Li, G.; Voss, N.; Gascoyne, R.; Zhou, Y. Aberrant expression of T-plastin in Sezary cells. Cancer Res. 2003, 63, 7122–7127. [Google Scholar]
- Yokobori, T.; Iinuma, H.; Shimamura, T.; Imoto, S.; Sugimachi, K.; Ishii, H.; Iwatsuki, M.; Ota, D.; Ohkuma, M.; Iwaya, T.; et al. Plastin3 is a novel marker for circulating tumor cells undergoing the epithelial-mesenchymal transition and is associated with colorectal cancer prognosis. Cancer Res. 2013, 73, 2059–2069. [Google Scholar] [CrossRef] [Green Version]
- Ueo, H.; Sugimachi, K.; Gorges, T.M.; Bartkowiak, K.; Yokobori, T.; Muller, V.; Shinden, Y.; Ueda, M.; Ueo, H.; Mori, M.; et al. Circulating tumour cell-derived plastin3 is a novel marker for predicting long-term prognosis in patients with breast cancer. Br. J. Cancer 2015, 112, 1519–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Rahman, M.A.; Chen, Z.G.; Shin, D.M. Multiple biological functions of Twist1 in various cancers. Oncotarget 2017, 8, 20380–20393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norozi, F.; Ahmadzadeh, A.; Shahjahani, M.; Shahrabi, S.; Saki, N. Twist as a new prognostic marker in hematological malignancies. Clin. Transl. Oncol. 2016, 18, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Goswami, M.; Duvic, M.; Dougherty, A.; Ni, X. Increased Twist expression in advanced stage of mycosis fungoides and Sezary syndrome. J. Cutan. Pathol. 2012, 39, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Beck, B.; Lapouge, G.; Rorive, S.; Drogat, B.; Desaedelaere, K.; Delafaille, S.; Dubois, C.; Salmon, I.; Willekens, K.; Marine, J.C.; et al. Different levels of Twist1 regulate skin tumor initiation, stemness, and progression. Cell Stem Cell 2015, 16, 67–79. [Google Scholar] [CrossRef] [Green Version]
- Pham, D.; Vincentz, J.W.; Firulli, A.B.; Kaplan, M.H. Twist1 regulates Ifng expression in Th1 cells by interfering with Runx3 function. J. Immunol. 2012, 189, 832–840. [Google Scholar] [CrossRef] [Green Version]
- Niesner, U.; Albrecht, I.; Janke, M.; Doebis, C.; Loddenkemper, C.; Lexberg, M.H.; Eulenburg, K.; Kreher, S.; Koeck, J.; Baumgrass, R.; et al. Autoregulation of Th1-mediated inflammation by twist1. J. Exp. Med. 2008, 205, 1889–1901. [Google Scholar] [CrossRef]
- Chong, B.F.; Dantzer, P.; Germeroth, T.; Hafner, M.; Wilson, A.J.; Xiao, G.; Wong, H.K. Induced Sezary syndrome PBMCs poorly express immune response genes up-regulated in stimulated memory T cells. J. Dermatol. Sci. 2010, 60, 8–20. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.K.; Gibson, H.; Hake, T.; Geyer, S.; Frederickson, J.; Marcucci, G.; Caligiuri, M.A.; Porcu, P.; Mishra, A. Promoter-Specific Hypomethylation Is Associated with Overexpression of PLS3, GATA6, and TWIST1 in the Sezary Syndrome. J. Investig. Dermatol. 2015, 135, 2084–2092. [Google Scholar] [CrossRef] [Green Version]
- Vermeer, M.H.; van Doorn, R.; Dijkman, R.; Mao, X.; Whittaker, S.; van Voorst Vader, P.C.; Gerritsen, M.J.; Geerts, M.L.; Gellrich, S.; Soderberg, O.; et al. Novel and highly recurrent chromosomal alterations in Sezary syndrome. Cancer Res. 2008, 68, 2689–2698. [Google Scholar] [CrossRef] [Green Version]
