On Neutrophil Extracellular Trap (NET) Removal: What We Know Thus Far and Why So Little


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. The NET Excitement
1.2. NETs Are Two-Faced
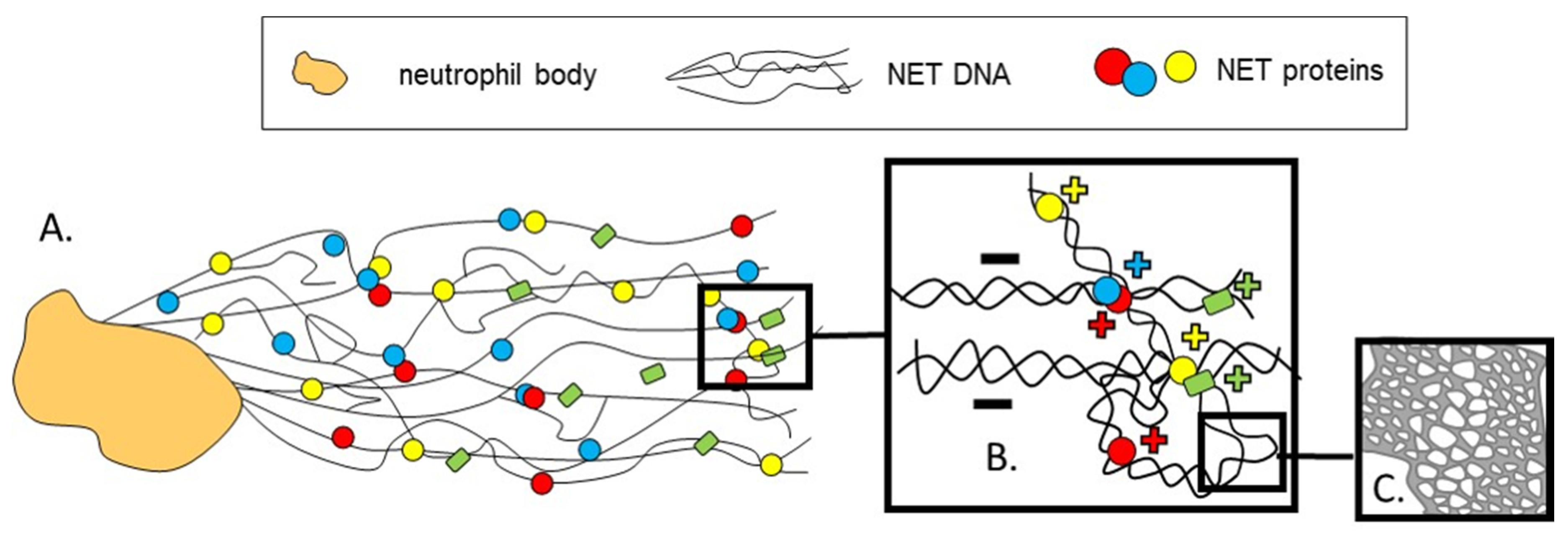
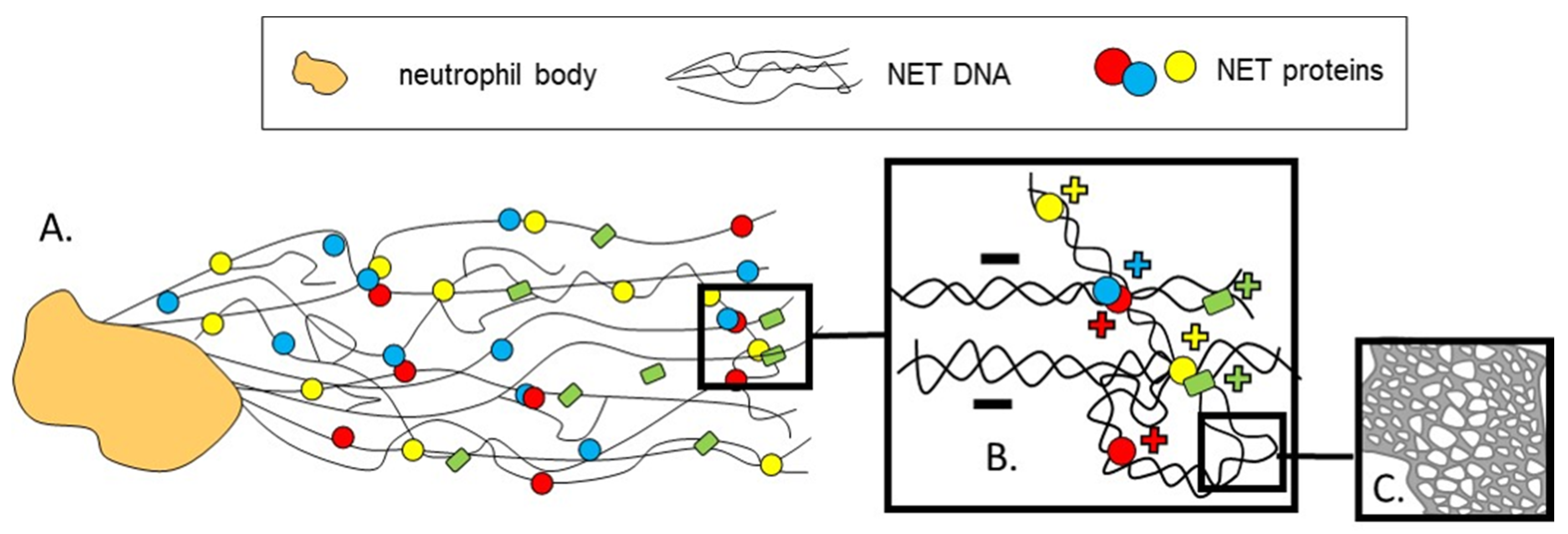
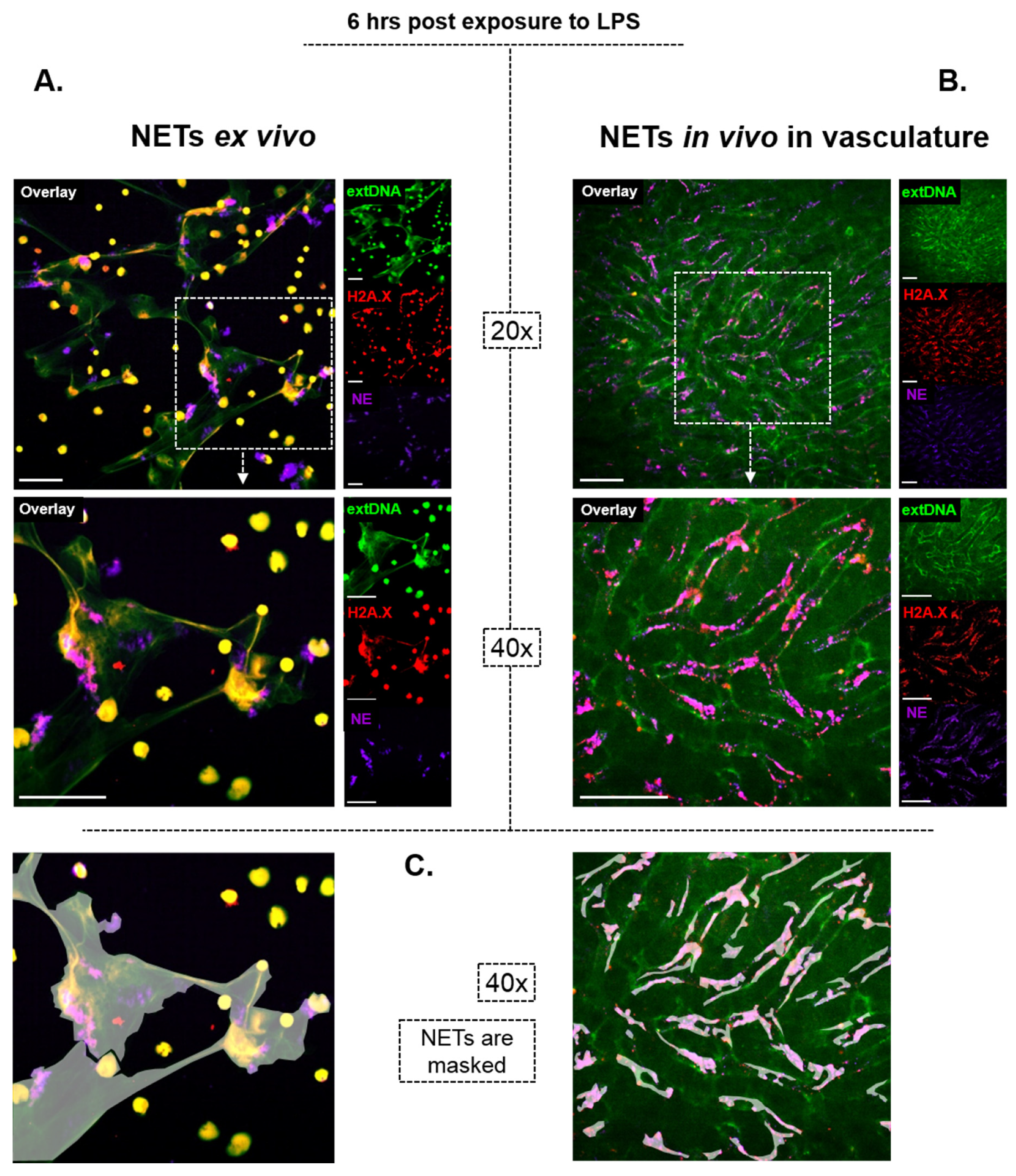
2. On How NETs Are Structured
3. On How NETs Cause Bystander Damage
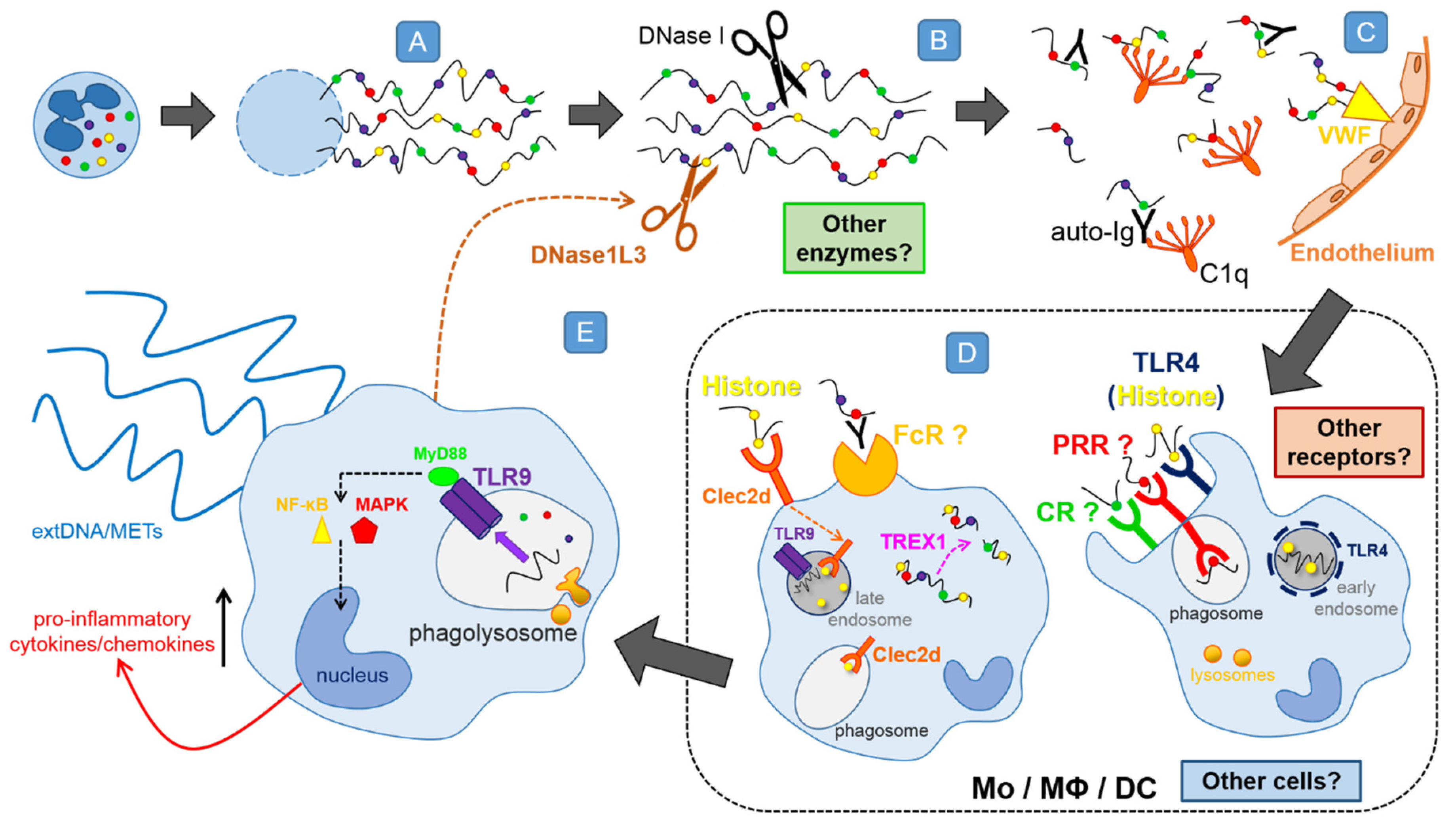
4. On How NETs Are Removed
4.1. DNases Dissolve the NET Backbone
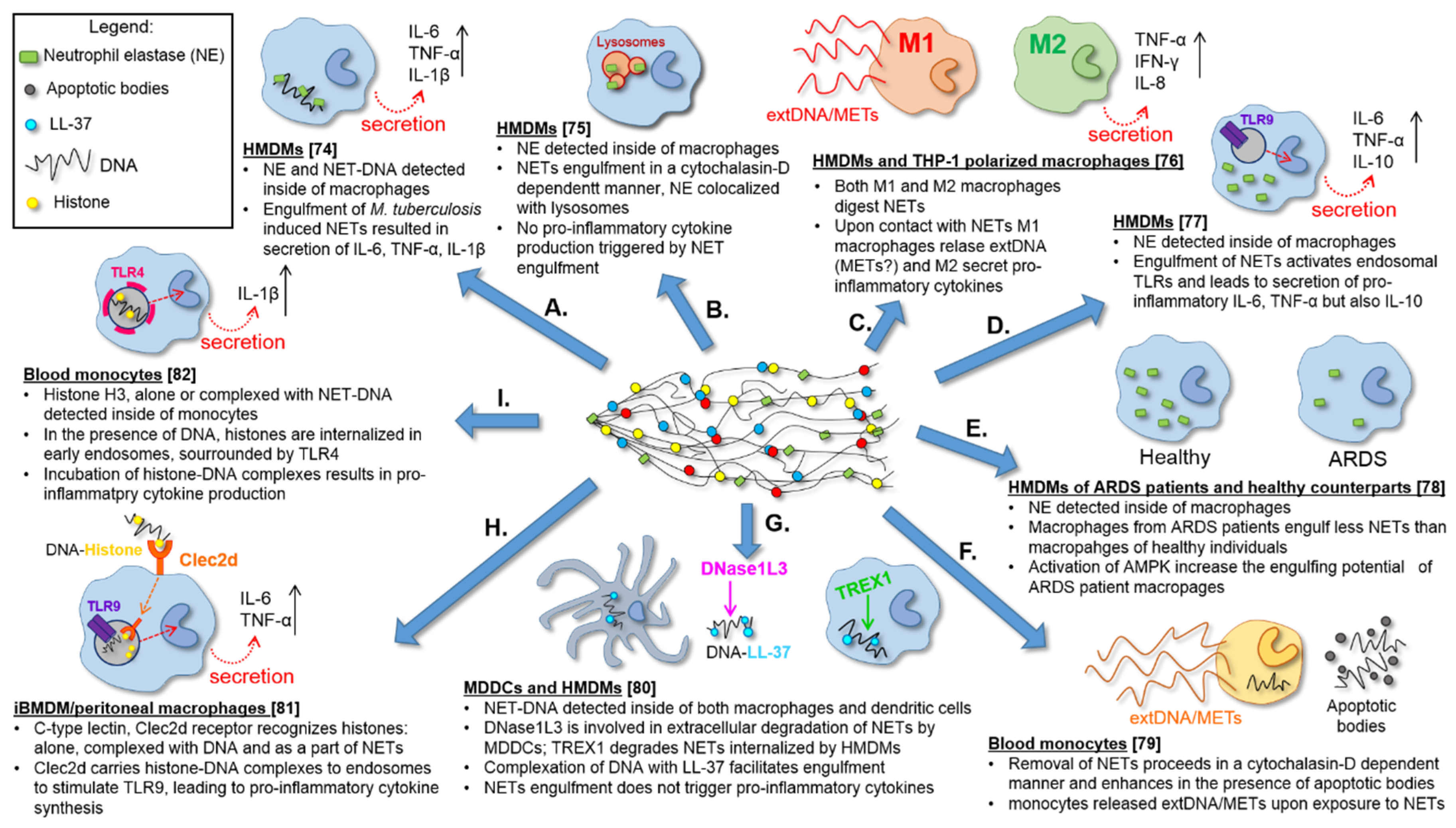
4.2. In Vitro Differentiated Macrophages Remove NET
4.3. Phagocytosis of NET Remnants
4.4. In Vitro Differentiated Dendritic Cells also Remove NETs
4.5. Phenotype of NET Engulfing Monocytes/Macrophages
4.6. Impact of NET Digestion on Macrophages—The Release of METs?
4.7. NET Components Facilitating or Attenuating Their Removal
4.8. Preliminary Data on Receptors and Pathways Involved
5. On Why NETs Are Not Removed Appropriately/Efficiently in Numerous Disorders
6. On Why We Know So Little about NET Removal
7. On How We Could Remove NETs Therapeutically
8. On Future Directions of Studies
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Brinkmann, V.; Reichard, U.; Goosman, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef] [PubMed]
- Urban, C.F.; Reichard, U.; Brinkmann, V.; Zychlinsky, A. Neutrophil extracellular traps capture and kill Candida albicans and hyphal forms. Cell. Microbiol. 2006, 8, 668–676. [Google Scholar] [CrossRef]
- Narasaraju, T.; Yang, E.; Samy, R.P.; Ng, H.H.; Poh, W.P.; Liew, A.A.; Phoon, M.C.; Van Rooijen, N.; Chow, V.T. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am. J. Pathol. 2011, 179, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Kichik, V.; Mondragón-Flores, R.; Mondragón-Castelán, M.; Gonzalez-Pozos, S.; Muñiz-Hernandez, S.; Rojas-Espinosa, O.; Chacón-Salinas, R.; Estrada-Parra, S.; Estrada-García, I. Neutrophil extracellular traps are induced by Mycobacterium tuberculosis. Tuberculosis 2009, 89, 29–37. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef]
- Buchanan, J.T.; Simpson, A.J.; Aziz, R.K.; Liu, G.Y.; Kristian, S.A.; Kotb, M.; Feramisco, J.; Nizet, V. DNase expression allows the pathogen group A Streptococcus to escape killing in neutrophil extracellular traps. Curr. Biol. 2006, 16, 396–400. [Google Scholar] [CrossRef] [Green Version]
- Beiter, K.; Wartha, F.; Albiger, B.; Normark, S.; Zychlinsky, A.; Henriques-Normark, B. An endonuclease allows Streptococcus pneumoniae to escape from neutrophil extracellular traps. Curr. Biol. 2006, 16, 401–407. [Google Scholar] [CrossRef] [Green Version]
- Urban, C.F.; Lourido, S.; Zychlinsky, A. How do microbes evade neutrophil killing? Cell. Microbiol. 2006, 8, 1687–1696. [Google Scholar] [CrossRef]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [Green Version]
- Metzler, K.D.; Fuchs, T.A.; Nauseef, W.M.; Reumaux, D.; Roesler, J.; Schulze, I.; Wahn, V.; Papayannopoulos, V.; Zychlinsky, A. Myeloperoxidase is required for neutrophil extracellular trap formation: Implications for innate immunity. Blood 2011, 117, 953–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Li, M.; Lindberg, M.R.; Kennett, M.J.; Xiong, N.; Wang, Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 2010, 207, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Handzlik, A.M.; Bystrzycka, W.; Cieloch, A.; Glodkowska, E. Nitric oxide and peroxynitrite trigger and enhance release of neutrophil extracellular traps. Cell. Mol. Life Sci. 2019, 77, 3059–3075. [Google Scholar] [CrossRef] [Green Version]
- Stojkov, D.; Amini, P.; Oberson, K.; Sokollik, C.; Duppenthaler, A.; Simon, H.U.; Yousefi, S. ROS and glutathionylation balance cytoskeletal dynamics in neutrophil extracellular trap formation. J. Cell Biol. 2017, 216, 4073–4090. [Google Scholar] [CrossRef] [PubMed]
- Amini, P.; Stojkov, D.; Felser, A.; Jackson, C.B.; Courage, C.; Schaller, A.; Gelman, L.; Soriano, M.E.; Nuoffer, J.M.; Scorrano, L.; et al. Neutrophil extracellular trap formation requires OPA1-dependent glycolytic ATP production. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Yousefi, S.; Mihalache, C.; Kozlowski, E.; Schmid, I.; Simon, H.U. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009, 16, 1438–1444. [Google Scholar] [CrossRef]
- Keshari, R.S.; Jyoti, A.; Kumar, S.; Dubey, M.; Verma, A.; Srinag, B.S.; Krishnamurthy, H.; Barthwal, M.K.; Dikshit, M. Neutrophil extracellular traps contain mitochondrial as well as nuclear DNA and exhibit inflammatory potential. Cytom. Part A 2012, 81, 238–247. [Google Scholar] [CrossRef]
