Eosinophils and Neutrophils—Molecular Differences Revealed by Spontaneous Raman, CARS and Fluorescence Microscopy

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Isolation of Eosinophils and Neutrophils from Blood
2.3. Spectroscopic Measurements
2.3.1. UV−Vis Absorption Spectroscopy
2.3.2. Raman Spectroscopy
2.3.3. Fluorescence Microscopy
2.3.4. Coherent Anti-Stokes Raman Scattering Microscopy (CARS)
3. Results
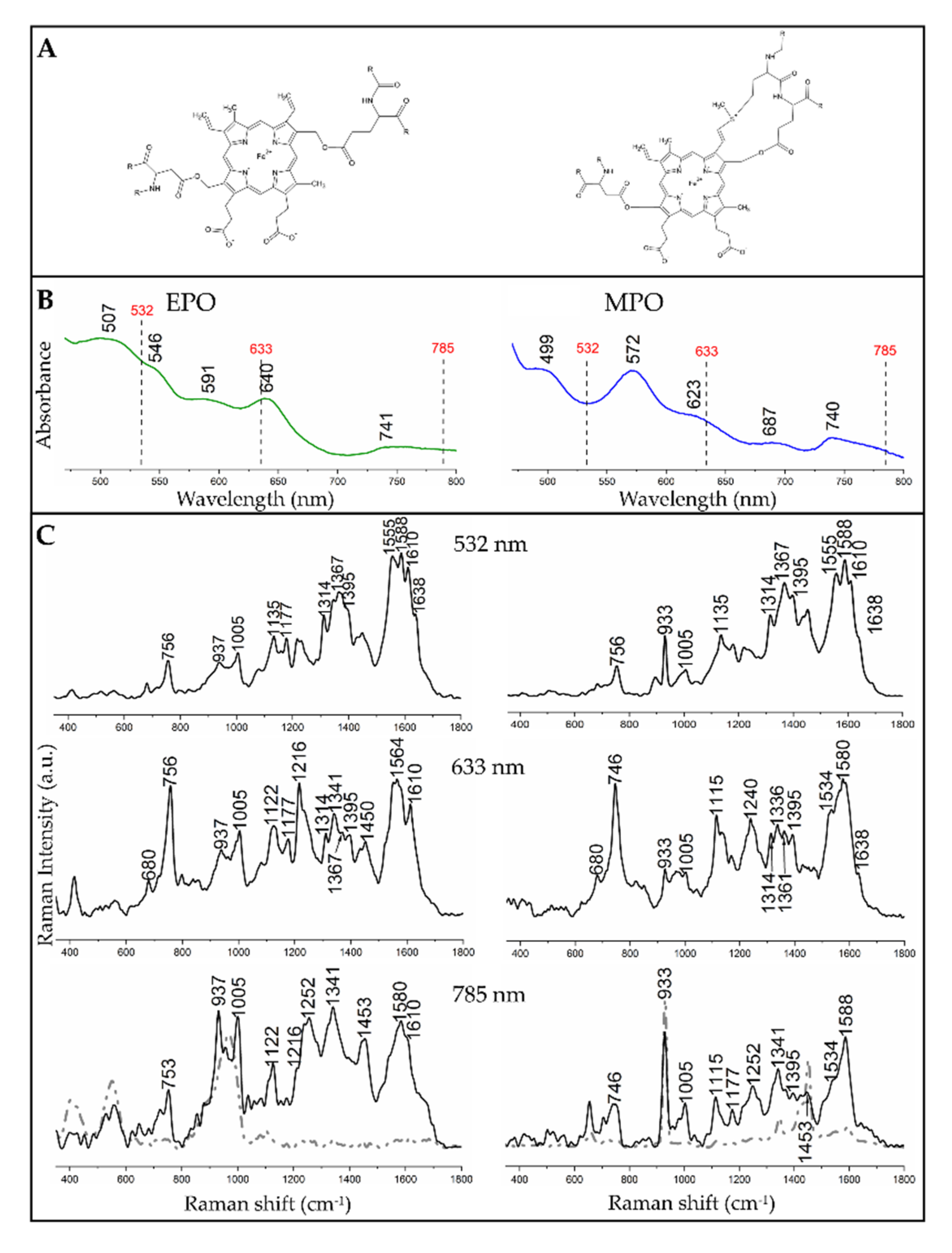
3.1. Resonance Raman Spectra of EPO and MPO in Solutions
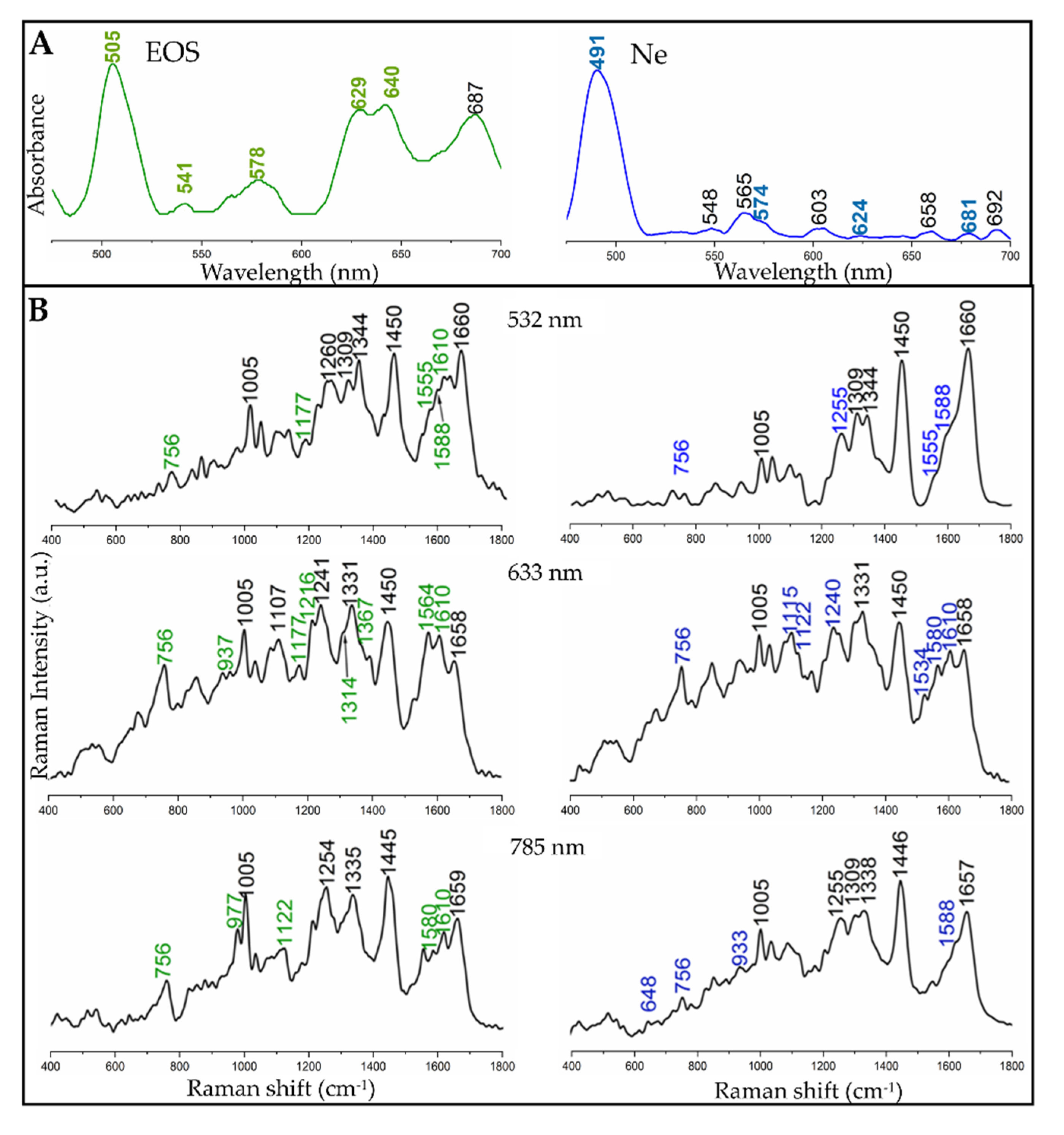
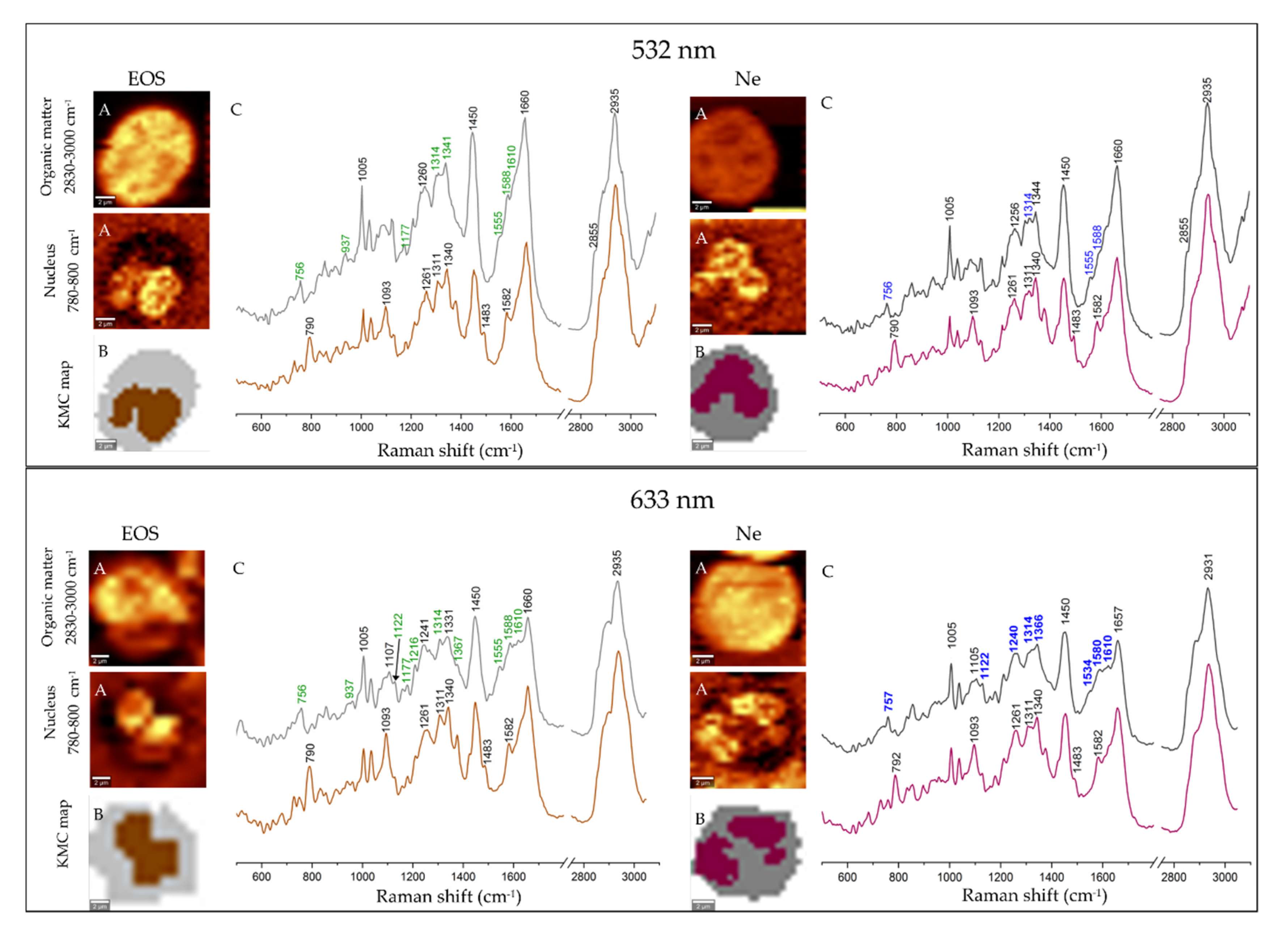
3.2. Electronic and Raman Features of Isolated Eosinophils and Neutrophils
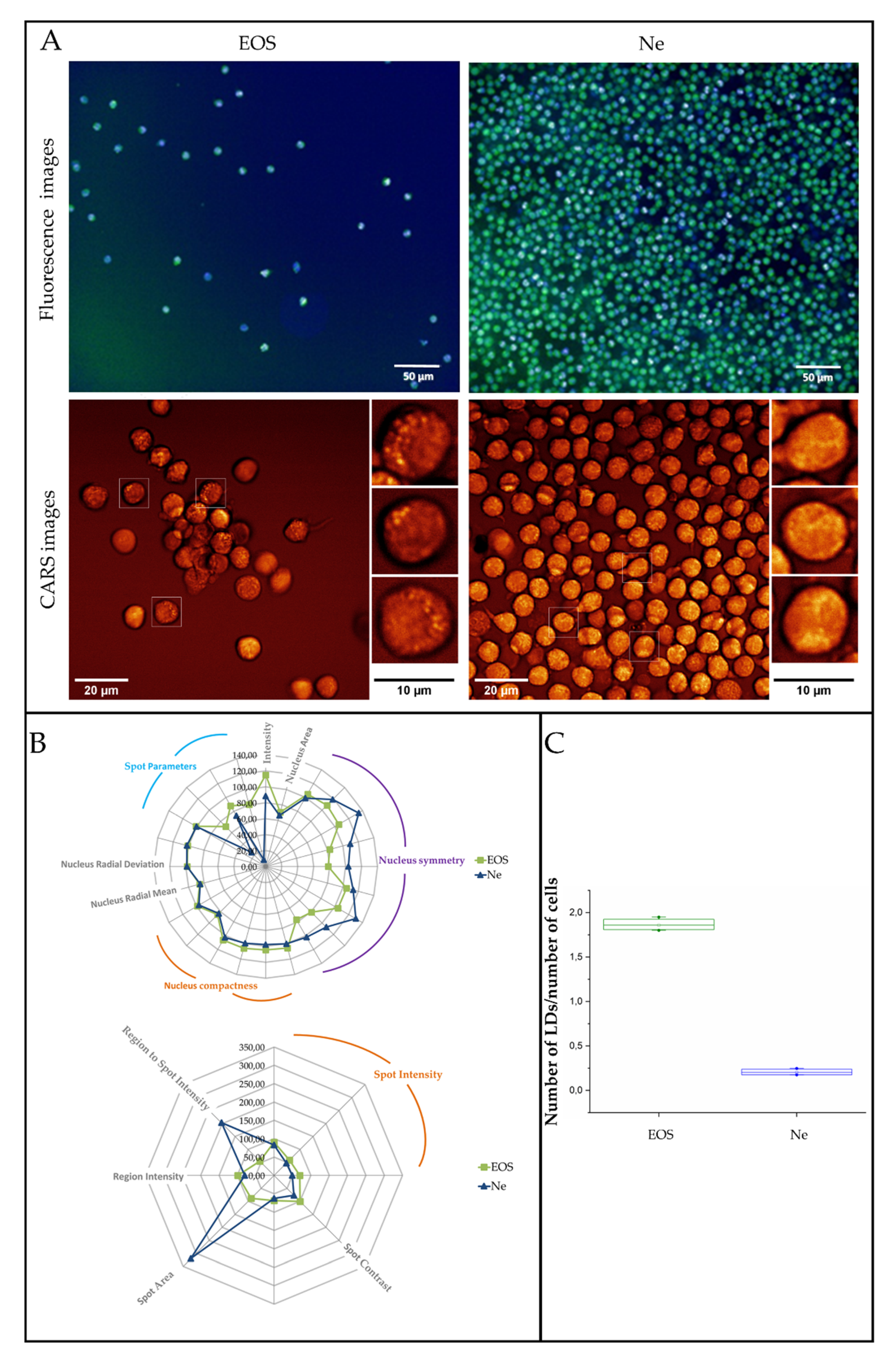
3.3. Fluorescence and Coherent Anti-Stokes Raman Scattering Imaging of Human Eosinophils and Neutrophils
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Maton, A.; Lahart, D.; Hopkins, J.; Warner, M.Q.; Johnson, S.; Wright, J.D. Cells: Building Blocks of Life, 3rd ed.; Prentice Hall science; Pearson Prentice Hall: Upper Saddle River, NJ, USA, 1997; ISBN 9780134232379. [Google Scholar]
- Kita, H. Eosinophils: Multifaceted biological properties and roles in health and disease. Immunol. Rev. 2011, 242, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Rothenberg, M.E.; Hogan, S.P. The Eosinophil. Annu. Rev. Immunol. 2006, 24, 147–174. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, H.F.; Dyer, K.D.; Foster, P.S. Eosinophils: Changing perspectives in health and disease. Nat. Rev. Immunol. 2012, 13, 9. [Google Scholar] [CrossRef] [PubMed]
- Giembycz, M.A.; Lindsay, M.A. Pharmacology of the Eosinophil. Pharmacol. Rev. 1999, 51, 213–340. [Google Scholar]