- Showe, L.C.; Fox, F.E.; Williams, D.; Au, K.; Niu, Z.; Rook, A.H. Depressed IL-12-mediated signal transduction in T cells from patients with Sezary syndrome is associated with the absence of IL-12 receptor beta 2 mRNA and highly reduced levels of STAT4. J. Immunol. 1999, 163, 4073–4079. [Google Scholar] [PubMed]
- Litvinov, I.V.; Cordeiro, B.; Fredholm, S.; Odum, N.; Zargham, H.; Huang, Y.; Zhou, Y.; Pehr, K.; Kupper, T.S.; Woetmann, A.; et al. Analysis of STAT4 expression in cutaneous T-cell lymphoma (CTCL) patients and patient-derived cell lines. Cell Cycle 2014, 13, 2975–2982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, J.M.; Schmidt, J.A.; Carson, K.R.; Musiek, A.C.; Mehta-Shah, N.; Payton, J.E. Novel cell adhesion/migration pathways are predictive markers of HDAC inhibitor resistance in cutaneous T cell lymphoma. EBioMedicine 2019, 46, 170–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopp, K.L.; Ralfkiaer, U.; Gjerdrum, L.M.; Helvad, R.; Pedersen, I.H.; Litman, T.; Jonson, L.; Hagedorn, P.H.; Krejsgaard, T.; Gniadecki, R.; et al. STAT5-mediated expression of oncogenic miR-155 in cutaneous T-cell lymphoma. Cell Cycle 2013, 12, 1939–1947. [Google Scholar] [CrossRef]
- Moyal, L.; Yehezkel, S.; Gorovitz, B.; Keren, A.; Gilhar, A.; Lubin, I.; Sherman, S.; Hodak, E. Oncogenic role of microRNA-155 in mycosis fungoides: An In Vitro and xenograft mouse model study. Br. J. Dermatol. 2017, 177, 791–800. [Google Scholar] [CrossRef]
- Ralfkiaer, U.; Hagedorn, P.H.; Bangsgaard, N.; Lovendorf, M.B.; Ahler, C.B.; Svensson, L.; Kopp, K.L.; Vennegaard, M.T.; Lauenborg, B.; Zibert, J.R.; et al. Diagnostic microRNA profiling in cutaneous T-cell lymphoma (CTCL). Blood 2011, 118, 5891–5900. [Google Scholar] [CrossRef]
- Tili, E.; Michaille, J.J.; Wernicke, D.; Alder, H.; Costinean, S.; Volinia, S.; Croce, C.M. Mutator activity induced by microRNA-155 (miR-155) links inflammation and cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 4908–4913. [Google Scholar] [CrossRef] [Green Version]
- Witten, L.; Slack, F.J. miR-155 as a novel clinical target for hematological malignancies. Carcinogenesis 2020, 41, 2–7. [Google Scholar] [CrossRef]
- Kasprzycka, M.; Zhang, Q.; Witkiewicz, A.; Marzec, M.; Potoczek, M.; Liu, X.; Wang, H.Y.; Milone, M.; Basu, S.; Mauger, J.; et al. Gamma c-signaling cytokines induce a regulatory T cell phenotype in malignant CD4+ T lymphocytes. J. Immunol. 2008, 181, 2506–2512. [Google Scholar] [CrossRef] [Green Version]
- Krejsgaard, T.; Odum, N.; Geisler, C.; Wasik, M.A.; Woetmann, A. Regulatory T cells and immunodeficiency in mycosis fungoides and Sezary syndrome. Leukemia 2012, 26, 424–432. [Google Scholar] [CrossRef] [Green Version]
- Jariwala, N.; Benoit, B.; Kossenkov, A.V.; Oetjen, L.K.; Whelan, T.M.; Cornejo, C.M.; Takeshita, J.; Kim, B.S.; Showe, L.C.; Wysocka, M.; et al. TIGIT and Helios Are Highly Expressed on CD4(+) T Cells in Sezary Syndrome Patients. J. Investig. Dermatol. 2017, 137, 257–260. [Google Scholar] [CrossRef] [Green Version]
