Oxidoreductases in Glycoprotein Glycosylation, Folding, and ERAD



Abstract
:1. Introduction
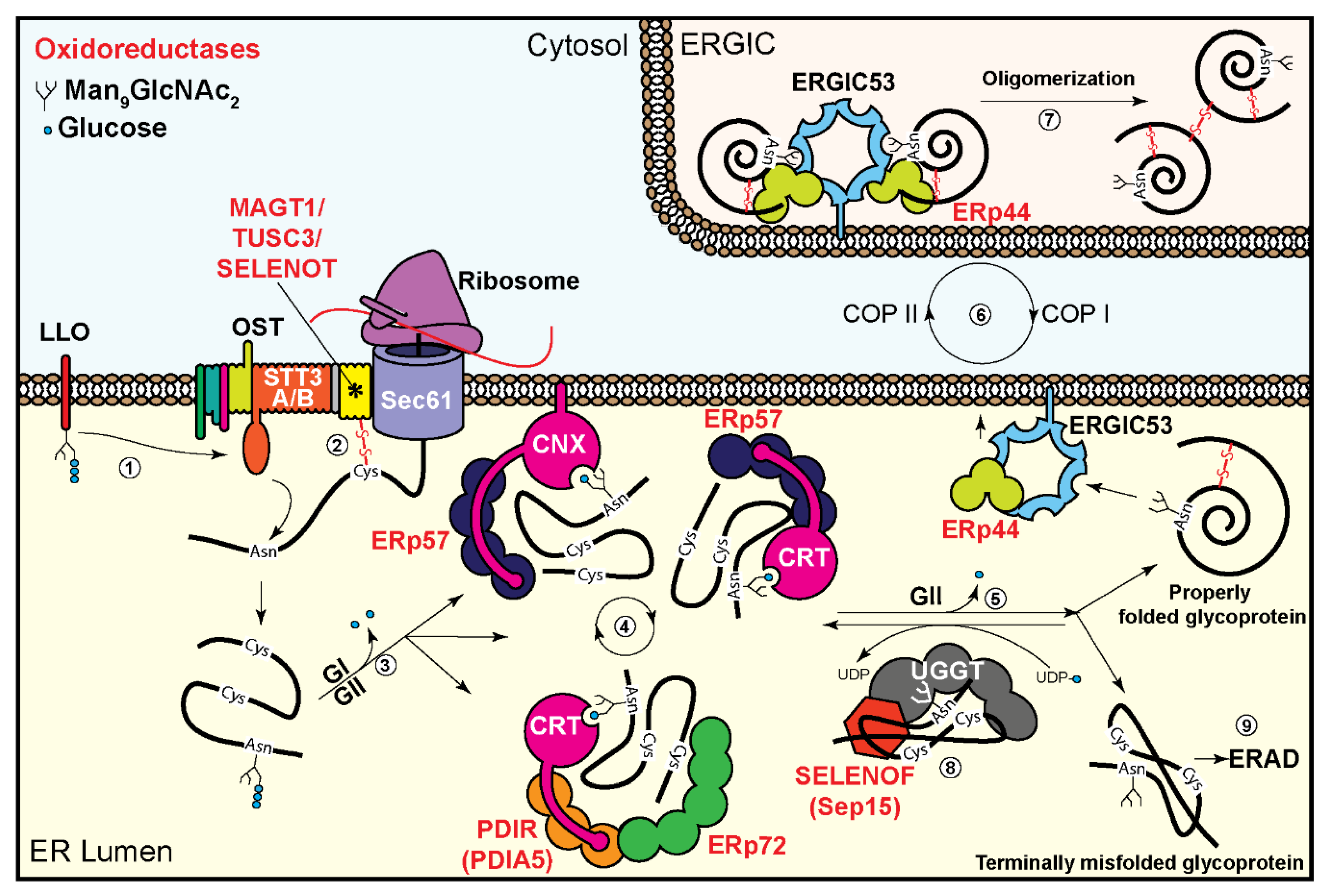
2. Oxidoreductases in the Oligosaccharyltransferase Complex
3. Oxidoreductases Associated with the Glycoprotein Folding Machinery
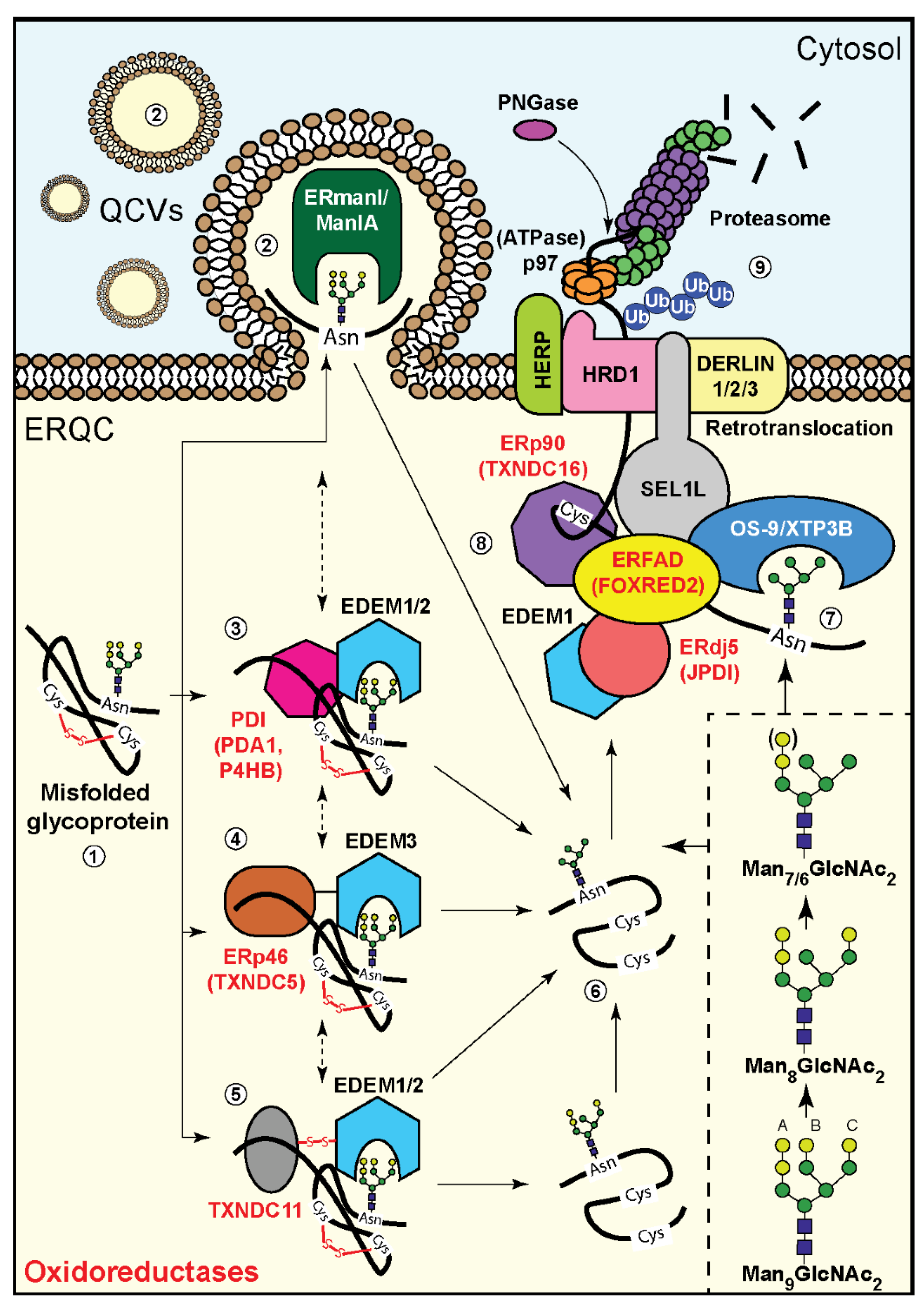
4. Involvement of Oxidoreductases in Mannose Trimming of Unfolded Glycoproteins
5. Oxidoreductases and Glycoprotein Targeting to ERAD
6. Concluding Remarks
Funding
Conflicts of Interest
References
- Bošnjak, I.; Bojović, V.; Šegvić-Bubić, T.; Bielen, A. Occurrence of protein disulfide bonds in different domains of life: A comparison of proteins from the Protein Data Bank. Protein. Eng. Des. Sel. 2014, 27, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P.J.; Bulleid, N.J. Mechanisms of Disulfide Bond Formation in Nascent Polypeptides Entering the Secretory Pathway. Cells 2020, 9, 1994. [Google Scholar] [CrossRef] [PubMed]
- Guerra, C.; Molinari, M. Thioredoxin-Related Transmembrane Proteins: TMX1 and Little Brothers TMX2, TMX3, TMX4 and TMX5. Cells 2020, 9, 2000. [Google Scholar] [CrossRef]
- Araki, K.; Inaba, K. Structure, mechanism, and evolution of Ero1 family enzymes. Antioxid. Redox. Signal. 2012, 16, 790–799. [Google Scholar] [CrossRef]
- Gross, E.; Sevier, C.S.; Heldman, N.; Vitu, E.; Bentzur, M.; Kaiser, C.A.; Thorpe, C.; Fass, D. Generating disulfides enzymatically: Reaction products and electron acceptors of the endoplasmic reticulum thiol oxidase Ero1p. Proc. Natl. Acad. Sci. USA 2006, 103, 299–304. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, V.D.; Saaranen, M.J.; Karala, A.R.; Lappi, A.K.; Wang, L.; Raykhel, I.B.; Alanen, H.I.; Salo, K.E.; Wang, C.C.; Ruddock, L.W. Two endoplasmic reticulum PDI peroxidases increase the efficiency of the use of peroxide during disulfide bond formation. J. Mol. Biol. 2011, 406, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Tavender, T.J.; Springate, J.J.; Bulleid, N.J. Recycling of peroxiredoxin IV provides a novel pathway for disulphide formation in the endoplasmic reticulum. EMBO J. 2010, 29, 4185–4197. [Google Scholar] [CrossRef] [Green Version]
- Zito, E.; Melo, E.P.; Yang, Y.; Wahlander, Å.; Neubert, T.A.; Ron, D. Oxidative protein folding by an endoplasmic reticulum-localized peroxiredoxin. Mol. Cell 2010, 40, 787–797. [Google Scholar] [CrossRef] [Green Version]
- Benham, A.M. The protein disulfide isomerase family: Key players in health and disease. Antioxid. Redox. Signal. 2012, 16, 781–789. [Google Scholar] [CrossRef]
- Fass, D.; Thorpe, C. Chemistry and Enzymology of Disulfide Cross-Linking in Proteins. Chem. Rev. 2018, 118, 1169–1198. [Google Scholar] [CrossRef]
- Hudson, D.A.; Gannon, S.A.; Thorpe, C. Oxidative protein folding: From thiol-disulfide exchange reactions to the redox poise of the endoplasmic reticulum. Free Radic. Biol. Med. 2015, 80, 171–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnér, E.S. Selenoproteins-What unique properties can arise with selenocysteine in place of cysteine? Exp. Cell Res. 2010, 316, 1296–1303. [Google Scholar] [CrossRef] [PubMed]
- Pitts, M.W.; Hoffmann, P.R. Endoplasmic reticulum-resident selenoproteins as regulators of calcium signaling and homeostasis. Cell Calcium 2018, 70, 76–86. [Google Scholar] [CrossRef]
- Lu, J.; Holmgren, A. Selenoproteins. J. Biol. Chem. 2009, 284, 723–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenkman, M.; Lederkremer, G.Z. Compartmentalization and Selective Tagging for Disposal of Misfolded Glycoproteins. Trends Biochem. Sci. 2019, 44, 827–836. [Google Scholar] [CrossRef] [PubMed]