- Brinkmann, V.; Zychlinsky, A. Beneficial suicide: Why neutrophils die to make NETs. Nat. Rev. Microbiol. 2007, 5, 577–582. [Google Scholar] [CrossRef]
- Steinberg, B.E.; Grinstein, S. Unconventional roles of the NADPH oxidase: Signaling, ion homeostasis, and cell death. Sci. STKE 2007, 2007, 1–3. [Google Scholar] [CrossRef]
- Yipp, B.G.; Petri, B.; Salina, D.; Jenne, C.N.; Scott, B.N.; Zbytnuik, L.D.; Pittman, K.; Asaduzzaman, M.; Wu, K.; Meijndert, H.C.; et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 2012, 18, 1386–1393. [Google Scholar] [CrossRef] [Green Version]
- Pilsczek, F.H.; Salina, D.; Poon, K.K.; Fahey, C.; Yipp, B.G.; Sibley, C.D.; Robbins, S.M.; Green, F.H.; Surette, M.G.; Sugai, M.; et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J. Immunol. 2010, 185, 7413–7425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Boeltz, S.; Amini, P.; Anders, H.J.; Andrade, F.; Bilyy, R.; Chatfield, S.; Cichon, I.; Clancy, D.M.; Desai, J.; Dumych, T.; et al. To NET or not to NET:current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death Differ. 2019, 26, 395–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azzouz, D.; Palaniyar, N. ApoNETosis: Discovery of a novel form of neutrophil death with concomitant apoptosis and NETosis. Cell Death Dis. 2018, 9, 839. [Google Scholar] [CrossRef] [Green Version]
- Kessenbrock, K.; Krumbholz, M.; Schönermarck, U.; Back, W.; Gross, W.L.; Werb, Z.; Gröne, H.-J.; Brinkmann, V.; Jenne, D.E. Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 2009, 15, 623–625. [Google Scholar] [CrossRef]
- Baker, V.S.; Imade, G.E.; Molta, N.B.; Tawde, P.; Pam, S.D.; Obadofin, M.O.; Sagay, S.A.; Egah, D.Z.; Iya, D.; Afolabi, B.B.; et al. Cytokine-associated neutrophil extracellular traps and antinuclear antibodies in Plasmodium falciparum infected children under six years of age. Malar. J. 2008, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kolaczkowska, E.; Jenne, C.N.; Surewaard, B.G.J.; Thanabalasuriar, A.; Lee, W.; Sanz, M.; Mowen, K.; Opdenakker, G.; Kubes, P. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat. Commun. 2015, 6, 6673. [Google Scholar] [CrossRef] [Green Version]
- Bruns, S.; Kniemeyer, O.; Hasenberg, M.; Aimanianda, V.; Nietzsche, S.; Thywien, A.; Jeron, A.; Latgé, J.P.; Brakhage, A.A.; Gunzer, M. Production of extracellular traps against aspergillus fumigatus in vitro and in infected lung tissue is dependent on invading neutrophils and influenced by hydrophobin roda. PLoS Pathog. 2010, 6, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Daßler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 2020, 217, 1–7. [Google Scholar] [CrossRef]
- Skendros, P.; Mitsios, A.; Chrysanthopoulou, A.; Mastellos, D.C.; Metallidis, S.; Rafailidis, P.; Ntinopoulou, M.; Sertaridou, E.; Tsironidou, V.; Tsigalou, C.; et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J. Clin. Investig. 2020. [Google Scholar] [CrossRef] [PubMed]
- Middleton, E.A.; He, X.-Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil Extracellular Traps (NETs) Contribute to Immunothrombosis in COVID-19 Acute Respiratory Distress Syndrome. Blood 2020. [Google Scholar] [CrossRef]
- Tomar, B.; Anders, H.J.; Desai, J.; Mulay, S.R. Neutrophils and Neutrophil Extracellular Traps Drive Necroinflammation in COVID-19. Cells 2020, 9, 1383. [Google Scholar] [CrossRef] [PubMed]
- Albrengues, J.; Shields, M.; Ng, D.; Park, C.G.; Ambrico, A.; Poindexter, M.; Upadhyay, P.; Uyeminami, D.; Pommier, A.; Küttner, V.; et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018, 361, eaao4227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakkim, A.; Fürnrohr, B.G.; Amann, K.; Laube, B.; Abed, U.A.; Brinkmann, V.; Herrmann, M.; Voll, R.E.; Zychlinsky, A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl. Acad. Sci. USA 2010, 107, 9813–9818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khandpur, R.; Carmona-Rivera, C.; Vivekanandan-Giri, A.; Gizinski, A.; Yalavarthi, S.; Knight, J.S.; Friday, S.; Li, S.; Patel, R.M.; Subramanian, V.; et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci. Transl. Med. 2013, 5, 178ra40. [Google Scholar] [CrossRef] [Green Version]
- Lande, R.; Ganguly, D.; Facchinetti, V.; Frasca, L.; Conrad, C.; Gregorio, J.; Meller, S.; Chamilos, G.; Sebasigari, R.; Riccieri, V.; et al. Neutrophils Activate Plasmacytoid Dendritic Cells by Releasing Self-DNA Peptide Complexes in Systemic Lupus Erythematosus. Sci. Transl. Med. 2011, 3, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Dwivedi, N.; Upadhyay, J.; Neeli, I.; Khan, S.; Pattanaik, D.; Myers, L.; Kirou, K.A.; Hellmich, B.; Knuckley, B.; Thompson, P.R.; et al. Felty’s syndrome autoantibodies bind to deiminated histones and neutrophil extracellular chromatin traps. Arthritis Rheum. 2012, 64, 982–992. [Google Scholar] [CrossRef] [Green Version]
