Tributyrin Attenuates Metabolic and Inflammatory Changes Associated with Obesity through a GPR109A-Dependent Mechanism

 , , , , , , , and
, , , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Experimental Design and Diets
2.3. Oxygen Consumption/Carbon Dioxide Production
2.4. Serum Analyses
2.5. Short-Chain Fatty Acid (SCFA) Measurements
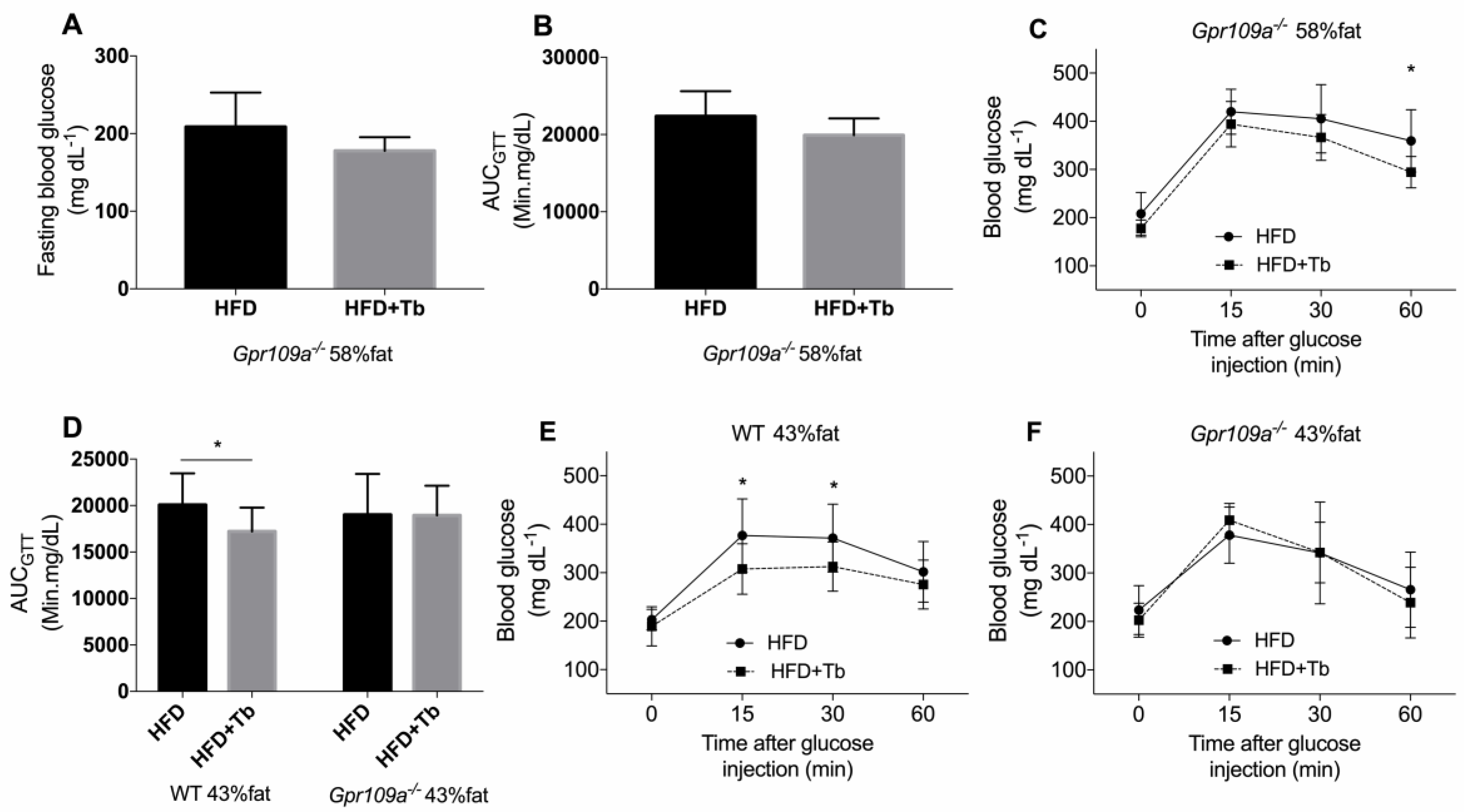
2.6. Glucose Tolerance Test (GTT)
2.7. Insulin Tolerance Test (ITT)
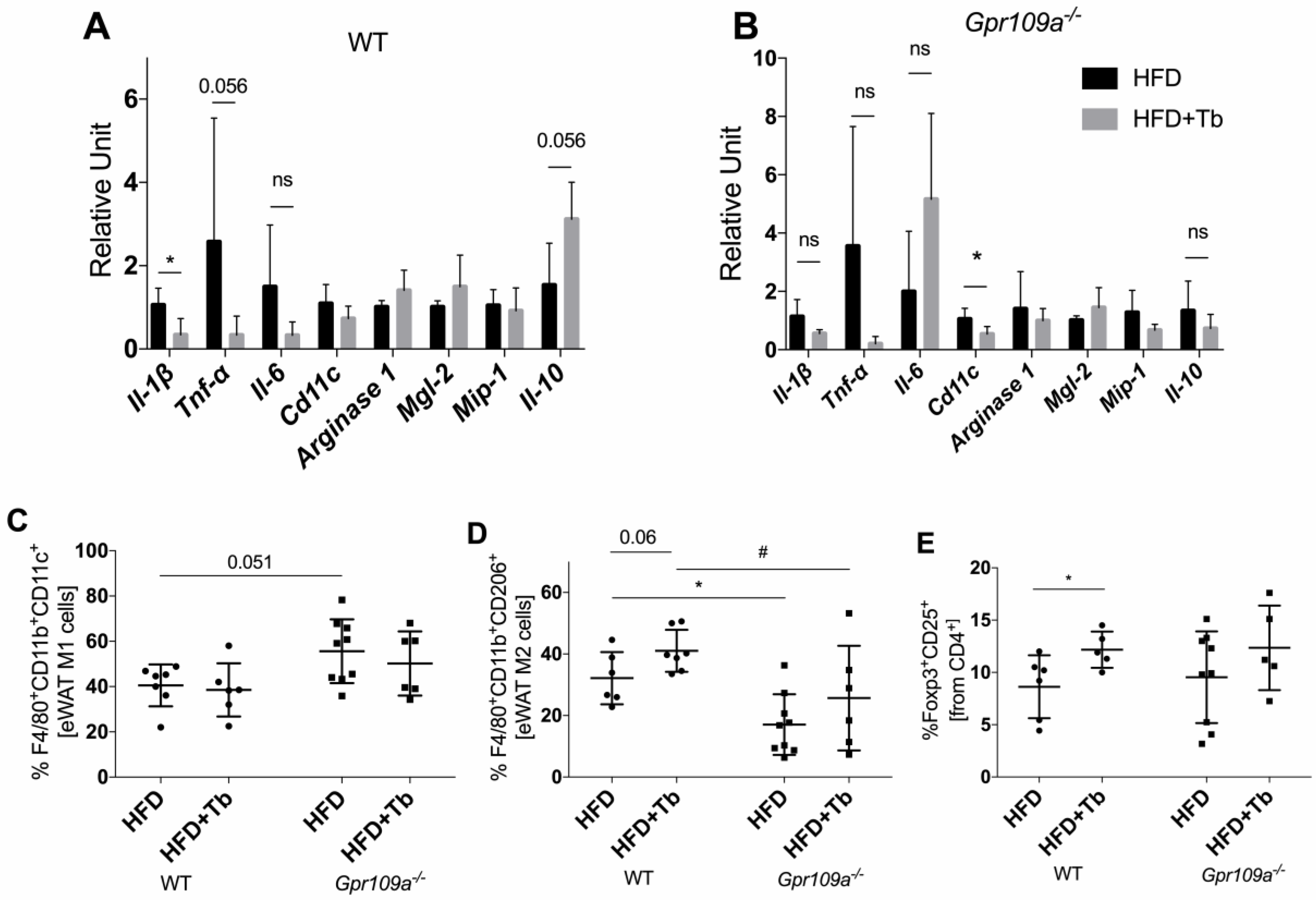
2.8. Immunophenotypic Characterization of Stromal Vascular Cells
2.9. Liver Analysis
2.10. Quantitative Reverse Transcription Polymerase Chain Reaction (RT-PCR)
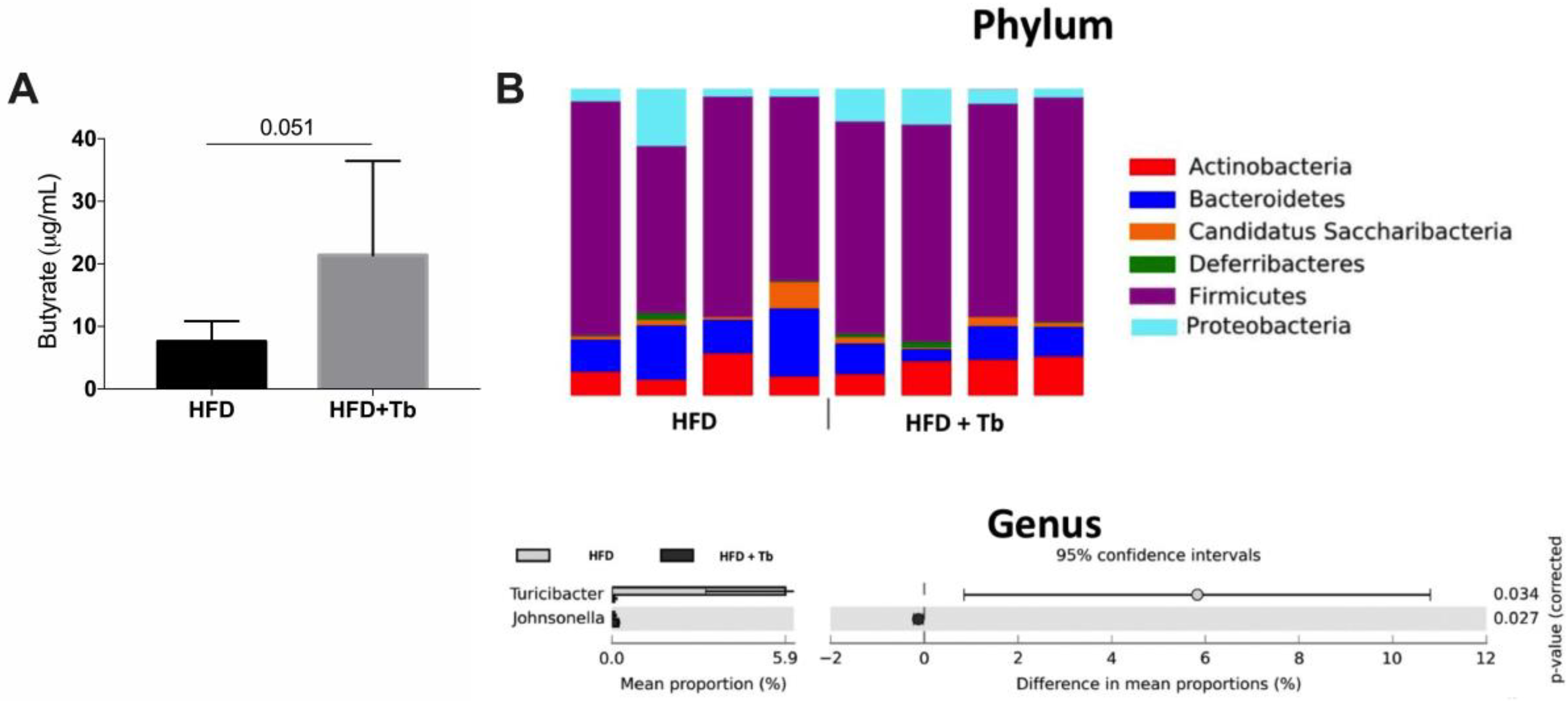