- Kornfeld, R.; Kornfeld, S. Assembly of asparagine-linked oligosaccharides. Annu. Rev. Biochem. 1985, 54, 631–664. [Google Scholar] [CrossRef] [PubMed]
- Aebi, M.; Bernasconi, R.; Clerc, S.; Molinari, M. N-glycan structures: Recognition and processing in the ER. Trends Biochem. Sci. 2010, 35, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Tannous, A.; Pisoni, G.B.; Hebert, D.N.; Molinari, M. N-linked sugar-regulated protein folding and quality control in the ER. Semin. Cell Dev. Biol. 2015, 41, 79–89. [Google Scholar] [CrossRef] [Green Version]
- Mohanty, S.; Chaudhary, B.P.; Zoetewey, D. Structural Insight into the Mechanism of N-Linked Glycosylation by Oligosaccharyltransferase. Biomolecules 2020, 10, 624. [Google Scholar] [CrossRef] [Green Version]
- Mohorko, E.; Glockshuber, R.; Aebi, M. Oligosaccharyltransferase: The central enzyme of N-linked protein glycosylation. J. Inherit. Metab. Dis. 2011, 34, 869–878. [Google Scholar] [CrossRef]
- Pfeffer, S.; Dudek, J.; Gogala, M.; Schorr, S.; Linxweiler, J.; Lang, S.; Becker, T.; Beckmann, R.; Zimmermann, R.; Förster, F. Structure of the mammalian oligosaccharyl-transferase complex in the native ER protein translocon. Nat. Commun. 2014, 5, 3072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braakman, I.; Bulleid, N.J. Protein folding and modification in the mammalian endoplasmic reticulum. Annu. Rev. Biochem. 2011, 80, 71–99. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Wang, T.; Zhao, G.; Kovach, A.; Li, H. The atomic structure of a eukaryotic oligosaccharyltransferase complex. Nature 2018, 555, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Canada, C.; Kelleher, D.J.; Gilmore, R. Cotranslational and posttranslational N-glycosylation of polypeptides by distinct mammalian OST isoforms. Cell 2009, 136, 272–283. [Google Scholar] [CrossRef] [Green Version]
- Dumax-Vorzet, A.; Roboti, P.; High, S. OST4 is a subunit of the mammalian oligosaccharyltransferase required for efficient N-glycosylation. J. Cell Sci. 2013, 126 Pt 12, 2595–2606. [Google Scholar] [CrossRef] [Green Version]
- Shrimal, S.; Cherepanova, N.A.; Gilmore, R. DC2 and KCP2 mediate the interaction between the oligosaccharyltransferase and the ER translocon. J. Cell Biol. 2017, 216, 3625–3638. [Google Scholar] [CrossRef] [PubMed]
- Roboti, P.; High, S. The oligosaccharyltransferase subunits OST48, DAD1 and KCP2 function as ubiquitous and selective modulators of mammalian N-glycosylation. J. Cell Sci. 2012, 125 Pt 14, 3474–3484. [Google Scholar] [CrossRef] [Green Version]
- Cherepanova, N.A.; Shrimal, S.; Gilmore, R. Oxidoreductase activity is necessary for N-glycosylation of cysteine-proximal acceptor sites in glycoproteins. J. Cell Biol. 2014, 206, 525–539. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Clapham, D.E. Mammalian MagT1 and TUSC3 are required for cellular magnesium uptake and vertebrate embryonic development. Proc. Natl. Acad. Sci. USA 2009, 106, 15750–15755. [Google Scholar] [CrossRef] [Green Version]
- Goytain, A.; Quamme, G.A. Identification and characterization of a novel mammalian Mg2+ transporter with channel-like properties. BMC Genom. 2005, 6, 48. [Google Scholar] [CrossRef] [Green Version]
- Matsuda-Lennikov, M.; Biancalana, M.; Zou, J.; Ravell, J.C.; Zheng, L.; Kanellopoulou, C.; Jiang, P.; Notarangelo, G.; Jing, H.; Masutani, E.; et al. Magnesium transporter 1 (MAGT1) deficiency causes selective defects in N-linked glycosylation and expression of immune-response genes. J. Biol. Chem. 2019, 294, 13638–13656. [Google Scholar] [CrossRef] [PubMed]