- Semeraro, F.; Ammollo, C.T.; Morrissey, J.H.; Dale, G.L.; Friese, P.; Esmon, N.L.; Esmon, C.T. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: Involvement of platelet TLR2 and TLR4. Blood 2011, 118, 1952–1961. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Wysocki, R.W.; Amoozgar, Z.; Maiorino, L.; Fein, M.R.; Jorns, J.; Schott, A.F.; Kinugasa-Katayama, Y.; Lee, Y.; Won, N.H.; et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci. Transl. Med. 2016, 8, 361ra138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires, R.H.; Felix, S.B.; Delcea, M. The architecture of neutrophil extracellular traps investigated by atomic force microscopy. Nanoscale 2016, 8, 14193–14202. [Google Scholar] [CrossRef]
- Urban, C.F.; Ermert, D.; Schmid, M.; Abu-Abed, U.; Goosmann, C.; Nacken, W.; Brinkmann, V.; Jungblut, P.R.; Zychlinsky, A. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 2009, 5, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinod, K.; Witsch, T.; Farley, K.; Gallant, M.; Remold-O’Donnell, E.; Wagner, D.D. Neutrophil elastase-deficient mice form neutrophil extracellular traps in an experimental model of deep vein thrombosis. J. Thromb. Haemost. 2016, 14, 551–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csomós, K.; Kristóf, E.; Jakob, B.; Csomós, I.; Kovács, G.; Rotem, O.; Hodrea, J.; Bagoly, Z.; Muszbek, L.; Balajthy, Z.; et al. Protein cross-linking by chlorinated polyamines and transglutamylation stabilizes neutrophil extracellular traps. Cell Death Dis. 2016, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Cichon, I.; Ortmann, W.; Bednarz, A.; Lenartowicz, M. Reduced Neutrophil Extracellular Trap (NET) Formation During Systemic Inflammation in Mice With Menkes Disease and Wilson Disease: Copper Requirement for NET Release. Front. Immunol. 2020, 10, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandel, P.; Metais, P. Les acides nucléiques du plasma sanguin chez l’homme. C. R. Seances Soc. Biol. Fil. 1948, 142, 241–243. [Google Scholar]
- Gould, T.J.; Lysov, Z.; Liaw, P.C. Extracellular DNA and histones: Double-edged swords in immunothrombosis. J. Thromb. Haemost. 2015, 13, S82–S91. [Google Scholar] [CrossRef]
- Ceppellini, R.; Polli, E.; Celada, F. A DNA-reacting factor in serum of a patient with lupus erythematosus diffusus. Proc. Soc. Exp. Biol. Med. 1957, 572–574. [Google Scholar] [CrossRef]
- Sur Chowdhury, C.; Hahn, S.; Hasler, P.; Hoesli, I.; Lapaire, O.; Giaglis, S. Elevated Levels of Total Cell-Free DNA in Maternal Serum Samples Arise from the Generation of Neutrophil Extracellular Traps. Fetal Diagn. Ther. 2016, 40, 263–267. [Google Scholar] [CrossRef]
- Kustanovich, A.; Schwartz, R.; Peretz, T.; Grinshpun, A. Life and death of circulating cell-free DNA. Cancer Biol. Ther. 2019, 20, 1057–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauthier, V.J.; Tyler, L.N.; Mannik, M. Blood clearance kinetics and liver uptake of mononucleosomes in mice. J. Immunol. 1996, 156, 1151–1156. [Google Scholar] [PubMed]
- Kumar, S.V.R.; Kulkarni, O.P.; Mulay, S.R.; Darisipudi, M.N.; Romoli, S.; Thomasova, D.; Scherbaum, C.R.; Hohenstein, B.; Hugo, C.; Müller, S.; et al. Neutrophil extracellular trap-related extracellular histones cause vascular necrosis in severe GN. J. Am. Soc. Nephrol. 2015, 26, 2399–2413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helseth, R.; Solheim, S.; Arnesen, H.; Seljeflot, I.; Opstad, T.B. The time course of markers of neutrophil extracellular traps in patients undergoing revascularisation for acute myocardial infarction or stable angina pectoris. Mediators Inflamm. 2016, 2016, 1–8. [Google Scholar] [CrossRef]
- Helseth, R.; Knudsen, E.C.; Eritsland, J.; Opstad, T.B.; Arnesen, H.; Andersen, G.Y.; Seljeflot, I. Glucose associated NETosis in patients with ST-elevation myocardial infarction: An observational study. BMC Cardiovasc. Disord. 2019, 19, 1–8. [Google Scholar] [CrossRef]
- Hamaguchi, S.; Hirose, T.; Matsumoto, N.; Akeda, Y.; Irisawa, T.; Seki, M.; Hosotsubo, H.; Yamamoto, K.; Tasaki, O.; Oishi, K.; et al. Neutrophil extracellular traps in bronchial aspirates: A quantitative analysis. Eur. Respir. J. 2014, 43, 1709–1718. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.K.; Kaplan, M.J. The role of neutrophils in the pathogenesis of systemic lupus erythematosus. Int. Immunopharmacol. 2015, 27, 448–453. [Google Scholar] [CrossRef] [Green Version]