2.11. Sequencing and Bioinformatics Analysis of Fecal 16S rRNA
2.12. Measurement of Hormones in Serum Samples
2.13. Statistical Analysis
3. Results
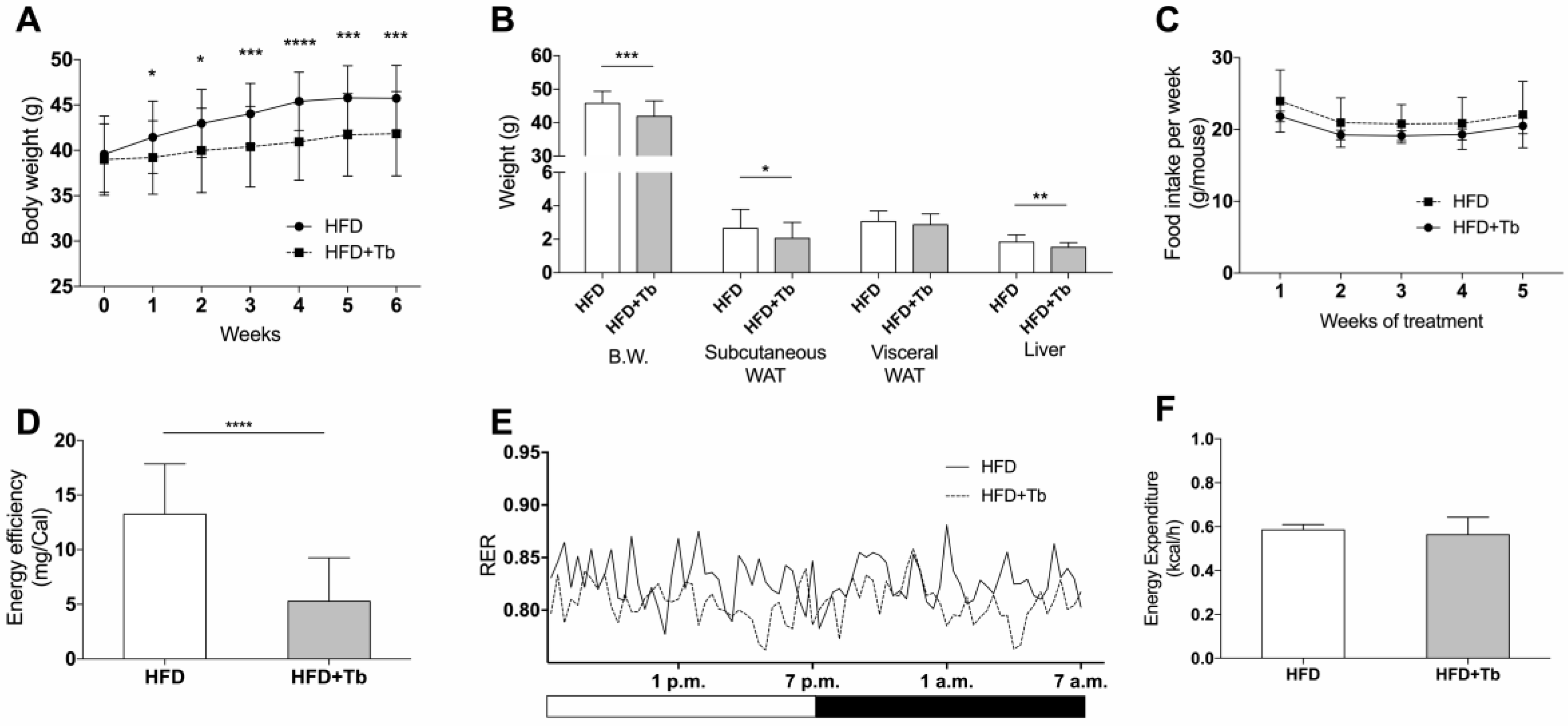
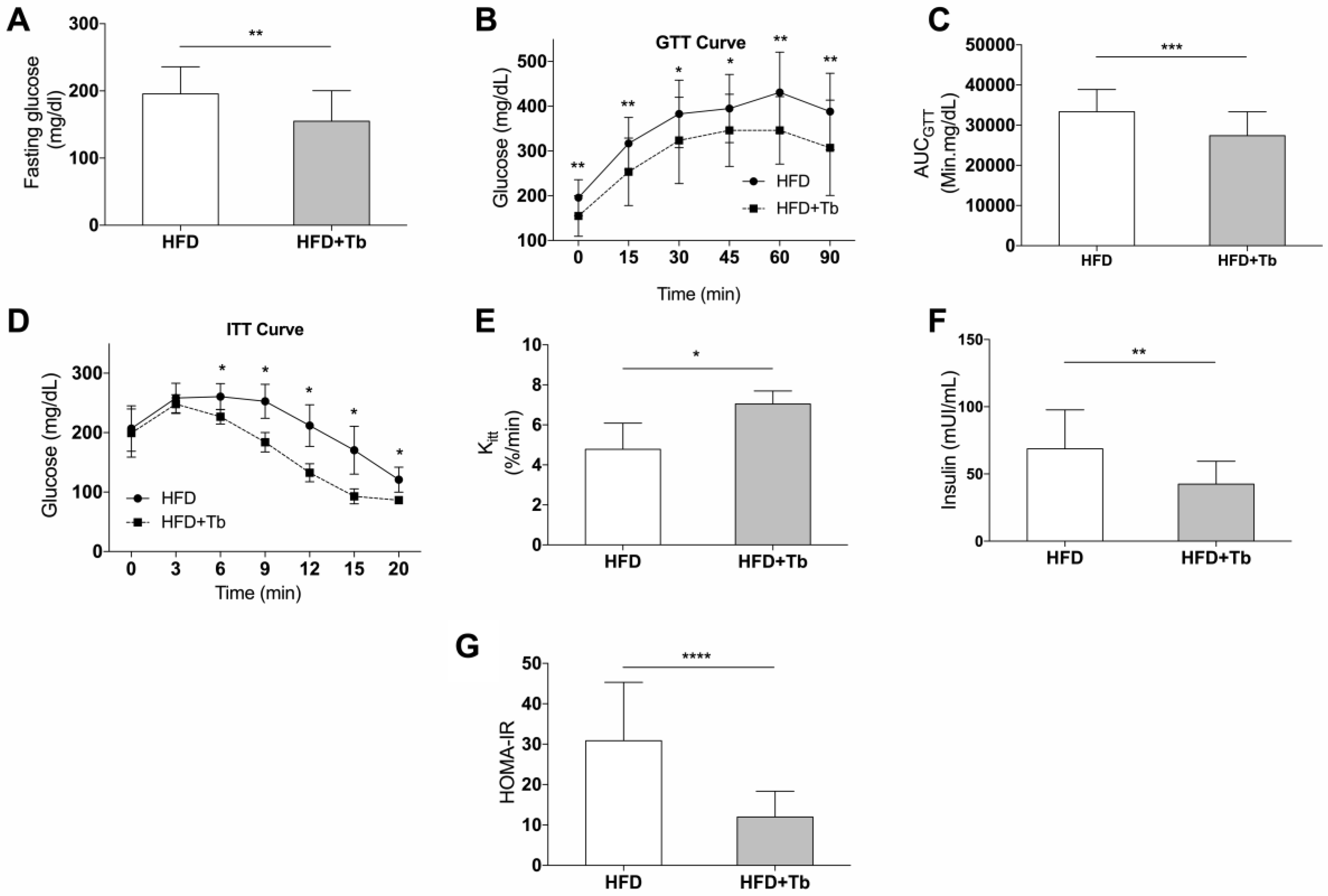
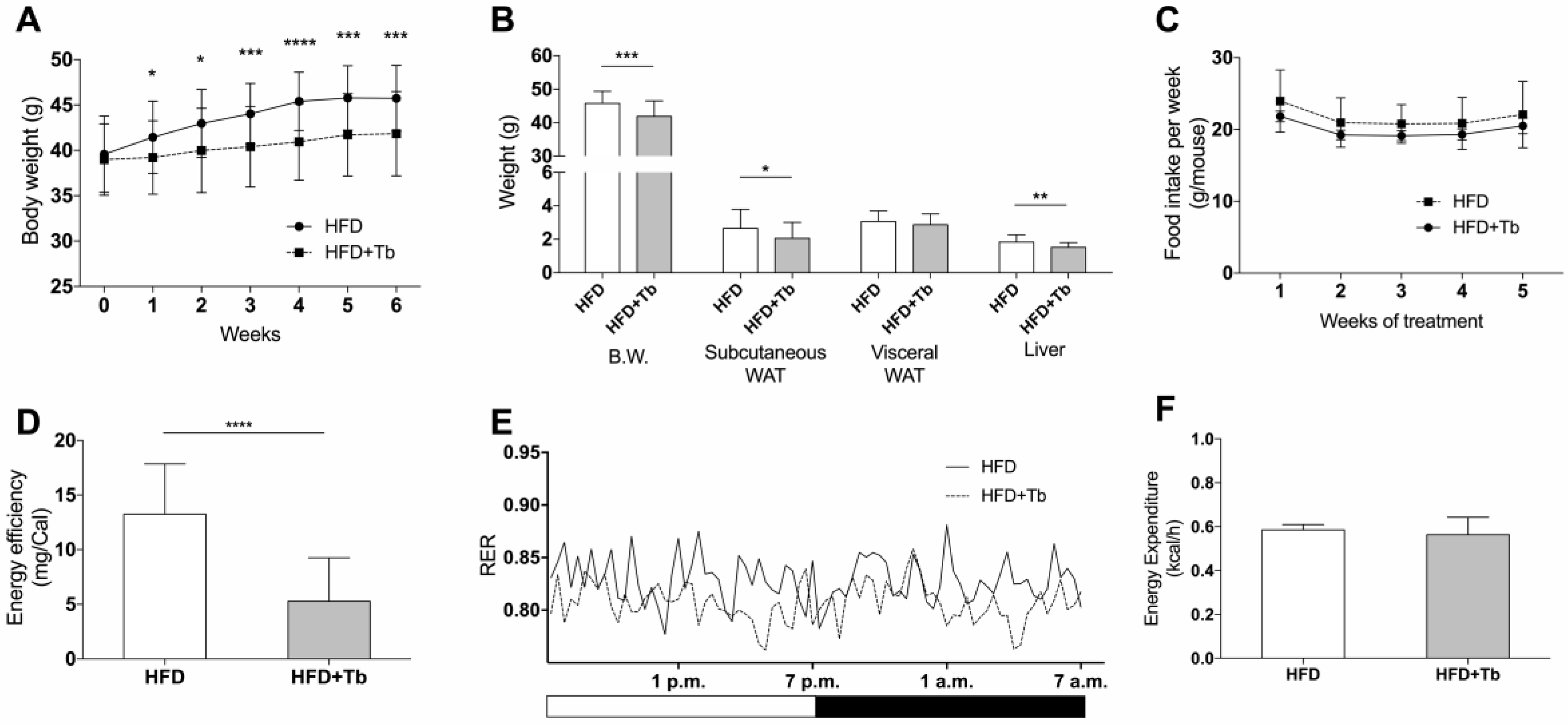
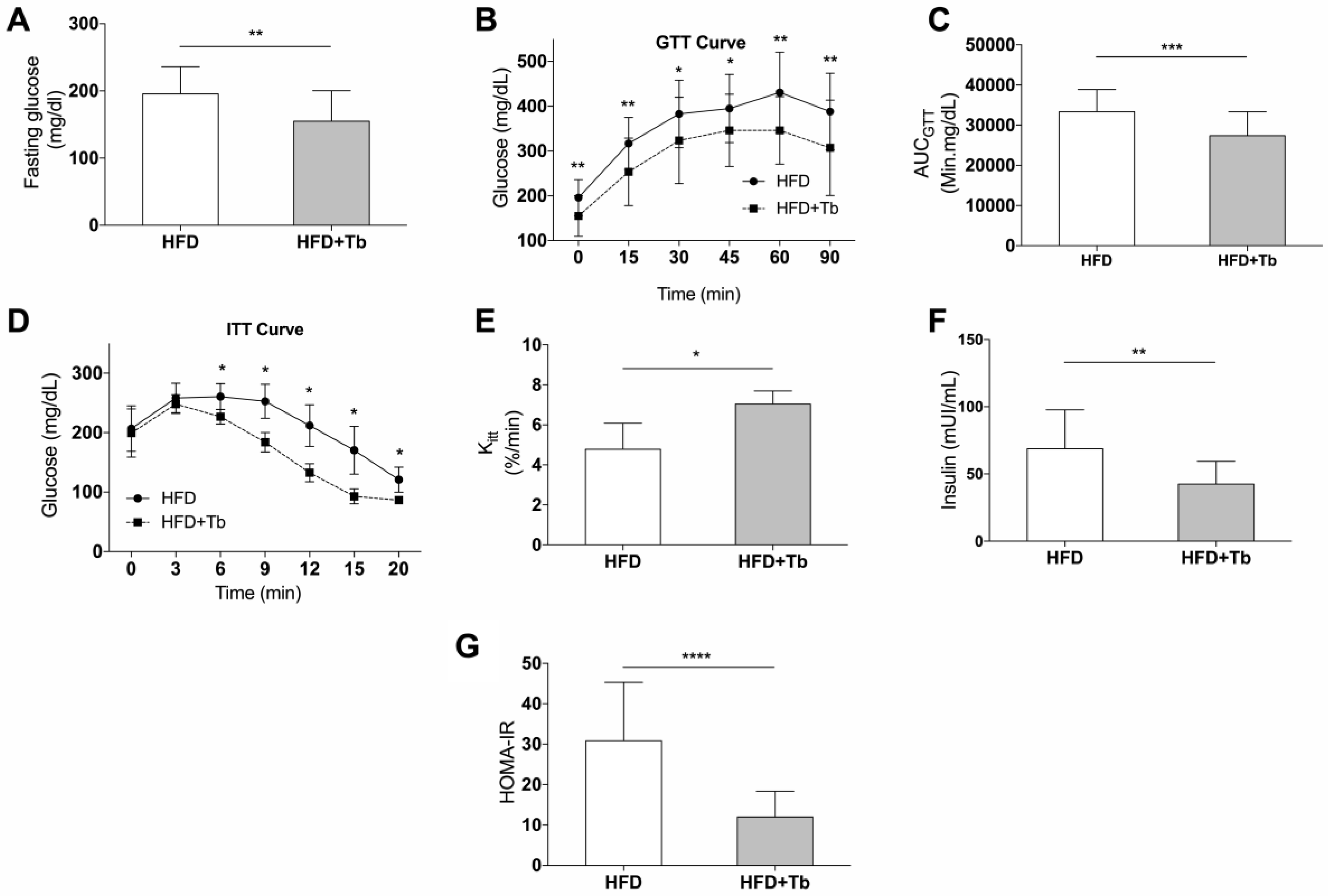
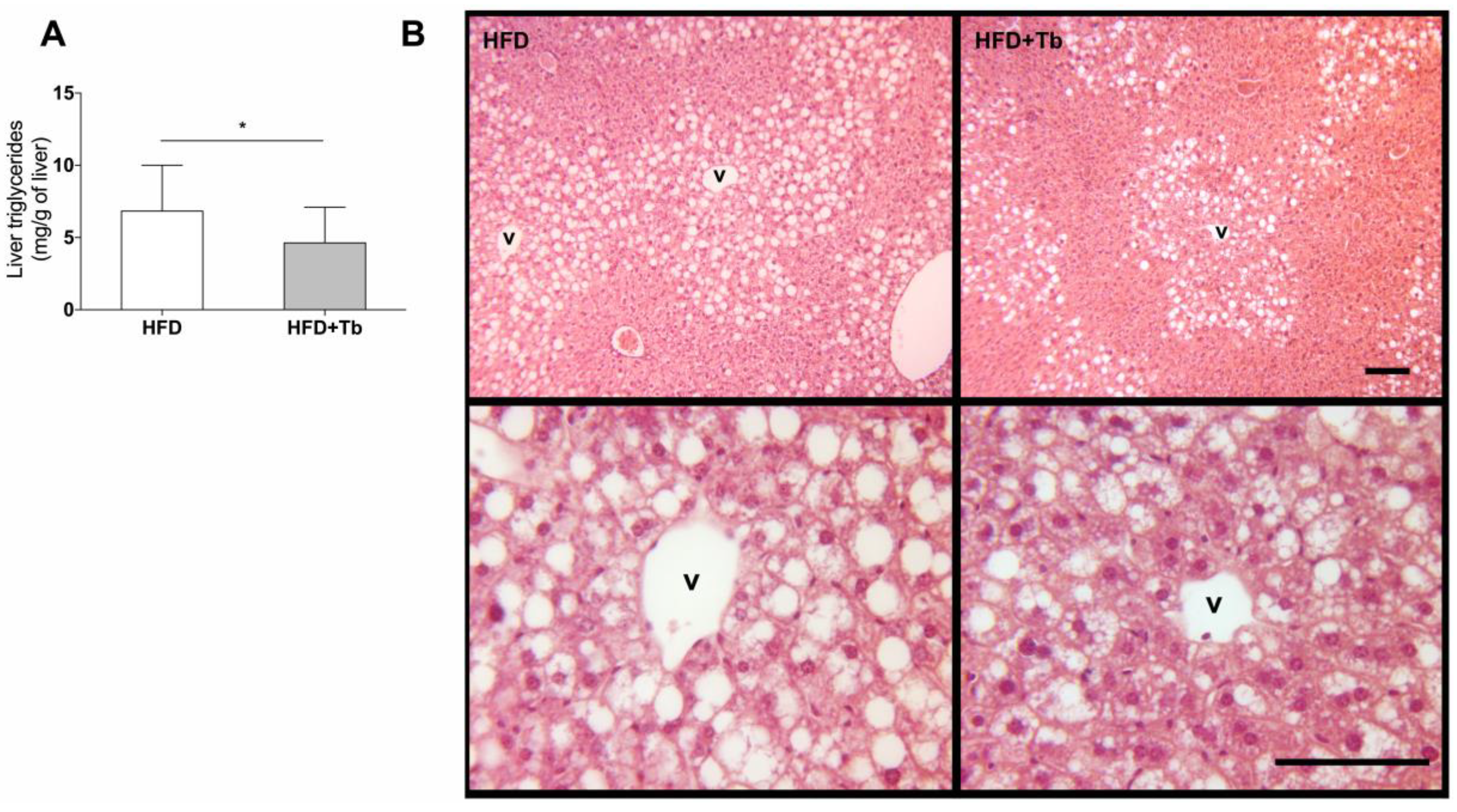