- Rygula, A.; Fernandes, R.F.; Grosicki, M.; Kukla, B.; Leszczenko, P.; Augustynska, D.; Cernescu, A.; Dorosz, A.; Malek, K.; Baranska, M. Raman imaging highlights biochemical heterogeneity of human eosinophils versus human eosinophilic leukaemia cell line. Br. J. Haematol. 2019, 186, 685–694. [Google Scholar] [CrossRef]
- Weller, P.F.; Spencer, L.A. Functions of tissue-resident eosinophils. Nat. Rev. Immunol. 2017, 17, 746–760. [Google Scholar] [CrossRef]
- Mayumi, M. EoL-1, A Human Eosinophilic Cell Line. Leuk. Lymphoma 1992, 7, 243–250. [Google Scholar] [CrossRef]
- Ramoji, A.; Neugebauer, U.; Bocklitz, T.; Foerster, M.; Kiehntopf, M.; Bauer, M.; Popp, J. Toward a Spectroscopic Hemogram: Raman Spectroscopic Differentiation of the Two Most Abundant Leukocytes from Peripheral Blood. Anal. Chem. 2012, 84, 5335–5342. [Google Scholar] [CrossRef]
- Melo, R.C.N.; Weller, P.F. Lipid droplets in leukocytes: Organelles linked to inflammatory responses. Exp. Cell Res. 2016, 340, 193–197. [Google Scholar] [CrossRef] [Green Version]
- Sibbett, S.S.; Klebanoff, S.J.; Hurst, J.K. Resonance Raman characterization of the heme prosthetic group in eosinophil peroxidase. FEBS Lett. 1985, 189, 271–275. [Google Scholar] [CrossRef] [Green Version]
- Rosales, C. Neutrophil: A Cell with Many Roles in Inflammation or Several Cell Types? Front. Physiol. 2018, 9, 113. [Google Scholar] [CrossRef] [PubMed]
- Melo, R.C.N.; Weller, P.F. Contemporary understanding of the secretory granules in human eosinophils. J. Leukoc. Biol. 2018, 104, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.J. Myeloperoxidase-derived oxidation: Mechanisms of biological damage and its prevention. J. Clin. Biochem. Nutr. 2010, 48, 8–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persson, T.; Andersson, P.; Bodelsson, M.; Laurell, M.; Malm, J.; Egesten, A. Bactericidal Activity of Human Eosinophilic Granulocytes against Escherichia coli. Infect. Immun. 2001, 69, 3591–3596. [Google Scholar] [CrossRef] [Green Version]