- YU, Y.; SU, K. Neutrophil Extracellular Traps and Systemic Lupus Erythematosus. J. Clin. Cell. Immunol. 2013, 4, 1–13. [Google Scholar] [CrossRef]
- Sur Chowdhury, C.; Giaglis, S.; Walker, U.A.; Buser, A.; Hahn, S.; Hasler, P. Enhanced neutrophil extracellular trap generation in rheumatoid arthritis: Analysis of underlying signal transduction pathways and potential diagnostic utility. Arthritis Res. Ther. 2014, 16, R122. [Google Scholar] [CrossRef] [Green Version]
- Komissarov, A.A.; Florova, G.; Idell, S. Effects of extracellular DNA on plasminogen activation and fibrinolysis. J. Biol. Chem. 2011, 286, 41949–41962. [Google Scholar] [CrossRef] [Green Version]
- Manzenreiter, R.; Kienberger, F.; Marcos, V.; Schilcher, K.; Krautgartner, W.D.; Obermayer, A.; Huml, M.; Stoiber, W.; Hector, A.; Griese, M.; et al. Ultrastructural characterization of cystic fibrosis sputum using atomic force and scanning electron microscopy. J. Cyst. Fibros. 2012, 11, 84–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiménez-alcázar, M.; Rangaswamy, C.; Panda, R.; Bitterling, J.; Simsek, Y.J.; Long, A.T.; Bilyy, R.; Krenn, V.; Renné, C.; Renné, T.; et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science 2017, 358, 1202–1206. [Google Scholar] [CrossRef] [Green Version]
- Brill, A.; Fuchs, T.A.; Savchenko, A.S.; Thomas, G.M.; Martinod, K.; de Meyer, S.F.; Bhandari, A.A.; Wagner, D.D. Neutrophil extracellular traps promote deep vein thrombosis in mice. J. Thromb. Haemost. 2012, 10, 136–144. [Google Scholar] [CrossRef] [Green Version]
- Almqvist, N.; Winkler, T.H.; Mårtensson, I.L. Autoantibodies: Focus on anti-DNA antibodies. Self/Nonself Immune Recognit. Signal. 2011, 2, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Herman, B.; Bibert, S.; Concord, E.; Dublet, B.; Weidenhaupt, M.; Vernet, T.; Gulino-Debrac, D. Identification of proteases involved in the proteolysis of vascular endothelium cadherin during neutrophil transmigration. J. Biol. Chem. 2003, 278, 14002–14012. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, P.A. The potential of neutrophil elastase inhibitors as anti-inflammatory therapies. Curr. Opin. Hematol. 2014, 21, 23–28. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.; Davis, R.P.; Kim, S.J.; Tse, M.; Esmon, C.T.; Kolaczkowska, E.; Jenne, C.N. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood 2017, 129, 1357–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Zhang, X.; Pelayo, R.; Monestier, M.; Ammollo, C.T.; Semeraro, F.; Taylor, F.B.; Esmon, N.L.; Lupu, F.; Esmon, C.T. Extracellular histones are major mediators of death in sepsis. Nat. Med. 2009, 15, 1318–1321. [Google Scholar] [CrossRef] [Green Version]
- Leffler, J.; Martin, M.; Gullstrand, B.; Tydén, H.; Lood, C.; Truedsson, L.; Bengtsson, A.A.; Blom, A.M. Neutrophil Extracellular Traps That Are Not Degraded in Systemic Lupus Erythematosus Activate Complement Exacerbating the Disease. J. Immunol. 2012, 188, 3522–3531. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wang, C.; Zhao, M.H.; Chen, M. Neutrophil extracellular traps can activate alternative complement pathways. Clin. Exp. Immunol. 2015, 181, 518–527. [Google Scholar] [CrossRef] [Green Version]
- Lachowicz-Scroggins, M.E.; Dunican, E.M.; Charbit, A.R.; Raymond, W.; Looney, M.R.; Peters, M.C.; Gordon, E.D.; Woodruff, P.G.; Lefrancais, E.; Phillips, B.R.; et al. Extracellular Dna, Neutrophil Extracellular Traps, and Inflammasome Activation in Severe Asthma. Am. J. Respir. Crit. Care Med. 2019, 199, 1076–1085. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.W.; Monteleone, M.; Boucher, D.; Sollberger, G.; Ramnath, D.; Condon, N.D.; von Pein, J.B.; Broz, P.; Sweet, M.J.; Schroder, K. Noncanonical inflammasome signaling elicits gasdermin D–dependent neutrophil extracellular traps. Sci. Immunol. 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fousert, E.; Toes, R.; Desai, J. Neutrophil Extracellular Traps (NETs) Take the Central Stage in Driving Autoimmune Responses. Cells 2020, 9, 915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braian, C.; Hogea, V.; Stendahl, O. Mycobacterium tuberculosis-induced neutrophil extracellular traps activate human macrophages. J. Innate Immun. 2013, 5, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Farrera, C.; Fadeel, B. Macrophage clearance of neutrophil extracellular traps is a silent process. J. Immunol. 2013, 191, 2647–2656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakazawa, D.; Shida, H.; Kusunoki, Y.; Miyoshi, A.; Nishio, S.; Tomaru, U.; Atsumi, T.; Ishizu, A. The responses of macrophages in interaction with neutrophils that undergo NETosis. J. Autoimmun. 2016, 67, 19–28. [Google Scholar] [CrossRef]