- Chaigne-Delalande, B.; Li, F.Y.; O’Connor, G.M.; Lukacs, M.J.; Jiang, P.; Zheng, L.; Shatzer, A.; Biancalana, M.; Pittaluga, S.; Matthews, H.F.; et al. Mg2+ regulates cytotoxic functions of NK and CD8 T cells in chronic EBV infection through NKG2D. Science 2013, 341, 186–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.Y.; Lenardo, M.J.; Chaigne-Delalande, B. Loss of MAGT1 abrogates the Mg2+ flux required for T cell signaling and leads to a novel human primary immunodeficiency. Magnes. Res. 2011, 24, S109–S114. [Google Scholar] [CrossRef] [PubMed]
- Blommaert, E.; Péanne, R.; Cherepanova, N.A.; Rymen, D.; Staels, F.; Jaeken, J.; Race, V.; Keldermans, L.; Souche, E.; Corveleyn, A.; et al. Mutations in MAGT1 lead to a glycosylation disorder with a variable phenotype. Proc. Natl. Acad. Sci. USA 2019, 116, 9865–9870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.Y.; Chaigne-Delalande, B.; Kanellopoulou, C.; Davis, J.C.; Matthews, H.F.; Douek, D.C.; Cohen, J.I.; Uzel, G.; Su, H.C.; Lenardo, M.J. Second messenger role for Mg2+ revealed by human T-cell immunodeficiency. Nature 2011, 475, 471–476. [Google Scholar] [CrossRef]
- Ravell, J.C.; Chauvin, S.D.; He, T.; Lenardo, M. An Update on XMEN Disease. J. Clin. Immunol. 2020, 40, 671–681. [Google Scholar] [CrossRef]
- Schwarz, M.; Knauer, R.; Lehle, L. Yeast oligosaccharyltransferase consists of two functionally distinct sub-complexes, specified by either the Ost3p or Ost6p subunit. FEBS Lett. 2005, 579, 6564–6568. [Google Scholar] [CrossRef] [Green Version]
- Schulz, B.L.; Stirnimann, C.U.; Grimshaw, J.P.; Brozzo, M.S.; Fritsch, F.; Mohorko, E.; Capitani, G.; Glockshuber, R.; Grütter, M.G.; Aebi, M. Oxidoreductase activity of oligosaccharyltransferase subunits Ost3p and Ost6p defines site-specific glycosylation efficiency. Proc. Natl. Acad. Sci. USA 2009, 106, 11061–11066. [Google Scholar] [CrossRef] [Green Version]
- Mohorko, E.; Owen, R.L.; Malojčić, G.; Brozzo, M.S.; Aebi, M.; Glockshuber, R. Structural basis of substrate specificity of human oligosaccharyl transferase subunit N33/Tusc3 and its role in regulating protein N-glycosylation. Structure 2014, 22, 590–601. [Google Scholar] [CrossRef] [Green Version]
- MacGrogan, D.; Levy, A.; Bova, G.S.; Isaacs, W.B.; Bookstein, R. Structure and methylation-associated silencing of a gene within a homozygously deleted region of human chromosome band 8p22. Genomics 1996, 35, 55–65. [Google Scholar] [CrossRef]
- Horak, P.; Tomasich, E.; Vaňhara, P.; Kratochvílová, K.; Anees, M.; Marhold, M.; Lemberger, C.E.; Gerschpacher, M.; Horvat, R.; Sibilia, M.; et al. TUSC3 loss alters the ER stress response and accelerates prostate cancer growth in vivo. Sci. Rep. 2014, 4, 3739. [Google Scholar] [CrossRef] [PubMed]
- Vaňhara, P.; Horak, P.; Pils, D.; Anees, M.; Petz, M.; Gregor, W.; Zeillinger, R.; Krainer, M. Loss of the oligosaccharyl transferase subunit TUSC3 promotes proliferation and migration of ovarian cancer cells. Int. J. Oncol. 2013, 42, 1383–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garshasbi, M.; Hadavi, V.; Habibi, H.; Kahrizi, K.; Kariminejad, R.; Behjati, F.; Tzschach, A.; Najmabadi, H.; Ropers, H.H.; Kuss, A.W. A defect in the TUSC3 gene is associated with autosomal recessive mental retardation. Am. J. Hum. Genet. 2008, 82, 1158–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slutsky, I.; Abumaria, N.; Wu, L.J.; Huang, C.; Zhang, L.; Li, B.; Zhao, X.; Govindarajan, A.; Zhao, M.G.; Zhuo, M.; et al. Enhancement of learning and memory by elevating brain magnesium. Neuron 2010, 65, 165–177. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Wan, W.; Zhang, Y.; Shang, W.; Pan, X.; Zhang, L.K.; Xiao, G. Comprehensive Interactome Analysis Reveals that STT3B Is Required for N-Glycosylation of Lassa Virus Glycoprotein. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.D.; Cherepanova, N.A.; Bozzacco, L.; MacDonald, M.R.; Gilmore, R.; Tai, A.W. Dengue Virus Hijacks a Noncanonical Oxidoreductase Function of a Cellular Oligosaccharyltransferase Complex. mBio 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Hamieh, A.; Cartier, D.; Abid, H.; Calas, A.; Burel, C.; Bucharles, C.; Jehan, C.; Grumolato, L.; Landry, M.; Lerouge, P.; et al. Selenoprotein T is a novel OST subunit that regulates UPR signaling and hormone secretion. EMBO Rep. 2017, 18, 1935–1946. [Google Scholar] [CrossRef]
- Pothion, H.; Jehan, C.; Tostivint, H.; Cartier, D.; Bucharles, C.; Falluel-Morel, A.; Boukhzar, L.; Anouar, Y.; Lihrmann, I. Selenoprotein T: An Essential Oxidoreductase Serving as a Guardian of Endoplasmic Reticulum Homeostasis. Antioxid. Redox. Signal 2020. [Google Scholar] [CrossRef]
- Jessop, C.E.; Watkins, R.H.; Simmons, J.J.; Tasab, M.; Bulleid, N.J.J. Protein disulphide isomerase family members show distinct substrate specificity: P5 is targeted to BiP client proteins. J. Cell Sci. 2009, 122 Pt 23, 4287–4295. [Google Scholar] [CrossRef] [Green Version]
- Hosoda, A.; Tokuda, M.; Akai, R.; Kohno, K.; Iwawaki, T. Positive contribution of ERdj5/JPDI to endoplasmic reticulum protein quality control in the salivary gland. Biochem J. 2009, 425, 117–125. [Google Scholar] [CrossRef] [Green Version]
- Frenkel, Z.; Shenkman, M.; Kondratyev, M.; Lederkremer, G.Z. Separate roles and different routing of calnexin and ERp57 in endoplasmic reticulum quality control revealed by interactions with asialoglycoprotein receptor chains. Mol. Biol. Cell 2004, 15, 2133–2142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozlov, G.; Gehring, K. Calnexin cycle—Structural features of the ER chaperone system. FEBS J. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zapun, A.; Darby, N.J.; Tessier, D.C.; Michalak, M.; Bergeron, J.J.; Thomas, D.Y. Enhanced catalysis of ribonuclease B folding by the interaction of calnexin or calreticulin with ERp57. J. Biol. Chem. 1998, 273, 6009–6012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frickel, E.M.; Frei, P.; Bouvier, M.; Stafford, W.F.; Helenius, A.; Glockshuber, R.; Ellgaard, L. ERp57 is a multifunctional thiol-disulfide oxidoreductase. J. Biol. Chem. 2004, 279, 18277–18287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliver, J.D.; van der Wal, F.J.; Bulleid, N.J.; High, S. Interaction of the thiol-dependent reductase ERp57 with nascent glycoproteins. Science 1997, 275, 86–88. [Google Scholar] [CrossRef] [PubMed]
- Vinaik, R.; Kozlov, G.; Gehring, K. Structure of the non-catalytic domain of the protein disulfide isomerase-related protein (PDIR) reveals function in protein binding. PLoS ONE 2013, 8, e62021. [Google Scholar] [CrossRef] [Green Version]
- Roversi, P.; Marti, L.; Caputo, A.T.; Alonzi, D.S.; Hill, J.C.; Dent, K.C.; Kumar, A.; Levasseur, M.D.; Lia, A.; Waksman, T.; et al. Interdomain conformational flexibility underpins the activity of UGGT, the eukaryotic glycoprotein secretion checkpoint. Proc. Natl. Acad. Sci. USA 2017, 114, 8544–8549. [Google Scholar] [CrossRef] [Green Version]
- Satoh, T.; Song, C.; Zhu, T.; Toshimori, T.; Murata, K.; Hayashi, Y.; Kamikubo, H.; Uchihashi, T.; Kato, K. Visualisation of a flexible modular structure of the ER folding-sensor enzyme UGGT. Sci. Rep. 2017, 7, 12142. [Google Scholar] [CrossRef] [Green Version]