- Linch, S.N.; Gold, J.A. The role of eosinophils in non-parasitic infections. Endocr. Metab. Immune Disord. Drug Targets 2011, 11, 165–172. [Google Scholar] [CrossRef] [Green Version]
- Klebanoff, S.J.; Coombs, R.W. Virucidal Effect of Stimulated Eosinophils on Human Immunodeficiency Virus Type 1. AIDS Res. Hum. Retroviruses 1996, 12, 25–29. [Google Scholar] [CrossRef]
- Puppels, G.J.; Garritsen, H.S.; Segers-Nolten, G.M.; de Mul, F.F.; Greve, J. Raman microspectroscopic approach to the study of human granulocytes. Biophys. J. 1991, 60, 1046–1056. [Google Scholar] [CrossRef] [Green Version]
- Salmaso, B.L.; Puppels, G.J.; Caspers, P.J.; Floris, R.; Wever, R.; Greve, J. Resonance Raman microspectroscopic characterization of eosinophil peroxidase in human eosinophilic granulocytes. Biophys. J. 1994, 67, 436–446. [Google Scholar] [CrossRef] [Green Version]
- Bozza, P.T.; Yu, W.; Weller, P.F. Mechanisms of Formation and Function of Eosinophil Lipid Bodies: Inducible Intracellular Sites Involved in Arachidonic Acid Metabolism. Mem. Inst. Oswaldo Cruz 1997, 92, 135–140. [Google Scholar] [CrossRef] [Green Version]
- Czamara, K.; Majzner, K.; Pacia, M.Z.; Kochan, K.; Kaczor, A.; Baranska, M. Raman spectroscopy of lipids: A review. J. Raman Spectrosc. 2015, 46, 4–20. [Google Scholar] [CrossRef]
- Wan, H.-C.; Melo, R.C.N.; Jin, Z.; Dvorak, A.M.; Weller, P.F. Roles and origins of leukocyte lipid bodies: Proteomic and ultrastructural studies. FASEB J. 2007, 21, 167–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bozza, P.T.; Bandeira-Melo, C. Mechanisms of leukocyte lipid body formation and function in