- Barrera-Vargas, A.; Gómez-Martín, D.; Carmona-Rivera, C.; Merayo-Chalico, J.; Torres-Ruiz, J.; Manna, Z.; Hasni, S.; Alcocer-Varela, J.; Kaplan, M.J. Differential ubiquitination in NETs regulates macrophage responses in systemic lupus erythematosus. Ann. Rheum. Dis. 2018, 77, 944–950. [Google Scholar] [CrossRef]
- Grégoire, M.; Uhel, F.; Lesouhaitier, M.; Gacouin, A.; Guirriec, M.; Mourcin, F.; Dumontet, E.; Chalin, A.; Samson, M.; Berthelot, L.L.; et al. Impaired efferocytosis and neutrophil extracellular trap clearance by macrophages in ARDS. Eur. Respir. J. 2018, 52, 1702590. [Google Scholar] [CrossRef]
- Haritha, V.H.; Seena, P.; Shaji, B.V.; Nithin, T.U.; Hazeena, V.N.; Anie, Y. Monocyte clearance of apoptotic neutrophils is unhindered in the presence of NETosis, but proteins of NET trigger ETosis in monocytes. Immunol. Lett. 2019, 207, 36–45. [Google Scholar] [CrossRef]
- Lazzaretto, B.; Fadeel, B. Intra- and Extracellular Degradation of Neutrophil Extracellular Traps by Macrophages and Dendritic Cells. J. Immunol. 2019, 203, 2276–2290. [Google Scholar] [CrossRef] [Green Version]
- Lai, J.J.; Cruz, F.M.; Rock, K.L. Immune Sensing of Cell Death through Recognition of Histone Sequences by C-Type Lectin-Receptor-2d Causes Inflammation and Tissue Injury. Immunity 2020, 52, 123–135.e6. [Google Scholar] [CrossRef] [PubMed]
- Tsourouktsoglou, T.D.; Warnatsch, A.; Ioannou, M.; Hoving, D.; Wang, Q.; Papayannopoulos, V. Histones, DNA, and Citrullination Promote Neutrophil Extracellular Trap Inflammation by Regulating the Localization and Activation of TLR4. Cell Rep. 2020, 31, 107602. [Google Scholar] [CrossRef]
- Mohanty, T.; Fisher, J.; Bakochi, A.; Neumann, A.; Cardoso, J.F.P.; Karlsson, C.A.Q.; Pavan, C.; Lundgaard, I.; Nilson, B.; Reinstrup, P.; et al. Neutrophil extracellular traps in the central nervous system hinder bacterial clearance during pneumococcal meningitis. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Shak, S. Aerosolized recombinant human DNase I for the treatment of cystic fibrosis. Chest 1995, 107, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Thammavongsa, V.; Missiakas, D.M.; Schneewind, O. Staphylococcus aureus Degrades Neutrophil Extracellular Traps to Promote Immune Cell Death. Science 2013, 342, 155–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Mayouf, S.M.; Sunker, A.; Abdwani, R.; Al Abrawi, S.; Almurshedi, F.; Alhashmi, N.; Al Sonbul, A.; Sewairi, W.; Qari, A.; Abdallah, E.; et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat. Genet. 2011, 43, 1186–1188. [Google Scholar] [CrossRef]
- Napirei, M.; Karsunky, H.; Zevnik, B.; Stephan, H.; Mannherz, H.G.; Möröy, T. Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat. Genet. 2000, 25, 177–181. [Google Scholar] [CrossRef]
- Martinod, K.; Demers, M.; Fuchs, T.A.; Wong, S.L.; Brill, A.; Gallant, M.; Hu, J.; Wang, Y.; Wagner, D.D. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc. Natl. Acad. Sci. USA 2013, 110, 8674–8679. [Google Scholar] [CrossRef] [Green Version]
- Lemansky, P.; Smolenova, E.; Wrocklage, C.; Hasilik, A. Neutrophil elastase is associated with serglycin on its way to lysosomes in U937 cells. Cell. Immunol. 2007, 246, 1–7. [Google Scholar] [CrossRef]
- Rodriguez, R.J.; White, R.R.; Senior, R.M.; Levine, E.A. Elastase release from human alveolar macrophages: Comparison between smokers and nonsmokers. Science 1977, 198, 313–314. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Grzybek, W.; van Rooijen, N.; Piccard, H.; Plytycz, B.; Arnold, B.; Opdenakker, G. Neutrophil elastase activity compensates for a genetic lack of matrix metalloproteinase-9 (MMP-9) in leukocyte infiltration in a model of experimental peritonitis. J. Leukoc. Biol. 2009, 85, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Dollery, C.M.; Owen, C.A.; Sukhova, G.K.; Krettek, A.; Shapiro, S.D.; Libby, P. Neutrophil elastase in human atherosclerotic plaques production by macrophages. Circulation 2003, 107, 2829–2836. [Google Scholar] [CrossRef] [PubMed]
- Bei, L.; Hu, T.; Qian, Z.M.; Shen, X. Extracellular Ca2+ regulates the respiratory burst of human neutrophils. Biochim. Biophys. Acta Mol. Cell Res. 1998, 1404, 475–483. [Google Scholar] [CrossRef] [Green Version]