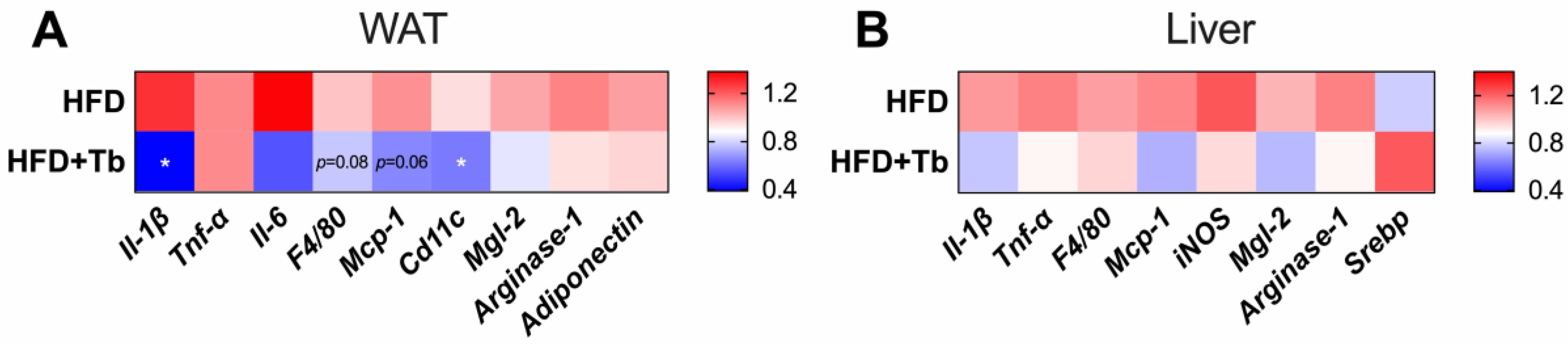
3.1. Tb Reverses the Biochemical and Metabolic Patterns Associated with Obesity
3.2. Tributyrin Increases Serum Concentrations of Butyrate Independently of the Gut Microbiota
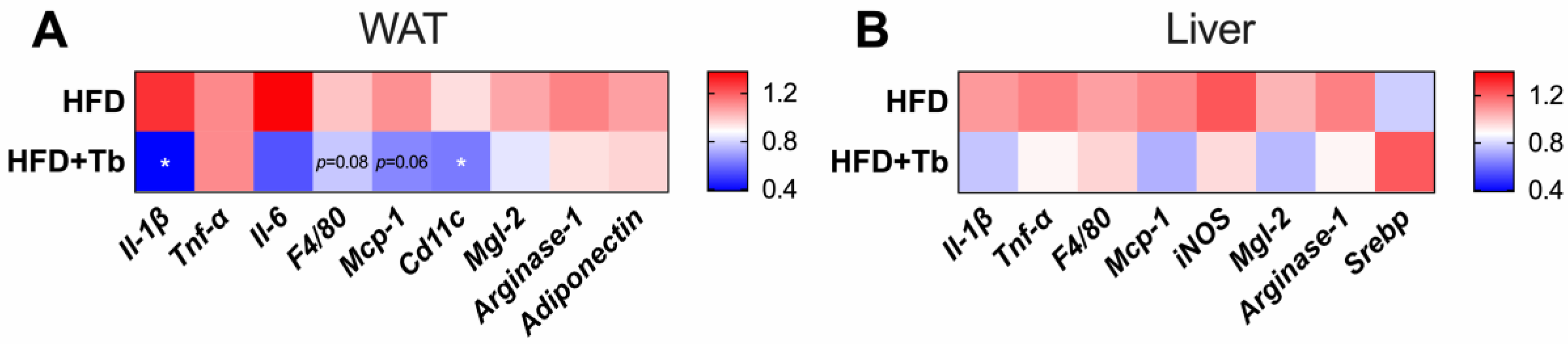
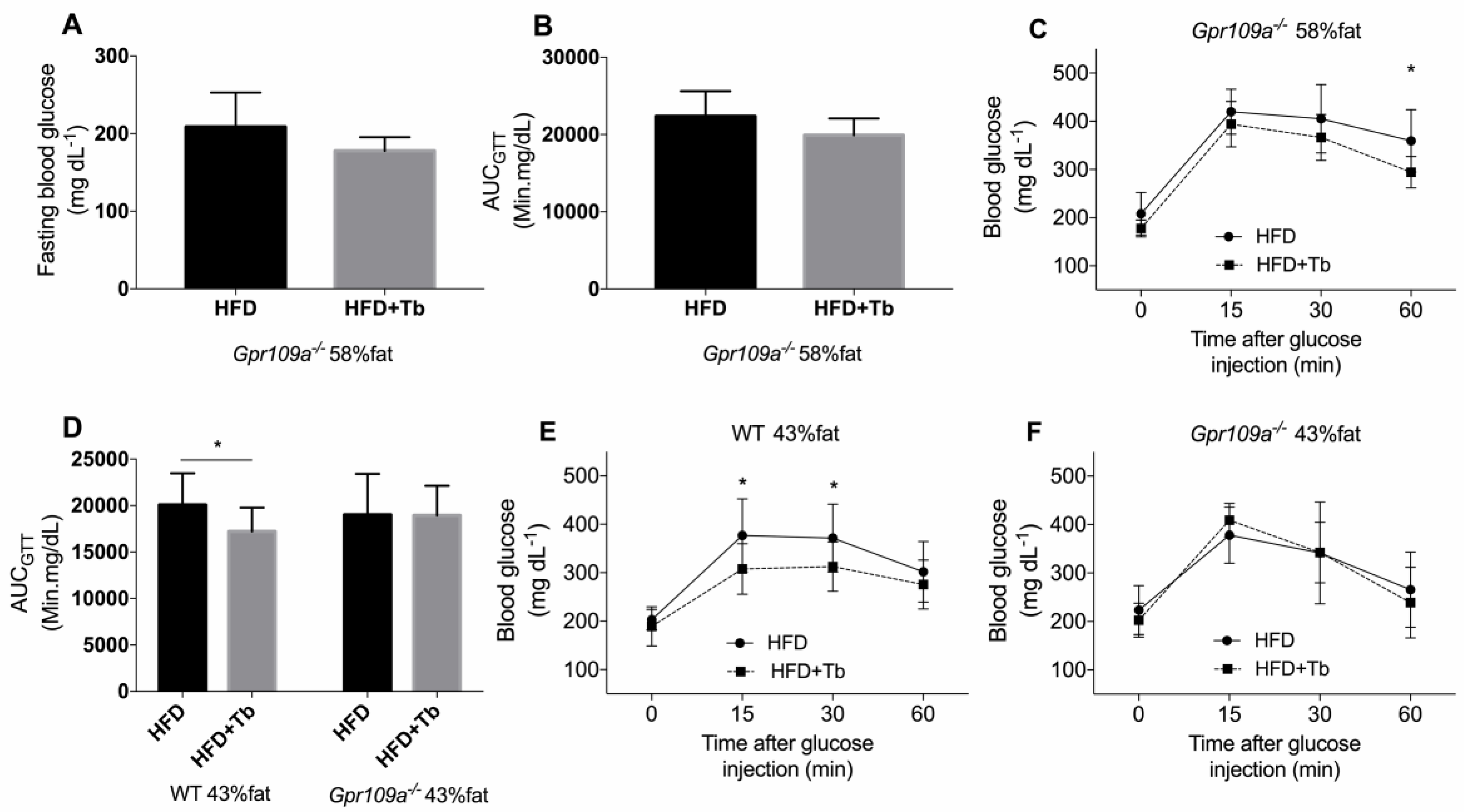
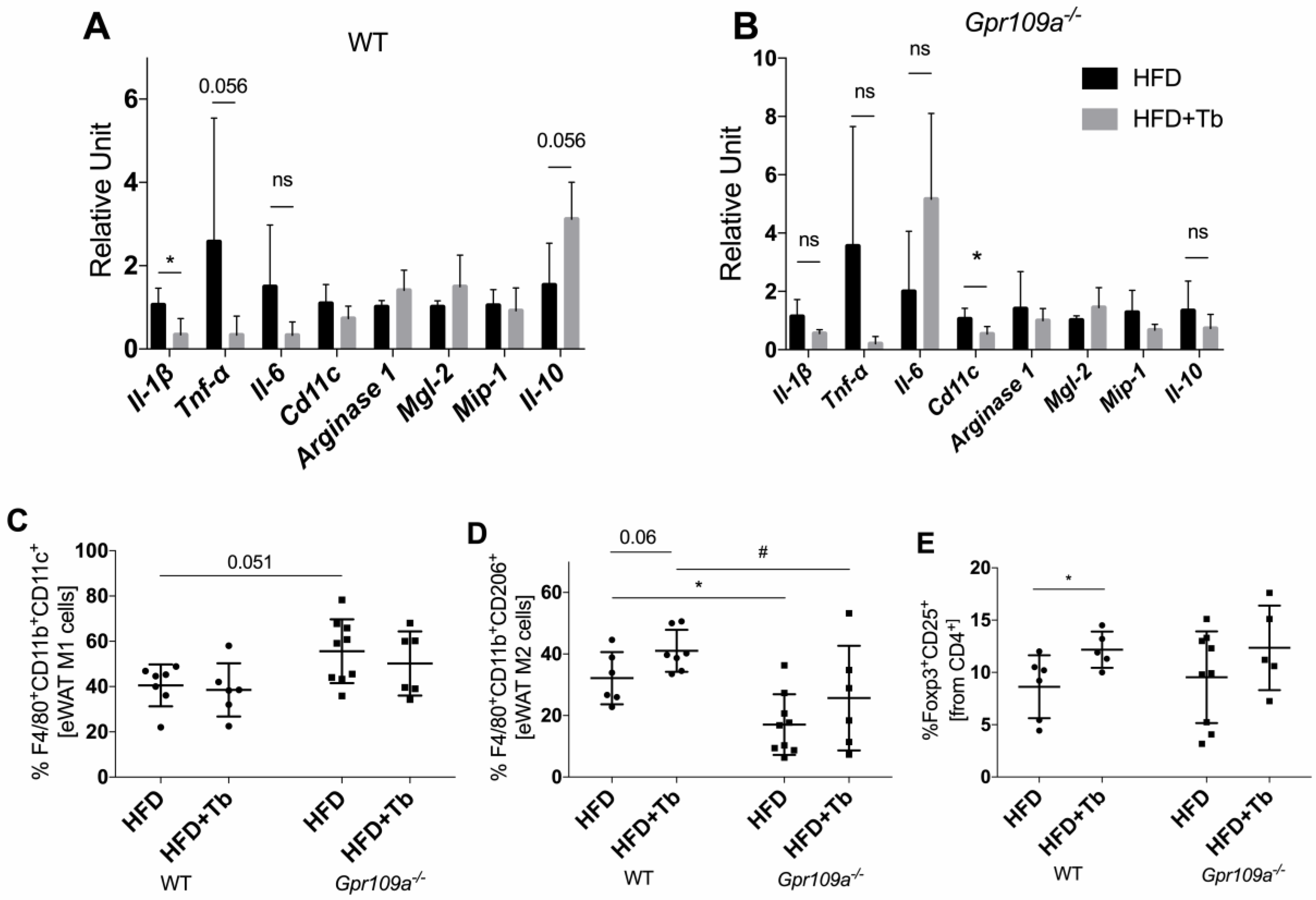
3.3. Tb-GPR109 Signaling Regulates Glucose Metabolism and Adipose Tissue Inflammation in Obese Mice
4. Discussion
5. Concluding Remarks and Perspectives
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- WHO. Fact Sheet. 2018. Available online: http://www.who.int/mediacentre/factsheets/fs311/en/ (accessed on 26 June 2020).