inflammation. Memórias do Inst. Oswaldo Cruz 2005, 100, 113–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melo, R.C.N.; Weller, P.F. Unraveling the complexity of lipid body organelles in human eosinophils. J. Leukoc. Biol. 2014, 96, 703–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melo, R.C.N.; D’Avila, H.; Wan, H.-C.; Bozza, P.T.; Dvorak, A.M.; Weller, P.F. Lipid Bodies in Inflammatory Cells: Structure, Function, and Current Imaging Techniques. J. Histochem. Cytochem. 2011, 59, 540–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Almeida, P.E.; Toledo, D.A.M.; Rodrigues, G.S.C.; D’Avila, H. Lipid Bodies as Sites of Prostaglandin E2 Synthesis During Chagas Disease: Impact in the Parasite Escape Mechanism. Front. Microbiol. 2018, 9, 499. [Google Scholar] [CrossRef] [Green Version]
- Listenberger, L.L.; Studer, A.M.; Brown, D.A.; Wolins, N.E. Fluorescent detection of lipid droplets and associated proteins. Curr. Protoc. Cell Biol. 2016, 2016, 4–31. [Google Scholar] [CrossRef]
- Brackmann, C.; Hillertz, P.; Pilon, M.; Enejder, A.; Hellerer, T.; Axa, C. Monitoring of lipid storage in Caenorhabditis elegans using coherent anti-Stokes Raman scattering (CARS) microscopy. Proc. Natl. Acad. Sci. USA 2007, 37, 14658–14663. [Google Scholar]
- Huff, T.B.; Cheng, J.X. In vivo coherent anti-Stokes Raman scattering imaging of sciatic nerve tissue. J. Microsc. 2007, 225, 175–182. [Google Scholar] [CrossRef] [Green Version]
- Folick, A.; Min, W.; Wang, M.C. Label-free imaging of lipid dynamics using Coherent Anti-stokes Raman Scattering (CARS) and Stimulated Raman Scattering (SRS) microscopy. Curr. Opin. Genet. Dev. 2011, 21, 585–590. [Google Scholar] [CrossRef] [Green Version]