- Newton, P.M.; Messing, R.O. The substrates and binding partners of protein kinase Cε. Biochem. J. 2010, 427, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, S.F. Structural Basis of Protein Kinase C Isoform Function. Physiol. Rev. 2008, 88, 1341–1378. [Google Scholar] [CrossRef] [Green Version]
- Parker, H.; Dragunow, M.; Hampton, M.B.; Kettle, A.J.; Winterbourn, C.C. Requirements for NADPH oxidase and myeloperoxidase in neutrophil extracellular trap formation differ depending on the stimulus. J. Leukoc. Biol. 2012, 92, 841–849. [Google Scholar] [CrossRef]
- Kenny, E.F.; Herzig, A.; Krüger, R.; Muth, A.; Mondal, S.; Thompson, P.R.; Brinkmann, V.; von Bernuth, H.; Zychlinsky, A. Diverse stimuli engage different neutrophil extracellular trap pathways. Elife 2017, 6, 1–21. [Google Scholar] [CrossRef]
- Petretto, A.; Bruschi, M.; Pratesi, F.; Croia, C.; Candiano, G.; Ghiggeri, G.; Migliorini, P. Neutrophil extracellular traps (NET) induced by different stimuli: A comparative proteomic analysis. PLoS ONE 2019, 14, 1–18. [Google Scholar] [CrossRef]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Maeß, M.B.; Wittig, B.; Cignarella, A.; Lorkowski, S. Reduced PMA enhances the responsiveness of transfected THP-1 macrophages to polarizing stimuli. J. Immunol. Methods 2014, 402, 76–81. [Google Scholar] [CrossRef]
- Park, E.K.; Jung, H.S.; Yang, H.I.; Yoo, M.C.; Kim, C.; Kim, K.S. Optimized THP-1 differentiation is required for the detection of responses to weak stimuli. Inflamm. Res. 2007, 56, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Pieterse, E.; Rother, N.; Yanginlar, C.; Gerretsen, J.; Boeltz, S.; Munoz, L.E.; Herrmann, M.; Pickkers, P.; Hilbrands, L.B.; Van Der Vlag, J. Cleaved N-terminal histone tails distinguish between NADPH oxidase (NOX)-dependent and NOX-independent pathways of neutrophil extracellular trap formation. Ann. Rheum. Dis. 2018, 77, 1790–1798. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wysocka, J.; Sayegh, J.; Lee, Y.H.; Pertin, J.R.; Leonelli, L.; Sonbuchner, L.S.; McDonald, C.H.; Cook, R.G.; Dou, Y.; et al. Human PAD4 regulates histone arginine methylation levels via demethylimination. Science 2004, 306, 279–283. [Google Scholar] [CrossRef]
- Mishra, N.; Schwerdtner, L.; Sams, K.; Mondal, S.; Ahmad, F.; Schmidt, R.E.; Coonrod, S.A.; Thompson, P.R.; Lerch, M.M.; Bossaller, L. Cutting Edge: Protein Arginine Deiminase 2 and 4 Regulate NLRP3 Inflammasome–Dependent IL-1β Maturation and ASC Speck Formation in Macrophages. J. Immunol. 2019, 203, 795–800. [Google Scholar] [CrossRef]
- Schaper, F.; de Leeuw, K.; Horst, G.; Bootsma, H.; Limburg, P.C.; Heeringa, P.; Bijl, M.; Westra, J. High mobility group box 1 skews macrophage polarization and negatively influences phagocytosis of apoptotic cells. Rheumatology (UK) 2016, 55, 2260–2270. [Google Scholar] [CrossRef] [Green Version]
- Pieterse, E.; Hofstra, J.; Berden, J.; Herrmann, M.; Dieker, J.; van der Vlag, J. Acetylated histones contribute to the immunostimulatory potential of neutrophil extracellular traps in systemic lupus erythematosus. Clin. Exp. Immunol. 2015, 179, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Chapman, E.A.; Lyon, M.; Simpson, D.; Mason, D.; Beynon, R.J.; Moots, R.J.; Wright, H.L. Caught in a trap? Proteomic analysis of neutrophil extracellular traps in rheumatoid arthritis and systemic lupus erythematosus. Front. Immunol. 2019, 10, 1–20. [Google Scholar] [CrossRef]
- Kilsgård, O.; Andersson, P.; Malmsten, M.; Nordin, S.L.; Linge, H.M.; Eliasson, M.; Sörenson, E.; Erjefält, J.S.; Bylund, J.; Olin, A.I.; et al. Peptidylarginine deiminases present in the airways during tobacco smoking and inflammation can citrullinate the host defense peptide LL-37, resulting in altered activities. Am. J. Respir. Cell Mol. Biol. 2012, 46, 240–248. [Google Scholar] [CrossRef]
- Hamam, H.J.; Palaniyar, N. Post-Translational Modifications in NETosis and NETs-Mediated Diseases. Biomolecules 2019, 9, 369. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.L.; Tangsombatvisit, S.; Rosenberg, J.M.; Mandelbaum, G.; Gillespie, E.C.; Gozani, O.P.; Alizadeh, A.A.; Utz, P.J. Specific post-translational histone modifications of neutrophil extracellular traps as immunogens and potential targets of lupus autoantibodies. Arthritis Res. Ther. 2012, 14, R25. [Google Scholar] [CrossRef] [Green Version]