- NCHS. National Center for Health Statistics. Health, United States, 2016: With Chartbook on Long-Term Trends in Health. In Cent Dis Control.; 2017; pp. 314–317. Available online: https://www.cdc.gov/nchs/data/hus/hus16.pdf#019 (accessed on 26 June 2020).
- Masters, R.K.; Reither, E.N.; Powers, D.A.; Yang, Y.C.; Burger, A.E.; Link, B.G. The Impact of Obesity on US Mortality Levels: The Importance of Age and Cohort Factors in Population Estimates. Am. J. Public Health 2013, 103, 1895–1901. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Saltiel, A.R. Inflammatory Links between Obesity and Metabolic Disease. J. Clin. Investig. 2011, 121, 2111–2117. [Google Scholar] [CrossRef] [Green Version]
- Saltiel, A.R.; Olefsky, J.M. Inflammatory Mechanisms Linking Obesity and Metabolic Disease. J. Clin. Investig. 2017, 3, 1271–1274. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Bäckhed, F.; Fulton, L.; Gordon, J.I. Diet-Induced Obesity Is Linked to Marked but Reversible Alterations in the Mouse Distal Gut Microbiome. Cell Host Microbe 2008, 3, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A Core Gut Microbiome in Obese and Lean Twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [Green Version]
- Ellekilde, M.; Selfjord, E.; Larsen, C.S.; Jakesevic, M.; Rune, I.; Tranberg, B.; Vogensen, F.K.; Nielsen, D.S.; Bahl, M.I.; Licht, T.R.; et al. Transfer of Gut Microbiota from Lean and Obese Mice to Antibiotic-Treated Mice. Sci. Rep. 2014, 4, 5922. [Google Scholar] [CrossRef] [Green Version]
- Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Cheng, J.; Duncan, A.E.; Kau, A.L.; Griffin, N.W.; Lombard, V.; Henrissat, B.; Bain, J.R.; et al. Gut Microbiota from Twins Discordant for Obesity Modulate Metabolism in Mice. Science 2013, 341, 1241214. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Ma, L.; Ma, Y.; Zhang, F.; Zhao, C.; Yongzhan, N. Insights into the role of gut microbiota in obesity: Pathogenesis, mechanisms, and therapeutic perspectives. Protein Cell. 2018, 9, 397–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrêa-Oliveira, R.; Fachi, J.L.; Vieira, A.; Sato, F.T.; Vinolo, M.A. Regulation of immune cell function by short-chain fatty acids. Clin. Transl. Immunol. 2016, 5, 73. [Google Scholar] [CrossRef] [PubMed]
- Marino, E.; Richards, J.L.; McLeod, K.H.; Stanley, D.; Yap, Y.A.; Knight, J.; McKenzie, C.; Kranich, J.; Oliveira, A.C.; Rossello, F.J.; et al. Gut microbial metabolites limit the frequency of autoimmune T cells and protect against type 1 diabetes. Nat. Immunol. 2017, 18, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Felizardo, R.J.F.; de Almeida, D.C.; Pereira, R.L.; Watanabe, I.K.M.; Doimo, N.T.S.; Ribeiro, W.R.; Cenedeze, M.A.; Hiyane, M.I.; Amano, M.T.; Braga, T.T.; et al. Gut microbial metabolite butyrate protects against proteinuric kidney disease through epigenetic—And GPR109a-mediated mechanisms. FASEB J. 2019, 33, 11894–11908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques, F.Z.; Nelson, E.; Chu, P.Y.; Horlock, D.; Fiedler, A.; Ziemann, M.; Tan, J.K.; Kuruppu, S.; Rajapakse, N.W.; El-Osta, A.; et al. High-Fiber Diet and Acetate Supplementation Change the Gut Microbiota and Prevent the Development of Hypertension and Heart Failure in Hypertensive Mice. Circulation 2017, 135, 964–977. [Google Scholar] [CrossRef] [PubMed]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Yu, D.; Schilter, H.C.; Rolph, M.S.; Mackay, F.; Artis, D.; et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009, 461, 1282–1286. [Google Scholar] [CrossRef] [PubMed]
- Macia, L.; Tan, J.; Vieira, A.T.; Leach, K.; Stanley, D.; Luong, S.; Maruya, M.; Ian McKenzie, C.; Hijikata, A.; Wong, C.; et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat. Commun. 2015, 6, 6734. [Google Scholar] [CrossRef] [Green Version]
- Tan, J.; McKenzie, C.; Vuillermin, P.J.; Goverse, G.; Vinuesa, C.G.; Mebius, R.E.; Macia, L.; Mackay, C.R. Dietary Fiber and Bacterial SCFA Enhance Oral Tolerance and Protect against Food Allergy through Diverse Cellular Pathways. Cell Rep. 2016, 15, 2809–2824. [Google Scholar] [CrossRef] [Green Version]
- Thorburn, A.N.; McKenzie, C.I.; Shen, S.; Stanley, D.; MacIa, L.; Mason, L.J.; Roberts, L.K.; Wong, C.H.Y.; Shim, R.; Robert, R.; et al. Evidence that asthma is a developmental origin disease influenced by maternal diet and bacterial metabolites. Nat. Commun. 2015, 6, 1–13. [Google Scholar] [CrossRef]
- Fachi, J.L.; Sécca, C.; Rodrigues, P.B.; de Mato, F.C.P.; Di Luccia, B.; Felipe, J.D.S.; Pral, L.P.; Rungue, M.; Rocha, V.D.M.; Sato, F.T.; et al. Acetate coordinates neutrophil and ILC3 responses against C. difficile through FFAR2. J. Exp. Med. 2020, 217, e20190489. [Google Scholar] [CrossRef]
- Zhao, Z.H.; Lai, J.K.L.; Qiao, L.; Fan, J.G. Role of gut microbial metabolites in nonalcoholic fatty liver disease. J. Dig. Dis. 2019, 20, 181–188. [Google Scholar] [CrossRef]
- Endo, H.; Niioka, M.; Kobayashi, N.; Tanaka, M.; Watanabe, T. Butyrate-Producing Probiotics Reduce Nonalcoholic Fatty Liver Disease Progression in Rats: New Insight into the Probiotics for the Gut-Liver Axis. PLoS ONE 2013, 8, e63388. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Lou, H.; Peng, Y.; Chen, S.; Zhang, Y.; Li, X. Comprehensive relationships between gut microbiome and faecal metabolome in individuals with type 2 diabetes and its complications. Endocrine 2019, 66, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Yanagi, K.; Cheng, C.; Alaniz, R.C.; Lee, K.; Jayaraman, A. Interactions between gut microbiota and non-alcoholic liver disease: The role of microbiota-derived metabolites. Pharmacol. Res. 2019, 141, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Den Besten, G.; Bleeker, A.; Gerding, A.; Van Eunen, K.; Havinga, R.; Van Dijk, T.H.; Oosterveer, M.H.; Jonker, J.W.; Groen, A.K.; Reijngoud, D.J.; et al. Short-chain fatty acids protect against high-fat diet-induced obesity via a pparg-dependent switch from lipogenesis to fat oxidation. Diabetes 2015, 64, 2398–2408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Fan, C.; Li, P.; Lu, Y.; Chang, X.; Qi, K. Short chain fatty acids prevent high-fat-diet-induced obesity in mice by regulating g protein-coupled receptors and gut Microbiota. Sci. Rep. 2016, 6, 37589. [Google Scholar] [CrossRef] [Green Version]