- Yim, S.H.; Everley, R.A.; Schildberg, F.A.; Lee, S.G.; Orsi, A.; Barbati, Z.R.; Karatepe, K.; Fomenko, D.E.; Tsuji, P.A.; Luo, H.R.; et al. Role of Selenof as a Gatekeeper of Secreted Disulfide-Rich Glycoproteins. Cell Rep. 2018, 23, 1387–1398. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.C.; Fujimori, D.G.; Weissman, J.S. Htm1p-Pdi1p is a folding-sensitive mannosidase that marks N-glycoproteins for ER-associated protein degradation. Proc. Natl. Acad. Sci. USA 2016, 113, E4015–E4024. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Tukachinsky, H.; Huang, C.H.; Jao, C.; Chu, Y.R.; Tang, H.Y.; Mueller, B.; Schulman, S.; Rapoport, T.A.; Salic, A. Processing and turnover of the Hedgehog protein in the endoplasmic reticulum. J. Cell Biol. 2011, 192, 825–838. [Google Scholar] [CrossRef] [Green Version]
- He, K.; Cunningham, C.N.; Manickam, N.; Liu, M.; Arvan, P.; Tsai, B. PDI reductase acts on Akita mutant proinsulin to initiate retrotranslocation along the Hrd1/Sel1L-p97 axis. Mol. Biol. Cell 2015, 26, 3413–3423. [Google Scholar] [CrossRef]
- Shenkman, M.; Ron, E.; Yehuda, R.; Benyair, R.; Khalaila, I.; Lederkremer, G.Z. Mannosidase activity of EDEM1 and EDEM2 depends on an unfolded state of their glycoprotein substrates. Commun. Biol. 2018, 1, 172. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, A.; Stephanowitz, H.; Krause, E.; Volkwein, C.; Hirsch, C.; Jarosch, E.; Sommer, T. A Complex of Htm1 and the Oxidoreductase Pdi1 Accelerates Degradation of Misfolded Glycoproteins. J. Biol. Chem. 2016, 291, 12195–12207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timms, R.T.; Menzies, S.A.; Tchasovnikarova, I.A.; Christensen, L.C.; Williamson, J.C.; Antrobus, R.; Dougan, G.; Ellgaard, L.; Lehner, P.J. Genetic dissection of mammalian ERAD through comparative haploid and CRISPR forward genetic screens. Nat. Commun. 2016, 7, 11786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, G.; Ninagawa, S.; Yagi, H.; Saito, T.; Ishikawa, T.; Sakuma, T.; Yamamoto, T.; Imami, K.; Ishihama, Y.; Kato, K.; et al. EDEM2 stably disulfide-bonded to TXNDC11 catalyzes the first mannose trimming step in mammalian glycoprotein ERAD. eLife 2020, 9, e53455. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Ito, S.; Wada, I.; Hosokawa, N. ER-resident protein 46 (ERp46) triggers the mannose-trimming activity of ER degradation-enhancing α-mannosidase-like protein 3 (EDEM3). J. Biol. Chem. 2018, 293, 10663–10674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamiya, Y.; Kamiya, D.; Yamamoto, K.; Nyfeler, B.; Hauri, H.P.; Kato, K. Molecular basis of sugar recognition by the human L-type lectins ERGIC-53, VIPL, and VIP36. J. Biol. Chem. 2008, 283, 1857–1861. [Google Scholar] [CrossRef] [Green Version]
- Anelli, T.; Ceppi, S.; Bergamelli, L.; Cortini, M.; Masciarelli, S.; Valetti, C.; Sitia, R. Sequential steps and checkpoints in the early exocytic compartment during secretory IgM biogenesis. EMBO J. 2007, 26, 4177–4188. [Google Scholar] [CrossRef] [Green Version]
- Cortini, M.; Sitia, R. ERp44 and ERGIC-53 synergize in coupling efficiency and fidelity of IgM polymerization and secretion. Traffic 2010, 11, 651–659. [Google Scholar] [CrossRef]
- Anelli, T.; Alessio, M.; Bachi, A.; Bergamelli, L.; Bertoli, G.; Camerini, S.; Mezghrani, A.; Ruffato, E.; Simmen, T.; Sitia, R. Thiol-mediated protein retention in the endoplasmic reticulum: The role of ERp44. EMBO J. 2003, 22, 5015–5022. [Google Scholar] [CrossRef] [PubMed]
- Mariappan, M.; Radhakrishnan, K.; Dierks, T.; Schmidt, B.; von Figura, K. ERp44 mediates a thiol-independent retention of formylglycine-generating enzyme in the endoplasmic reticulum. J. Biol. Chem. 2008, 283, 6375–6383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakihana, T.; Araki, K.; Vavassori, S.; Iemura, S.; Cortini, M.; Fagioli, C.; Natsume, T.; Sitia, R.; Nagata, K. Dynamic regulation of Ero1α and peroxiredoxin 4 localization in the secretory pathway. J. Biol. Chem. 2013, 288, 29586–29594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, S.; Amagai, Y.; Sannino, S.; Tempio, T.; Anelli, T.; Harayama, M.; Masui, S.; Sorrentino, I.; Yamada, M.; Sitia, R.; et al. Zinc regulates ERp44-dependent protein quality control in the early secretory pathway. Nat. Commun. 2019, 10, 603. [Google Scholar] [CrossRef] [PubMed]