- Heid, J.B.; Schmidt, A.; Oberle, N.; Goerdt, S.; Krammer, P.H.; Suri-Payer, E.; Klemke, C.D. FOXP3+CD25- tumor cells with regulatory function in Sezary syndrome. J. Investig. Dermatol. 2009, 129, 2875–2885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capriotti, E.; Vonderheid, E.C.; Thoburn, C.J.; Wasik, M.A.; Bahler, D.W.; Hess, A.D. Expression of T-plastin, FoxP3 and other tumor-associated markers by leukemic T-cells of cutaneous T-cell lymphoma. Leuk. Lymphoma 2008, 49, 1190–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Querfeld, C.; Leung, S.; Myskowski, P.L.; Curran, S.A.; Goldman, D.A.; Heller, G.; Wu, X.; Kil, S.H.; Sharma, S.; Finn, K.J.; et al. Primary T Cells from Cutaneous T-cell Lymphoma Skin Explants Display an Exhausted Immune Checkpoint Profile. Cancer Immunol. Res. 2018, 6, 900–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swainson, L.A.; Mold, J.E.; Bajpai, U.D.; McCune, J.M. Expression of the autoimmune susceptibility gene FcRL3 on human regulatory T cells is associated with dysfunction and high levels of programmed cell death-1. J. Immunol. 2010, 184, 3639–3647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joller, N.; Lozano, E.; Burkett, P.R.; Patel, B.; Xiao, S.; Zhu, C.; Xia, J.; Tan, T.G.; Sefik, E.; Yajnik, V.; et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity 2014, 40, 569–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harjunpaa, H.; Guillerey, C. TIGIT as an emerging immune checkpoint. Clin. Exp. Immunol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Bin Dhuban, K.; d’Hennezel, E.; Nashi, E.; Bar-Or, A.; Rieder, S.; Shevach, E.M.; Nagata, S.; Piccirillo, C.A. Coexpression of TIGIT and FCRL3 identifies Helios+ human memory regulatory T cells. J. Immunol. 2015, 194, 3687–3696. [Google Scholar] [CrossRef] [Green Version]
- Anzengruber, F.; Ignatova, D.; Schlaepfer, T.; Chang, Y.T.; French, L.E.; Pascolo, S.; Contassot, E.; Bobrowicz, M.; Hoetzenecker, W.; Guenova, E. Divergent LAG-3 versus BTLA, TIGIT, and FCRL3 expression in Sezary syndrome. Leuk. Lymphoma 2019, 60, 1899–1907. [Google Scholar] [CrossRef] [Green Version]
- Aliahmad, P.; Kaye, J. Development of all CD4 T lineages requires nuclear factor TOX. J. Exp. Med. 2008, 205, 245–256. [Google Scholar] [CrossRef]
- Ando, M.; Ito, M.; Srirat, T.; Kondo, T.; Yoshimura, A. Memory T cell, exhaustion, and tumor immunity. Immunol. Med. 2019, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Su, M.W.; Jiang, X.; Zhou, Y. Evidence of an oncogenic role of aberrant TOX activation in cutaneous T-cell lymphoma. Blood 2015, 125, 1435–1443. [Google Scholar] [CrossRef]
- Lefrancois, P.; Xie, P.; Wang, L.; Tetzlaff, M.T.; Moreau, L.; Watters, A.K.; Netchiporouk, E.; Provost, N.; Gilbert, M.; Ni, X.; et al. Gene expression profiling and immune cell-type deconvolution highlight robust disease progression and survival markers in multiple cohorts of CTCL patients. Oncoimmunology 2018, 7, e1467856. [Google Scholar] [CrossRef] [Green Version]