- Saini, V.; Marchese, A.; Majetschak, M. CXC chemokine receptor 4 is a cell surface receptor for extracellular ubiquitin. J. Biol. Chem. 2010, 285, 15566–15576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zumerle, S.; Calı, B.; Munari, F.; Angioni, R.; Di Virgilio, F.; Molon, B.; Viola, A. Intercellular Calcium Signaling Induced by ATP Potentiates Macrophage Phagocytosis. Cell Rep. 2019, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Martin, M.; Leffler, J.; Blom, A.M. Annexin A2 and A5 serve as new ligands for C1q on apoptotic cells. J. Biol. Chem. 2012, 287, 33733–33744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neeli, I.; Radic, M. Opposition between PKC isoforms regulates histone deimination and neutrophil extracellular chromatin release. Front. Immunol. 2013, 4, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Kužnik, A.; Benčina, M.; Švajger, U.; Jeras, M.; Rozman, B.; Jerala, R. Mechanism of Endosomal TLR Inhibition by Antimalarial Drugs and Imidazoquinolines. J. Immunol. 2011, 186, 4794–4804. [Google Scholar] [CrossRef] [Green Version]
- Roh, J.S.; Sohn, D.H. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw. 2018, 18, 1–14. [Google Scholar] [CrossRef]
- Czaikoski, P.G.; Mota, J.M.S.C.; Nascimento, D.C.; Sônego, F.; Castanheira, F.V.E.S.; Melo, P.H.; Scortegagna, G.T.; Silva, R.L.; Barroso-Sousa, R.; Souto, F.O.; et al. Neutrophil extracellular traps induce organ damage during experimental and clinical sepsis. PLoS ONE 2016, 11, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Mitsios, A.; Arampatzioglou, A.; Arelaki, S.; Mitroulis, I.; Ritis, K. NETopathies? Unraveling the Dark Side of Old Diseases through Neutrophils. Front. Immunol. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Joshi, M.B.; Philippova, M.; Erne, P.; Hasler, P.; Hahn, S.; Resink, T.J. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett. 2010, 584, 3193–3197. [Google Scholar] [CrossRef] [Green Version]
- Caudrillier, A.; Kessenbrock, K.; Gilliss, B.M.; Nguyen, J.X.; Marques, M.B.; Monestier, M.; Toy, P.; Werb, Z.; Looney, M.R. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J. Clin. Investig. 2012, 122, 2661–2671. [Google Scholar] [CrossRef]
- Yoshida, M.; Sasaki, M.; Sugisaki, K.; Yamaguchi, Y.; Yamada, M. Neutrophil extracellular trap components in fibrinoid necrosis of the kidney with myeloperoxidase-ANCA-associated vasculitis. Clin. Kidney J. 2013, 6, 308–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.C.-S.; Yu, H.-S.; Yen, F.-L.; Lin, C.-L.; Chen, G.-S.; Lan, C.-C.E. Neutrophil extracellular trap formation is increased in psoriasis and induces human β-defensin-2 production in epidermal keratinocytes. Sci. Rep. 2016, 6, 31119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanecko, S.; Laskowski, M. Studies of the Specificity of Deoxyribonuclease I. III. Hydrolysis of Chains Carrying a Monoesterified Phosphate on Carbon 5′. J. Biol. Chem. 1961, 236, 3312–3316. [Google Scholar]
- Donis-Maturano, L.; Sánchez-Torres, L.E.; Cerbulo-Vázquez, A.; Chacón-Salinas, R.; García-Romo, G.S.; Orozco-Uribe, M.C.; Yam-Puc, J.C.; González-Jiménez, M.A.; Paredes-Vivas, Y.L.; Calderón-Amador, J.; et al. Prolonged exposure to neutrophil extracellular traps can induce mitochondrial damage in macrophages and dendritic cells. Springerplus 2015, 4, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Zhao, Y.; Lai, D.; Zhang, P.; Yang, Y.; Li, Y.; Fei, K.; Jiang, G.; Fan, J. Neutrophil extracellular traps promote macrophage pyroptosis in sepsis article. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- McDonald, B.; Urrutia, R.; Yipp, B.G.; Jenne, C.N.; Kubes, P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe 2012, 12, 324–333. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Nishi, H.; Travers, R.; Tsuboi, N.; Martinod, K.; Wagner, D.D.; Stan, R.; Croce, K.; Mayadas, T.N. Endocytosis of soluble immune complexes leads to their clearance by FcgRIIIB but induces neutrophil extracellular traps via FcgRIIA in vivo. Blood 2016, 120, 4421–4432. [Google Scholar] [CrossRef] [Green Version]
- Maueröder, C.; Kienhöfer, D.; Hahn, J.; Schauer, C.; Manger, B.; Schett, G.; Herrmann, M.; Hoffmann, M.H. How neutrophil extracellular traps orchestrate the local immune response in gout. J. Mol. Med. 2015, 93, 727–734. [Google Scholar] [CrossRef]
- Hahn, J.; Schauer, C.; Czegley, C.; Kling, L.; Petru, L.; Schmid, B.; Weidner, D.; Reinwald, C.; Biermann, M.H.C.; Blunder, S.; et al. Aggregated neutrophil extracellular traps resolve inflammation by proteolysis of cytokines and chemokines and protection from antiproteases. FASEB J. 2019, 33, 1401–1414. [Google Scholar] [CrossRef]
- Neumann, A.; Brogden, G.; von Köckritz-Blickwede, M. Extracellular Traps: An Ancient Weapon of Multiple Kingdoms. Biology 2020, 9, 34. [Google Scholar] [CrossRef] [Green Version]