- Christianson, J.C.; Shaler, T.A.; Tyler, R.E.; Kopito, R.R. OS-9 and GRP94 deliver mutant alpha1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat. Cell Biol. 2008, 10, 272–282. [Google Scholar] [CrossRef] [Green Version]
- Groisman, B.; Shenkman, M.; Ron, E.; Lederkremer, G.Z. Mannose trimming is required for delivery of a glycoprotein from EDEM1 to XTP3-B and to late endoplasmic reticulum-associated degradation steps. J. Biol. Chem. 2011, 286, 1292–1300. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, N.; Kamiya, Y.; Kamiya, D.; Kato, K.; Nagata, K. Human OS-9, a lectin required for glycoprotein endoplasmic reticulum-associated degradation, recognizes mannose-trimmed N-glycans. J. Biol. Chem. 2009, 284, 17061–17068. [Google Scholar] [CrossRef] [Green Version]
- Riemer, J.; Hansen, H.G.; Appenzeller-Herzog, C.; Johansson, L.; Ellgaard, L. Identification of the PDI-family member ERp90 as an interaction partner of ERFAD. PLoS ONE 2011, 6, e17037. [Google Scholar] [CrossRef] [Green Version]
- Hagiwara, M.; Maegawa, K.; Suzuki, M.; Ushioda, R.; Araki, K.; Matsumoto, Y.; Hoseki, J.; Nagata, K.; Inaba, K. Structural basis of an ERAD pathway mediated by the ER-resident protein disulfide reductase ERdj5. Mol. Cell 2011, 41, 432–444. [Google Scholar] [CrossRef]
- Maegawa, K.I.; Watanabe, S.; Noi, K.; Okumura, M.; Amagai, Y.; Inoue, M.; Ushioda, R.; Nagata, K.; Ogura, T.; Inaba, K. The Highly Dynamic Nature of ERdj5 Is Key to Efficient Elimination of Aberrant Protein Oligomers through ER-Associated Degradation. Structure 2017, 25, 846–857.e4. [Google Scholar] [CrossRef] [Green Version]
- Ushioda, R.; Hoseki, J.; Araki, K.; Jansen, G.; Thomas, D.Y.; Nagata, K. ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science 2008, 321, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Ellgaard, L.; McCaul, N.; Chatsisvili, A.; Braakman, I. Co- and Post-Translational Protein Folding in the ER. Traffic 2016, 17, 615–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutkevich, L.A.; Williams, D.B. Participation of lectin chaperones and thiol oxidoreductases in protein folding within the endoplasmic reticulum. Curr. Opin. Cell Biol. 2011, 23, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Benyair, R.; Ron, E.; Lederkremer, G.Z. Protein quality control, retention, and degradation at the endoplasmic reticulum. Int. Rev. Cell Mol. Biol. 2011, 292, 197–280. [Google Scholar] [PubMed]
- Caramelo, J.J.; Parodi, A.J. A sweet code for glycoprotein folding. FEBS Lett. 2015, 589, 3379–3387. [Google Scholar] [CrossRef] [Green Version]
- Lederkremer, G.Z. Glycoprotein folding, quality control and ER-associated degradation. Curr. Opin. Struct. Biol. 2009, 19, 515–523. [Google Scholar] [CrossRef]
- Avezov, E.; Frenkel, Z.; Ehrlich, M.; Herscovics, A.; Lederkremer, G.Z. Endoplasmic reticulum (ER) mannosidase I is compartmentalized and required for N-glycan trimming to Man5-6GlcNAc2 in glycoprotein ER-associated degradation. Mol. Biol. Cell 2008, 19, 216–225. [Google Scholar] [CrossRef] [Green Version]