- Le, T.T.; Yue, S.; Cheng, J.X. Shedding new light on lipid biology with coherent anti-Stokes Raman scattering microscopy. J. Lipid Res. 2010, 51, 3091–3102. [Google Scholar] [CrossRef] [Green Version]
- Nan, X.; Cheng, J.X.; Xie, X.S. Vibrational imaging of lipid droplets in live fibroblast cells with coherent anti-Stokes Raman scattering microscopy. J. Lipid Res. 2003, 44, 2202–2208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, P.O.; Botstein, D.; Lapidot, M.; Pilpel, Y.; Mazo, A.; Hodgson, J.W.; Petruk, S.; Sedkov, Y.; Brock, H.W.; Timmons, J.A.; et al. Label-Free Biomedical Imaging with. Science 2008, 1857, 1857–1861. [Google Scholar] [CrossRef] [Green Version]
- Cui, S.; Zhang, S.; Yue, S. Raman Spectroscopy and Imaging for Cancer Diagnosis. J. Healthc. Eng. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Grosicki, M.; Wójcik, T.; Chlopicki, S.; Kieć-Kononowicz, K. In vitro study of histamine and histamine receptor ligands influence on the adhesion of purified human eosinophils to endothelium. Eur. J. Pharmacol. 2016, 777, 49–59. [Google Scholar] [CrossRef]
- Heuke, S.; Vogler, N.; Meyer, T.; Akimov, D.; Kluschke, F.; Röwert-Huber, H.-J.; Lademann, J.; Dietzek, B.; Popp, J. Detection and Discrimination of Non-Melanoma Skin Cancer by Multimodal Imaging. Healthc. 2013, 1, 64–83. [Google Scholar] [CrossRef] [Green Version]
- Wever, R.; Hamers, M.N.; de Graaf, C.J.; Weening, R.S.; Roos, D.; Johnston, R.B. Characterization of the Peroxidase in Human Eosinophils BT—Biochemistry and Function of Phagocytes; Rossi, F., Patriarca, P., Eds.; Springer: Boston, MA, USA, 1982; pp. 501–508. ISBN 978-1-4684-8088-7. [Google Scholar]
- Dayer, M.; Moosavi-Movahedi, A.; Dayer, M. Band Assignment in Hemoglobin Porphyrin Ring Spectrum: Using Four-Orbital Model of Gouterman. Protein Pept. Lett. 2010, 17, 473–479. [Google Scholar] [CrossRef] [Green Version]