- Narducci, M.G.; Arcelli, D.; Picchio, M.C.; Lazzeri, C.; Pagani, E.; Sampogna, F.; Scala, E.; Fadda, P.; Cristofoletti, C.; Facchiano, A.; et al. MicroRNA profiling reveals that miR-21, miR486 and miR-214 are upregulated and involved in cell survival in Sezary syndrome. Cell Death Dis. 2011, 2, e151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tessema, M.; Yingling, C.M.; Picchi, M.A.; Wu, G.; Ryba, T.; Lin, Y.; Bungum, A.O.; Edell, E.S.; Spira, A.; Belinsky, S.A. ANK1 Methylation regulates expression of MicroRNA-486-5p and discriminates lung tumors by histology and smoking status. Cancer Lett. 2017, 410, 191–200. [Google Scholar] [CrossRef]
- Bianchi, E.; Bulgarelli, J.; Ruberti, S.; Rontauroli, S.; Sacchi, G.; Norfo, R.; Pennucci, V.; Zini, R.; Salati, S.; Prudente, Z.; et al. MYB controls erythroid versus megakaryocyte lineage fate decision through the miR-486-3p-mediated downregulation of MAF. Cell Death Differ. 2015, 22, 1906–1921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, A.E.; Lu, W.T.; Godfrey, J.D.; Antonov, A.V.; Paicu, C.; Moxon, S.; Dalmay, T.; Wilczynska, A.; Muller, P.A.; Bushell, M. The cytoskeleton adaptor protein ankyrin-1 is upregulated by p53 following DNA damage and alters cell migration. Cell Death Dis. 2016, 7, e2184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Yan, B. Multiple roles and regulatory mechanisms of the transcription factor GATA6 in human cancers. Clin. Genet. 2020, 97, 64–72. [Google Scholar] [CrossRef] [Green Version]
- Kamijo, H.; Miyagaki, T.; Shishido-Takahashi, N.; Nakajima, R.; Oka, T.; Suga, H.; Sugaya, M.; Sato, S. Aberrant CD137 ligand expression induced by GATA6 overexpression promotes tumor progression in cutaneous T-cell lymphoma. Blood 2018, 132, 1922–1935. [Google Scholar] [CrossRef] [Green Version]
- Parra, M. Class IIa HDACs—New insights into their functions in physiology and pathology. FEBS J. 2015, 282, 1736–1744. [Google Scholar] [CrossRef]
- Gil, V.S.; Bhagat, G.; Howell, L.; Zhang, J.; Kim, C.H.; Stengel, S.; Vega, F.; Zelent, A.; Petrie, K. Deregulated expression of HDAC9 in B cells promotes development of lymphoproliferative disease and lymphoma in mice. Dis. Model Mech. 2016, 9, 1483–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutt, D.M.; Roth, D.M.; Vignaud, H.; Cullin, C.; Bouchecareilh, M. The histone deacetylase inhibitor, Vorinostat, represses hypoxia inducible factor 1 alpha expression through translational inhibition. PLoS ONE 2014, 9, e106224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linares, A.; Assou, S.; Lapierre, M.; Thouennon, E.; Duraffourd, C.; Fromaget, C.; Boulahtouf, A.; Tian, G.; Ji, J.; Sahin, O.; et al. Increased expression of the HDAC9 gene is associated with antiestrogen resistance of breast cancers. Mol. Oncol. 2019, 13, 1534–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milde, T.; Oehme, I.; Korshunov, A.; Kopp-Schneider, A.; Remke, M.; Northcott, P.; Deubzer, H.E.; Lodrini, M.; Taylor, M.D.; von Deimling, A.; et al. HDAC5 and HDAC9 in medulloblastoma: Novel markers for risk stratification and role in tumor cell growth. Clin. Cancer Res. 2010, 16, 3240–3252. [Google Scholar] [CrossRef] [Green Version]