- Frenkel, Z.; Gregory, W.; Kornfeld, S.; Lederkremer, G.Z. Endoplasmic reticulum-associated degradation of mammalian glycoproteins involves sugar chain trimming to Man6-5GlcNAc2. J. Biol. Chem. 2003, 278, 34119–34124. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, N.; Tremblay, L.O.; Sleno, B.; Kamiya, Y.; Wada, I.; Nagata, K.; Kato, K.; Herscovics, A. EDEM1 accelerates the trimming of alpha1,2-linked mannose on the C branch of N-glycans. Glycobiology 2010, 20, 567–575. [Google Scholar] [CrossRef] [Green Version]
- Benyair, R.; Ogen-Shtern, N.; Lederkremer, G.Z. Glycan regulation of ER-associated degradation through compartmentalization. Semin. Cell Dev. Biol. 2015, 41, 99–109. [Google Scholar] [CrossRef]
- Herscovics, A.; Romero, P.A.; Tremblay, L.O. The specificity of the yeast and human class I ER alpha 1,2-mannosidases involved in ER quality control is not as strict previously reported. Glycobiology 2002, 12, 14G–15G. [Google Scholar] [PubMed]
- Benyair, R.; Ogen-Shtern, N.; Mazkereth, N.; Shai, B.; Ehrlich, M.; Lederkremer, G.Z. Mammalian ER mannosidase I resides in quality control vesicles, where it encounters its glycoprotein substrates. Mol. Biol. Cell 2015, 26, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Aikawa, J.; Matsuo, I.; Ito, Y. In vitro mannose trimming property of human ER alpha-1,2 mannosidase I. Glycocon. J. 2012, 29, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Marin, M.B.; Ghenea, S.; Spiridon, L.N.; Chiritoiu, G.N.; Petrescu, A.J.; Petrescu, S.M. Tyrosinase degradation is prevented when EDEM1 lacks the intrinsically disordered region. PLoS ONE 2012, 7, e42998. [Google Scholar] [CrossRef] [Green Version]
- Ron, E.; Shenkman, M.; Groisman, B.; Izenshtein, Y.; Leitman, J.; Lederkremer, G.Z. Bypass of glycan-dependent glycoprotein delivery to ERAD by up-regulated EDEM1. Mol. Biol. Cell 2011, 22, 3945–3954. [Google Scholar] [CrossRef]
- Shenkman, M.; Groisman, B.; Ron, E.; Avezov, E.; Hendershot, L.M.; Lederkremer, G.Z. A Shared Endoplasmic Reticulum-associated Degradation Pathway Involving the EDEM1 Protein for Glycosylated and Nonglycosylated Proteins. J. Biol. Chem. 2013, 288, 2167–2178. [Google Scholar] [CrossRef] [Green Version]
- Sakoh-Nakatogawa, M.; Nishikawa, S.; Endo, T. Roles of protein-disulfide isomerase-mediated disulfide bond formation of yeast Mnl1p in endoplasmic reticulum-associated degradation. J. Biol. Chem. 2009, 284, 11815–11825. [Google Scholar] [CrossRef] [Green Version]
- Sokolowska, I.; Pilka, E.S.; Sandvig, K.; Wegrzyn, G.; Slominska-Wojewodzka, M. Hydrophobicity of protein determinants influences the recognition of substrates by EDEM1 and EDEM2 in human cells. BMC Cell Biol. 2015, 16, 1. [Google Scholar] [CrossRef] [Green Version]
- Lamriben, L.; Oster, M.E.; Tamura, T.; Tian, W.; Yang, Z.; Clausen, H.; Hebert, D.N. EDEM1’s mannosidase-like domain binds ERAD client proteins in a redox-sensitive manner and possesses catalytic activity. J. Biol. Chem. 2018, 293, 13932–13945. [Google Scholar] [CrossRef] [Green Version]
- Ogen-Shtern, N.; Avezov, E.; Shenkman, M.; Benyair, R.; Lederkremer, G.Z. Mannosidase IA is in Quality Control Vesicles and Participates in Glycoprotein Targeting to ERAD. J. Mol. Biol. 2016, 428, 3194–3205. [Google Scholar] [CrossRef]
- Sun, Z.; Brodsky, J.I. Protein quality control in the secretory pathway. J. Cell Biol. 2019, 218, 3171–3187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ushioda, R.; Miyamoto, A.; Inoue, M.; Watanabe, S.; Okumura, M.; Maegawa, K.I.; Uegaki, K.; Fujii, S.; Fukuda, Y.; Umitsu, M.; et al. Redox-assisted regulation of Ca2+ homeostasis in the endoplasmic reticulum by disulfide reductase ERdj5. Proc. Natl. Acad. Sci. USA 2016, 113, E6055–E6063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarosch, E.; Taxis, C.; Volkwein, C.; Bordallo, J.; Finley, D.; Wolf, D.H.; Sommer, T. Protein dislocation from the ER requires