- Bakkenist, A.R.J.; Wever, R.; Vulsma, T.; Plat, H.; Van Gelder, B.F. Isolation procedure and some properties of myeloperoxidase from human leucocytes. Biochim. Biophys. Acta-Enzymol. 1978, 524, 45–54. [Google Scholar] [CrossRef]
- Brogioni, S.; Feis, A.; Marzocchi, M.P.; Zederbauer, M.; Furtmüller, P.G.; Obinger, C.; Smulevich, G. Resonance Raman assignment of myeloperoxidase and the selected mutants Asp94Val and Met243Thr. Effect of the heme distortion. J. Raman Spectrosc. 2006, 37, 263–276. [Google Scholar] [CrossRef]
- Sijtsema, N.M.; Otto, C.; Segers-Nolten, G.M.J.; Verhoeven, A.J.; Greve, J. Resonance Raman Microspectroscopy of Myeloperoxidase and Cytochrome b558 in Human Neutrophilic Granulocytes. Biophys. J. 1998, 74, 3250–3255. [Google Scholar] [CrossRef] [Green Version]
- Atkins, C.G.; Buckley, K.; Blades, M.W.; Turner, R.F.B. Raman Spectroscopy of Blood and Blood Components. Appl. Spectrosc. 2017, 71, 767–793. [Google Scholar] [CrossRef]
- Kiryu, C.; Kaneda, M.; Shiraishi, T.; Tsuda, M.; Inana, I.; Sakiyama, T.; Fujinaga, T.; Nishida, A.; Kakinuma, K. Spectrophotometric determination of neutrophil cytochrome b558 of chronic granulomatous disease. Pediatr. Int. 1998, 40, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Staniszewska-Slezak, E.; Malek, K.; Baranska, M. Complementary analysis of tissue homogenates composition obtained by Vis and NIR laser excitations and Raman spectroscopy. Spectrochim. Acta-Part A Mol. Biomol. Spectrosc. 2015, 147, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Majzner, K.; Chlopicki, S.; Baranska, M. Lipid droplets formation in human endothelial cells in response to polyunsaturated fatty acids and 1-methyl-nicotinamide (MNA); confocal Raman imaging and fluorescence