- Stoeger, T.; Gerlach, M.; Morimoto, R.I.; Nunes Amaral, L.A. Large-scale investigation of the reasons why potentially important genes are ignored. PLoS Biol. 2018, 16, e2006643. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Disease Type | Skin Inflammation | Lympho-Proliferation | Malignancy |
|---|---|---|---|
| Sézary syndrome | Th2 | clonal | + |
| L-HES | Th2 | frequently clonal | − |
| Atopic Dermatitis | Th2 | reactive | − |
| Psoriasis | Th1, Th17 | reactive | − |
| Contact Dermatitis | Th1, Th2 or Th17 | reactive | − |
| Clinical Features | Sézary Syndrome | Lymphocytic-Variant HES |
| Classification | lymphoma, stage IV | benign lymphoproliferation |
| Diagnostic criteria | Sézary cells > 1000/µL (or, CD4/CD8 ratio ≥ 10, CD4+CD7− cells ≥ 40%, CD4+CD26− cells ≥ 30%), with identical T-cell clone in blood + skin [6,55] | Rule out other causes of HES. Blood eosinophilia >1500/µL, abnormal T cells with no standardized threshold, frequent T-cell clonality, T cells secrete IL-5 [25,35,56]. |
| Clinical course | moderately aggressive | indolent |
| Skin and physical symptoms | widespread erythroderma, pruritus, lymphadenopathy | limited erythroderma, urticaria, pruritus [24,27] |
| Residence of T cells | blood, skin, lymph node | blood, skin, lymph node, soft tissue [25] |
| Skin pathology | epidermotropic Sézary cells with cerebriform nuclei, eosinophils in some patients | abundant eosinophils, and perivascular, dermal infiltrate of small-medium size T cells with irregular nucleus and scarce cytoplasm [25,35] |
| Eosinophilia | some patients, late, moderate complications uncommon [29] >700/µL poor prognostic indicator [46] | all patients, early, severe, can cause organ damage |
| First line therapy | Systemic immunomodulation: ECP combined with interferons or other systemic (bexarotene, romidepsin, low dose methotrexate) and/or skin-directed (topicals, TSEBT) therapy [57,58] | systemic corticosteroids [35,59,60] |
| Second line therapy | Targeted and immune enhancing/sparing therapies preferred: mogamulizumab. romidepsin, alemtuzumab, intermediate dose methotrexate. Refractory disease: clinical trials, allogeneic HCT, chemotherapy [57,58] | IFN-α + glucocorticoids Steroid-sparing: mepolizumab, alemtuzumab, mycophenolate mofetil, cyclosporin, methotrexate, JAK kinase inhibitors (ruxolitinib, tofacitinib) [35,59,60] and imatinib, despite lack of FIP1L1-PDFGRA fusion, for patients who fail other tharapies [61] |
| Follow up | Monitor complete blood count with differential, liver function, LDH, flow cytometry for Sezary cells [33] in blood, physical examination for nodes, viscera and skin infections [62]. | monitor T-cell lymphoma risk with lymphocyte counts, PB flow cytometry, BM cytogenetics [59] |
| Progression symptoms | Increases in pruritus, erythroderma, or skin tumor burden, enlarging lymph nodes, visceral organ involvement, immune suppression [63] | 10–25% progress to T-cell lymphoma, cytogenetic changes Nonspecific symptoms: rapid increase in lymphocytosis, lymph node involvement, infiltrative nodules [25] |