- Loureiro, A.; Pais, C.; Sampaio, P. Relevance of Macrophage Extracellular Traps in C. albicans Killing. Front. Immunol. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.-W. The Role of ESX-1 in Mycobacterium tuberculosis Pathogenesis. Microbiol. Spectr. 2017, 5, 1–8. [Google Scholar] [CrossRef]
- Wong, K.W.; Jacobs, W.R. Mycobacterium tuberculosis exploits human interferon γ to stimulate macrophage extracellular trap formation and necrosis. J. Infect. Dis. 2013, 208, 109–119. [Google Scholar] [CrossRef] [Green Version]
- Branzk, N.; Lubojemska, A.; Hardison, S.E.; Wang, Q.; Gutierrez, M.G.; Brown, G.D.; Papayannopoulos, V. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat. Immunol. 2014, 15, 1017–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homa, J.; Ortmann, W.; Kolaczkowska, E. Conservative mechanisms of extracellular trap formation by annelida eisenia andrei: Serine protease activity requirement. PLoS ONE 2016, 11, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Bonne-Année, S.; Kerepesi, L.A.; Hess, J.A.; Wesolowski, J.; Paumet, F.; Lok, J.B.; Nolan, T.J.; Abraham, D. Extracellular traps are associated with human and mouse neutrophil and macrophage mediated killing of larval Strongyloides stercoralis. Microbes Infect. 2014, 16, 502–511. [Google Scholar] [CrossRef] [Green Version]
- King, P.T.; Sharma, R.; O’Sullivan, K.; Selemidis, S.; Lim, S.; Radhakrishna, N.; Lo, C.; Prasad, J.; Callaghan, J.; McLaughlin, P.; et al. Nontypeable haemophilus influenzae induces sustained lung oxidative stress and protease expression. PLoS ONE 2015, 10, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Yost, C.C.; Schwertz, H.; Cody, M.J.; Wallace, J.A.; Campbell, R.A.; Vieira-De-Abreu, A.; Araujo, C.V.; Schubert, S.; Harris, E.S.; Rowley, J.W.; et al. Neonatal NET-inhibitory factor and related peptides inhibit neutrophil extracellular trap formation. J. Clin. Investig. 2016, 126, 3783–3798. [Google Scholar] [CrossRef]
- Okubo, K.; Kamiya, M.; Urano, Y.; Nishi, H.; Herter, J.M.; Mayadas, T.; Hirohama, D.; Suzuki, K.; Kawakami, H.; Tanaka, M.; et al. Lactoferrin Suppresses Neutrophil Extracellular Traps Release in Inflammation. EBioMedicine 2016, 10, 204–215. [Google Scholar] [CrossRef] [Green Version]
- Van Der Wal, A.; Norde, W.; Zehnder, A.J.B.; Lyklema, J. Determination of the total charge in the cell walls of Gram-positive bacteria. Colloids Surfaces B Biointerfaces 1997, 9, 81–100. [Google Scholar] [CrossRef]
- Gottenbos, B.; Grijpma, D.W.; van der Mei, H.C.; Feijen, J.; Busscher, H.J. Antimicrobial effects of positively charged surfaces on adhering Gram-positive and Gram-negative bacteria. J. Antimicrob. Chemother. 2001, 48, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Bartneck, M.; Keul, H.A.; Gabriele, Z.K.; Groll, J. Phagocytosis independent extracellular nanoparticle clearance by human immune cells. Nano Lett. 2010, 10, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, G. Nucleic Acid Immunity, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2017; Volume 133.100. [Google Scholar]
- Moss, T.; Leblanc, B. DNA-Protein Interactions: Principles and Protocols Methods in Molecular Biology, 4th ed.; Springer: Berlin/Heidelberg, Germany, 2015; ISBN 1603270140. [Google Scholar]
- Napirei, M.; Ludwig, S.; Mezrhab, J.; Klo, T.; Mannherz, H.G. Murine serum nucleases—Contrasting effects of plasmin and heparin on the activities of DNase1 and DNase1-like 3 (DNase1l3). FEBS J. 2009, 276, 1059–1073. [Google Scholar] [CrossRef] [PubMed]
- Scieszka, D.; Lin, Y.; Li, W.; Choudhury, S.; Yu, Y.; Freire, M. Netome: The Molecular Characterization of Neutrophil Extracellular Traps (NETs). bioRxiv 2020. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santocki, M.; Kolaczkowska, E. On Neutrophil Extracellular Trap (NET) Removal: What We Know Thus Far and Why So Little. Cells 2020, 9, 2079. https://doi.org/10.3390/cells9092079
Santocki M, Kolaczkowska E. On Neutrophil Extracellular Trap (NET) Removal: What We Know Thus Far and Why So Little. Cells. 2020; 9(9):2079. https://doi.org/10.3390/cells9092079
Chicago/Turabian StyleSantocki, Michal, and Elzbieta Kolaczkowska. 2020. "On Neutrophil Extracellular Trap (NET) Removal: What We Know Thus Far and Why So Little" Cells 9, no. 9: 2079. https://doi.org/10.3390/cells9092079
APA StyleSantocki, M., & Kolaczkowska, E. (2020). On Neutrophil Extracellular Trap (NET) Removal: What We Know Thus Far and Why So Little. Cells, 9(9), 2079. https://doi.org/10.3390/cells9092079