microscopy studies. J. Biophotonics 2016, 9, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Szafraniec, E.; Kus, E.; Wislocka, A.; Kukla, B.; Sierka, E.; Untereiner, V.; Sockalingum, G.D.; Chlopicki, S.; Baranska, M. Raman spectroscopy–based insight into lipid droplets presence and contents in liver sinusoidal endothelial cells and hepatocytes. J. Biophotonics 2019, 12. [Google Scholar] [CrossRef]
- Asher, S.A. UV Resonance Raman Studies of Molecular Structure and Dynamics: Applications in Physical and Biophysical Chemistry. Annu. Rev. Phys. Chem. 1988, 39, 537–588. [Google Scholar] [CrossRef]
- Czernuszewicz, R.S.; Zaczek, M.B. Resonance Raman Spectroscopy. Encycl. Inorg. Bioinorg. Chem. 2008. [Google Scholar] [CrossRef]
- Strekas, T.C.; Packer, A.J.; Spiro, T.G. Resonance Raman spectra of ferri-hemoglobin fluoride: Three scattering regimes. J. Raman Spectrosc. 1973, 1, 197–206. [Google Scholar] [CrossRef]
- Asher, S.A.; Vickery, L.E.; Schuster, T.M.; Sauer, K. Resonance Raman spectra of methemoglobin derivatives. Selective enhancement of axial ligand vibrations and lack of an effect of inositol hexaphosphate. Biochemistry 1977, 16, 5849–5856. [Google Scholar] [CrossRef] [Green Version]
- Wood, B.R.; Tait, B.; McNaughton, D. Micro-Raman characterisation of the R to T state transition of haemoglobin within a single living erythrocyte. Biochim. Biophys. Acta-Mol. Cell Res. 2001, 1539, 58–70. [Google Scholar] [CrossRef] [Green Version]
- Muniz, V.S.; Weller, P.F.; Neves, J.S. Eosinophil crystalloid granules: Structure, function, and beyond. J. Leukoc. Biol. 2012, 92, 281–288. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 532 nm | 633 nm | 785 nm | Assignment of Modes with Their Symmetry |
|---|---|---|---|
| EPO/MPO | EPO/MPO | EPO/MPO | |