| Molecular Features | Sézary Syndrome | Lymphocytic-VariantHES |
| T-cell phenotype | memory T cell with heterogeneous molecular phenotype [43,64] | memory T cell [30,42] |
| T-cell surface antigens | CD3+/−CD4+, CD7 and/or CD26 loss CLA+, CCR7+, CCR4+, CCR10+ [65,66] | CD3−CD4+CD7−CD5++, CD3+CD4+CD7−, or, CD3+CD4−CD8− [23,35] |
| Cytokines | Th2 (IL-4, IL-5, IL-13), suppressive (IL-10), autocrine or paracrine growth stimulation (IL-15, IL-16, IL-32) [67,68] | Th2 (IL-4, IL-5, IL-13) [28,42] |
| Molecular drivers | Mutations in pathways related to DNA damage repair (TP53), apoptosis, (FAS), cell cycle (MYC, RB1), epigenetic modulators (DNMT3A, TET2), JAK/STAT (JAK3, STAT3, STAT5B), ARID1A, NF-κB (NFKB2, CARD11), TCR-signaling (CD28, PLCG1) [37,38,69,70] | IL-5, GATA3, JAK/STAT, IL17RB, TGFβ signaling [30,53] |
| Genetic abnormalities | Frequent SNV and CNV, C > T transitions consistent with UV damage, recurrent 10q and 17p deletions, recurrent 8q and 17q amplifications [37,38]; gene fusions [68] | Seldom reported, partial 6q deletion and other karyotype abnormalities [23] |
| Meta-Analysis Study | Sézary Patients | Healthy Donors | BID | Technology |
| Moerman-Herzog et al. [22] | n = 3 CD3+CD4+CD45RO+ | n = 3 CD3+CD4+CD45RO+ | n.a. | microarray |
| Prior Study | Sézary Patients | Healthy Donors | BID | Technology |
| Fanok et al. [73] | n = 8 CD3+CD4+CD7− and/or CD3+CD4+CD26− | n =4 CD3+CD4+CD45RO+ | n.a. | RNAseq |
| Wang et al. [68] | n = 22 CD3+CD4+ | n = 5 CD3+CD4+ | n.a. | RNAseq |
| Wysocka et al. [78] | n = 6 CD3+CD4+ | n = 3 CD3+CD4+ | n.a. | microarray |
| Wang et al. [72] | n = 6 CD3+CD4+CD7- | n = 9 CD3+CD4+ | n.a. | microarray |
| Booken et al. [16] | n = 10 PBMC | n =10 PBMC | n.a. | microarray |
| Hahtola et al. [17] | n = 4 PBMC | n = 5 PBMC | n.a. | microarray |
| van Doorn et al. [14] | n = 10 CD3+CD4+ | n = 3 CD3+CD4+ | n = 5 CD3+CD4+ | microarray |
| Kari et al. [71] | n = 18 >60% CD4+ | n = 12 Th2-skewed PBMC | n.a. | microarray |
| Prior Study | Sézary Malignant Cells | Patient-Matched Non-Malignant Cells | BID | Technology |
| Borcherding et al. [74] | n = 1 CD3+CD4+CD5brightSSChi | n = 1 CD3+CD4+CD5intSSCint | n.a. | scRNAseq |
| Lee et al. [75] | n = 3 CD3+CD4+Vβ+ | n = 3 CD3+CD4+Vβ− | n.a. | RNAseq |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moerman-Herzog, A.; Mehdi, S.J.; Wong, H.K. Gene Expression Comparison between Sézary Syndrome and Lymphocytic-Variant Hypereosinophilic Syndrome Refines Biomarkers for Sézary Syndrome. Cells 2020, 9, 1992. https://doi.org/10.3390/cells9091992
Moerman-Herzog A, Mehdi SJ, Wong HK. Gene Expression Comparison between Sézary Syndrome and Lymphocytic-Variant Hypereosinophilic Syndrome Refines Biomarkers for Sézary Syndrome. Cells. 2020; 9(9):1992. https://doi.org/10.3390/cells9091992
Chicago/Turabian StyleMoerman-Herzog, Andrea, Syed J. Mehdi, and Henry K. Wong. 2020. "Gene Expression Comparison between Sézary Syndrome and Lymphocytic-Variant Hypereosinophilic Syndrome Refines Biomarkers for Sézary Syndrome" Cells 9, no. 9: 1992. https://doi.org/10.3390/cells9091992