- Canfora, E.E.; Jocken, J.W.; Blaak, E.E. Short-chain fatty acids in control of body weight and insulin sensitivity. Nat. Rev. Endocrinol. 2015, 11, 577–591. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Yin, J.; Zhang, J.; Ward, R.E.; Martin, R.J.; Lefevre, M.; Cefalu, W.T.; Ye, J. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 2009, 58, 1509–1517. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.; Mach, N. The crosstalk between the gut microbiota and mitochondria during exercise. Front. Physiol. 2017, 8, 319. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, F.; Ding, X.; Wu, G.; Lam, Y.Y.; Wang, X.; Fu, H.; Xue, X.; Lu, C.; Ma, J.; et al. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science 2018, 359, 1151–1156. [Google Scholar] [CrossRef] [Green Version]
- De Groot, P.F.; Belzer, C.; Aydin, Ö.; Levin, E.; Levels, J.H.; Aalvink, S.; Boot, F.; Holleman, F.; Van Raalte, D.H.; Scheithauer, T.P.; et al. Distinct fecal and oral microbiota composition in human type 1 diabetes, an observational study. PLoS ONE 2017, 12, e0188475. [Google Scholar] [CrossRef]
- Huang, J.; Pearson, J.A.; Peng, J.; Hu, Y.; Sha, S.; Xing, Y.; Huang, G.; Li, X.; Hu, F.; Xie, Z.; et al. Gut microbial metabolites alter IgA immunity in type 1 diabetes. JCI Insight 2020, 5, e135718. [Google Scholar] [CrossRef]
- Galisteo, M.; Duarte, J.; Zarzuelo, A. Effects of Dietary Fibers on Disturbances Clustered in the Metabolic Syndrome. J. Nutr. Biochem. 2008, 19, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, A.C.; Ostman, E.M.; Knudsen, K.E.B.; Holst, J.J.; Bjorck, I.M.E. A Cereal-Based Evening Meal Rich in Indigestible Carbohydrates Increases Plasma Butyrate the Next Morning. J. Nutr. 2010, 140, 1932–1936. [Google Scholar] [CrossRef] [PubMed]
- Tarini, J.; Wolever, T.M.S. The Fermentable Fibre Inulin Increases Postprandial Serum Short-Chain Fatty Acids and Reduces Free-Fatty Acids and Ghrelin in Healthy Subjects. Appl. Physiol. Nutr. Metab. 2010, 35, 9–16. [Google Scholar] [CrossRef]
- Lin, H.V.; Frassetto, A.; Kowalik, E.J.; Nawrocki, A.R.; Lu, M.M.; Kosinski, J.R.; Hubert, J.A.; Szeto, D.; Yao, X.; Forrest, G.; et al. Butyrate and propionate protect against diet-induced obesity and regulate gut hormones via free fatty acid receptor 3-independent mechanisms. PLoS ONE 2012, 7, e35240. [Google Scholar] [CrossRef]
- Li, Z.; Yi, C.X.; Katiraei, S.; Kooijman, S.; Zhou, E.; Chung, C.K.; Gao, Y.; Van Den Heuvel, J.K.; Meijer, O.C.; Berbée, J.F.P.; et al. Butyrate reduces appetite and activates brown adipose tissue via the gut-brain neural circuit. Gut 2018, 67, 1269–1279. [Google Scholar] [CrossRef] [Green Version]
- Fang, W.; Xue, H.; Chen, X.; Chen, K.; Ling, W. Supplementation with Sodium Butyrate Modulates the Composition of the Gut Microbiota and Ameliorates High-Fat Diet-Induced Obesity in Mice. J. Nutr. 2019, 149, 747–754. [Google Scholar] [CrossRef]
- Vinolo, M.A.R.; Rodrigues, H.G.; Festuccia, W.T.; Crisma, A.R.; Alves, V.S.; Martins, A.R.; Amaral, C.L.; Fiamoncini, J.; Hirabara, S.M.; Sato, F.T.; et al. Tributyrin attenuates obesity-associated inflammation and insulin resistance in high-fat-fed mice. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E272–E282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egorin, M.J.; Yuan, Z.M.; Sentz, D.L.; Plaisance, K.; Eiseman, J.L. Plasma pharmacokinetics of butyrate after intravenous administration of sodium butyrate or oral administration of tributyrin or sodium butyrate to mice and rats. Cancer Chemother. Pharmacol. 1999, 43, 445–453. [Google Scholar] [CrossRef]
- Tunaru, S.; Kero, J.; Schaub, A.; Wufka, C.; Blaukat, A.; Pfeffer, K.; Offermanns, S. PUMA-G and HM74 are receptors for nicotinic acid and mediate its anti-lipolytic effect. Nat. Med. 2003, 9, 352–355. [Google Scholar] [CrossRef]
- Masi, L.N.; Crisma, A.R.; Martins, A.R.; do Amaral, C.L.; Torres, R.P.; Mancini Filho, J.; Hirabara, S.M.; Curi, R. Inflammatory state of periaortic adipose tissue in mice under obesogenic dietary regimens. J. Nutr. Intermed. Metab. 2016, 6, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, research0034.1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmieder, R.; Edwards, R. Quality Control and Preprocessing of Metagenomic Datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly Accurate OTU Sequences from Microbial Amplicon Reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and Clustering Orders of Magnitude Faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Pylro, V.S.; Roesch, L.F.W.; Morais, D.K.; Clark, I.M.; Hirsch, P.R.; Tótola, M.R. Data Analysis for 16S Microbial Profiling from Different Benchtop Sequencing Platforms. J. Microbiol. Methods 2014, 107, 30–37. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. Correspondence QIIME Allows Analysis of High- Throughput Community Sequencing Data Intensity Normalization Improves Color Calling in SOLiD Sequencing. Nat. Publ. Gr. 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian Classifier for Rapid Assignment of RRNA Sequences into the New Bacterial Taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [Green Version]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical Analysis of Taxonomic and Functional Profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [Green Version]
- Speakman, J.R. Use of high-fat diets to study rodent obesity as a model of human obesity. Int. J. Obes. 2019, 43, 1491–1492. [Google Scholar] [CrossRef] [Green Version]
- Murphy, E.F.; Cotter, P.D.; Healy, S.; Marques, T.M.; O’Sullivan, O.; Fouhy, F.; Clarke, S.F.; O’Toole, P.W.; Quigley, E.M.; Stanton, C.; et al. Composition and energy harvesting capacity of the gut microbiota: Relationship to diet, obesity and time in mouse models. Gut 2010, 59, 1635–1642. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.T.; Nieuwdorp, M.; Bäckhed, F. Microbial modulation of insulin sensitivity. Cell Metab. 2014, 20, 753–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaiss, C.A.; Itav, S.; Rothschild, D.; Meijer, M.T.; Levy, M.; Moresi, C.; Dohnalová, L.; Braverman, S.; Rozin, S.; Malitsky, S.; et al. Persistent microbiome alterations modulate the rate of post-dieting weight regain. Nature 2016, 540, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Fragiadakis, G.K.; Wastyk, H.C.; Robinson, J.L.; Sonnenburg, E.D.; Sonnenburg, J.L.; Gardner, C.D. Long-term dietary intervention reveals resilience of the gut microbiota despite changes in diet and weight. Am. J. Clin. Nutr. 2020, 111, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- Cresci, G.A.; Glueck, B.; McMullen, M.R.; Xin, W.; Allende, D.; Nagy, L.E. Prophylactic tributyrin treatment mitigates chronic-binge ethanol-induced intestinal barrier and liver injury. J. Gastroenterol. Hepatol. 2017, 32, 1587–1597. [Google Scholar] [CrossRef]