| 756/756 | 756/746 | 756/746 | ν15 (B1g) |
| 937/933 | 937/933 | 937/933 | ν46, (Eu) |
| 1005/1005 | 1005/1005 | 1005/1005 | Phenylalanine |
| -/- | 1122/1115 | 1122/1115 | ν22 (A2g) |
| 1135/1135 | -/- | -/- | ν14 (B1g) |
| 1177/- | 1177/- | -/1177 | ν30 (B2g) |
| -/- | 1216/- | -/- | ν13 (B1g) |
| -/- | -/1240 | 1252/1252 | ν42 (Eu) |
| 1314/1314 | 1314/1314 | -/- | ν21 (A2g) |
| -/- | 1341/1336 | 1341/1341-/- | ν41 (Eu)/δs(=CH2) |
| 1367/1367 | 1367/1361 | -/- | ν4 (A1g) |
| 1395/1395 | 1395/1395 | -/1395 | ν40 (Eu) |
| -/- | -/- | 1453/1453 | CH2/CH3, δs(=CH2) |
| -/- | -/1534 | -/1534 | unknown |
| 1555/1555 | -/- | -/- | ν11 (B1g) |
| -/- | 1564/- | -/- | ν19 (A2g) |
| 1588/1588 | -/1580 | 1580/1588 | ν2 (A1g) |
| 1610/1610 | 1610/- | 1610/- | ν(C = C) |
| 1638/1638 | -/1638 | -/- | ν10 (B1g) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dorosz, A.; Grosicki, M.; Dybas, J.; Matuszyk, E.; Rodewald, M.; Meyer, T.; Popp, J.; Malek, K.; Baranska, M. Eosinophils and Neutrophils—Molecular Differences Revealed by Spontaneous Raman, CARS and Fluorescence Microscopy. Cells 2020, 9, 2041. https://doi.org/10.3390/cells9092041
Dorosz A, Grosicki M, Dybas J, Matuszyk E, Rodewald M, Meyer T, Popp J, Malek K, Baranska M. Eosinophils and Neutrophils—Molecular Differences Revealed by Spontaneous Raman, CARS and Fluorescence Microscopy. Cells. 2020; 9(9):2041. https://doi.org/10.3390/cells9092041
Chicago/Turabian StyleDorosz, Aleksandra, Marek Grosicki, Jakub Dybas, Ewelina Matuszyk, Marko Rodewald, Tobias Meyer, Jürgen Popp, Kamilla Malek, and Malgorzata Baranska. 2020. "Eosinophils and Neutrophils—Molecular Differences Revealed by Spontaneous Raman, CARS and Fluorescence Microscopy" Cells 9, no. 9: 2041. https://doi.org/10.3390/cells9092041