- Donde, H.; Ghare, S.; Joshi-Barve, S.; Zhang, J.W.; Vadhanam, M.V.; Gobejishvili, L.; Lorkiewicz, P.; Srivastava, S.; McClain, C.J.; Barve, S. Tributyrin Inhibits Ethanol-Induced Epigenetic Repression of CPT-1A and Attenuates Hepatic Steatosis and Injury. CMGH 2020, 9, 569–585. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, M.; Sakaki, H.; Usami, M.; Iizuka, N.; Shuno, K.; Aoyama, M.; Usami, Y. Oral administration of tributyrin increases concentration of butyrate in the portal vein and prevents lipopolysaccharide-induced liver injury in rats. Clin. Nutr. 2011, 30, 252–258. [Google Scholar] [CrossRef]
- Liu, J.; Han, L.; Zhu, L.; Yu, Y. Free Fatty Acids, Not Triglycerides, Are Associated with Non-Alcoholic Liver Injury Progression in High Fat Diet Induced Obese Rats. Lipids Health Dis. 2016, 15, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Ge, H.; Li, X.; Weiszmann, J.; Wang, P.; Baribault, H.; Chen, J.-L.; Tian, H.; Li, Y. Activation of G Protein-Coupled Receptor 43 in Adipocytes Leads to Inhibition of Lipolysis and Suppression of Plasma Free Fatty Acids. Endocrinology 2008, 149, 4519–4526. [Google Scholar] [CrossRef] [Green Version]
- Ohira, H.; Tsutsui, W.; Mamoto, R.; Yamaguchi, S.; Nishida, M.; Ito, M.; Fujioka, Y. Butyrate Attenuates Lipolysis in Adipocytes Co-Cultured with Macrophages through Non-Prostaglandin E2–Mediated and Prostaglandin E2–Mediated Pathways. Lipids Health Dis. 2016, 15, 213. [Google Scholar] [CrossRef] [Green Version]
- Mollica, M.P.; Raso, G.M.; Cavaliere, G.; Trinchese, G.; De Filippo, C.; Aceto, S.; Prisco, M.; Pirozzi, C.; Di Guida, F.; Lama, A.; et al. Butyrate Regulates Liver Mitochondrial Function, Efficiency, and Dynamic, in Insulin Resistant Obese Mice. Diabetes 2017, 66, 1405–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feingold, K.R.; Moser, A.; Shigenaga, J.K.; Grunfeld, C. Inflammation Stimulates Niacin Receptor (GPR109A/HCA2) Expression in Adipose Tissue and Macrophages. J. Lipid Res. 2014, 55, 2501–2508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, N.; Gurav, A.; Sivaprakasam, S.; Brady, E.; Padia, R.; Shi, H.; Thangaraju, M.; Prasad, P.D.; Manicassamy, S.; Munn, D.H.; et al. Activation of Gpr109a, Receptor for Niacin and the Commensal Metabolite Butyrate, Suppresses Colonic Inflammation and Carcinogenesis. Immunity 2014, 40, 128–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobbins, R.L.; Shearn, S.P.; Byerly, R.L.; Gao, F.F.; Mahar, K.M.; Napolitano, A.; Nachbaur, G.J.; Le Monnier de Gouville, A.C. GSK256073, a Selective Agonist of G-Protein Coupled Receptor 109A (GPR109A) Reduces Serum Glucose in Subjects with Type 2 Diabetes Mellitus. Diabetes Obes. Metab. 2013, 15, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Cao, Z.; Lai, X.; Wang, W.; Guo, Z.; Yan, L.; Wang, Y.; Shi, Y.; Zhou, N. Niacin fine-tunes energy homeostasis through canonical GPR109A signaling. FASEB J. 2019, 33, 4765–4779. [Google Scholar] [CrossRef] [PubMed]
- Feuerer, M.; Herrero, L.; Cipolletta, D.; Naaz, A.; Wong, J.; Nayer, A.; Lee, J.; Goldfine, A.B.; Benoist, C.; Shoelson, S.; et al. Lean, but Not Obese, Fat Is Enriched for a Unique Population of Regulatory T Cells That Affect Metabolic Parameters. Nat. Med. 2009, 15, 930–939. [Google Scholar] [CrossRef]
- Zeng, H.; Chi, H. Metabolic Control of Regulatory T Cell Development and Function. Trends Immunol. 2015, 36, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; Van Der Veeken, J.; deRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef]
- Edelman, M.J.; Bauer, K.; Khanwani, S.; Tait, N.; Trepel, J.; Karp, J.; Nemieboka, N.; Chung, E.J.; Van Echo, D. Clinical and pharmacologic study of tributyrin: An oral butyrate prodrug. Cancer Chemother. Pharmacol. 2003, 51, 439–444. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HFD | HFD+Tb | |
|---|---|---|
| TAG (mg/dL) | 64.4 ± 15.3 (n = 28) | 54.7 ± 15.8 * (n = 30) |
| Cholesterol (mg/dL) | 196.0 ± 33.9 (n = 25) | 197.0 ± 36.3 (n = 26) |
| LDL (mg/dL) | 48.4 ± 12.8 (n = 17) | 48.1 ± 14.5 (n = 18) |
| HDL (mg/dL) | 41.9 ± 8.5 (n = 16) | 41.8 ± 6.2 (n = 16) |
| NEFA (mM) | 1.160 ± 0.221 (n = 16) | 0.900 ± 0.224 * (n = 17) |
| AST (U/mL) | 17.19 ± 8.05 (n = 8) | 16.53 ± 6.30 (n = 9) |
| ALT (U/mL) | 11.77 ± 3.04 (n = 7) | 7.85 ± 2.55 * (n = 9) |
| WT | Gpr109a−/− | |||
|---|---|---|---|---|
| HFD (n = 12, 13) | HFD+Tb (n = 13, 14) | HFD (n = 8, 9) | HFD+Tb (n = 9) | |
| TAG (mg/dL) | 47.8 ± 11.3 | 55.1 ± 15.4 | 101.4 ± 45.1 | 64.3 ± 23.7 * |
| Cholesterol (mg/dL) | 125.3 ± 56.2 | 150.7 ± 59.8 | 177.5 ± 19.5 | 144.6 ± 42.8 |
| LDL (mg/dL) | 86.9 ± 43.0 | 105.5 ± 48.0 | 117.6 ± 12.9 | 100.2 ± 31.5 |
| HDL (mg/dL) | 28.1 ± 13.3 | 34.2 ± 13.9 | 36.1 ± 10.8 | 31.7 ± 14.5 |
| NEFA (mM) | 0.54 ± 0.12 | 0.40 ± 0.07 *** | 0.56 ± 0.10 | 0.46 ± 0.10 * |
| AST (U/mL) | 12.29 ± 2.13 | 12.71 ± 1.73 | 12.81 ± 2.10 | 11.83 ± 2.02 |
| ALT (U/mL) | 8.20 ± 1.48 | 8.21 ± 1.18 | 10.11 ± 1.94 | 10.04 ± 1.63 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sato, F.T.; Yap, Y.A.; Crisma, A.R.; Portovedo, M.; Murata, G.M.; Hirabara, S.M.; Ribeiro, W.R.; Marcantonio Ferreira, C.; Cruz, M.M.; Pereira, J.N.B.; et al. Tributyrin Attenuates Metabolic and Inflammatory Changes Associated with Obesity through a GPR109A-Dependent Mechanism. Cells 2020, 9, 2007. https://doi.org/10.3390/cells9092007
Sato FT, Yap YA, Crisma AR, Portovedo M, Murata GM, Hirabara SM, Ribeiro WR, Marcantonio Ferreira C, Cruz MM, Pereira JNB, et al. Tributyrin Attenuates Metabolic and Inflammatory Changes Associated with Obesity through a GPR109A-Dependent Mechanism. Cells. 2020; 9(9):2007. https://doi.org/10.3390/cells9092007
Chicago/Turabian StyleSato, Fabio Takeo, Yu Anne Yap, Amanda Rabello Crisma, Mariana Portovedo, Gilson Masahiro Murata, Sandro Massao Hirabara, Willian Rodrigues Ribeiro, Caroline Marcantonio Ferreira, Maysa Mariana Cruz, Joice Naiara Bertaglia Pereira, and et al. 2020. "Tributyrin Attenuates Metabolic and Inflammatory Changes Associated with Obesity through a GPR109A-Dependent Mechanism" Cells 9, no. 9: 2007. https://doi.org/10.3390/cells9092007
APA StyleSato, F. T., Yap, Y. A., Crisma, A. R., Portovedo, M., Murata, G. M., Hirabara, S. M., Ribeiro, W. R., Marcantonio Ferreira, C., Cruz, M. M., Pereira, J. N. B., Payolla, T. B., Guima, S. E. S., Thomas, A. M., Setubal, J. C., Alonso-Vale, M. I. C., Santos, M. F., Curi, R., Marino, E., & Vinolo, M. A. R. (2020). Tributyrin Attenuates Metabolic and Inflammatory Changes Associated with Obesity through a GPR109A-Dependent Mechanism. Cells, 9(9), 2007. https://doi.org/10.3390/cells9092007