polyubiquitination and the AAA-ATPase Cdc48. Nat. Cell Biol. 2002, 4, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Bordallo, J.; Plemper, R.K.; Finger, A.; Wolf, D.H. Der3p/Hrd1p is required for endoplasmic reticulum-associated degradation of misfolded lumenal and integral membrane proteins. Mol. Biol. Cell 1998, 9, 209–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bays, N.W.; Gardner, R.G.; Seelig, L.P.; Joazeiro, C.A.; Hampton, R.Y. Hrd1p/Der3p is a membrane-anchored ubiquitin ligase required for ER-associated degradation. Nat. Cell Biol. 2001, 3, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Baldridge, R.D.; Rapoport, T.A. Autoubiquitination of the Hrd1 Ligase Triggers Protein Retrotranslocation in ERAD. Cell 2016, 166, 394–407. [Google Scholar] [CrossRef] [Green Version]
- Kadowaki, H.; Satrimafitrah, P.; Takami, Y.; Nishitoh, H. Molecular mechanism of ER stress-induced pre-emptive quality control involving association of the translocon, Derlin-1, and HRD1. Sci. Rep. 2018, 8, 7317. [Google Scholar] [CrossRef] [Green Version]
- Leitman, J.; Shenkman, M.; Gofman, Y.; Shtern, N.O.; Ben-Tal, N.; Hendershot, L.M.; Lederkremer, G.Z. Herp coordinates compartmentalization and recruitment of HRD1 and misfolded proteins for ERAD. Mol. Biol. Cell 2014, 25, 1050–1060. [Google Scholar] [CrossRef]
- Ye, Y.; Tang, W.K.; Zhang, T.; Xia, D. A Mighty “Protein Extractor” of the Cell: Structure and Function of the p97/CDC48 ATPase. Front. Mol. Biosci. 2017, 4, 39. [Google Scholar] [CrossRef]
- Alexandru, G.; Graumann, J.; Smith, G.T.; Kolawa, N.J.; Fang, R.; Deshaies, R.J. UBXD7 binds multiple ubiquitin ligases and implicates p97 in HIF1alpha turnover. Cell 2008, 134, 804–816. [Google Scholar] [CrossRef] [Green Version]
- Allen, M.D.; Buchberger, A.; Bycroft, M. The PUB domain functions as a p97 binding module in human peptide N-glycanase. J. Biol. Chem. 2006, 281, 25502–25508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, S.G.; Khare, M.; Ramani, R.; Watts, G.D.; Simon, M.; Osann, K.E.; Donkervoort, S.; Dec, E.; Nalbandian, A.; Platt, J.; et al. Genotype-phenotype studies of VCP-associated inclusion body myopathy with Paget disease of bone and/or frontotemporal dementia. Clin. Genet. 2013, 83, 422–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bebeacua, C.; Förster, A.; McKeown, C.; Meyer, H.H.; Zhang, X.; Freemont, P.S. Distinct conformations of the protein complex p97-Ufd1-Npl4 revealed by electron cryomicroscopy. Proc. Natl. Acad. Sci. USA 2012, 109, 1098–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, P.; Stanley, A.M.; Rapoport, T.A. Retrotranslocation of a misfolded luminal ER protein by the ubiquitin-ligase Hrd1p. Cell 2010, 143, 579–591. [Google Scholar] [CrossRef] [Green Version]
- Ellgaard, L.; Sevier, C.S.; Bulleid, N.J. How Are Proteins Reduced in the Endoplasmic Reticulum? Trends Biochem. Sci. 2018, 43, 32–43. [Google Scholar] [CrossRef] [Green Version]
- Oka, O.B.; Pringle, M.A.; Schopp, I.M.; Braakman, I.; Bulleid, N.J. ERdj5 is the ER reductase that catalyzes the removal of non-native disulfides and correct folding of the LDL receptor. Mol. Cell 2013, 50, 793–804. [Google Scholar] [CrossRef] [Green Version]
- Grubb, S.; Guo, L.; Fisher, E.A.; Brodsky, J.L. Protein disulfide isomerases contribute differentially to the endoplasmic reticulum-associated degradation of apolipoprotein B and other substrates. Mol. Biol. Cell 2012, 23, 520–532. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Proteins | Interacting Oxidoreductases | Trx-Like Motifs |
|---|---|---|
| Glycosylation (OST Complex) | ||
| STT3A | SELENOT (SELT) [47,48] | Active: Trxl1-CVSU |
| STT3B | MAGT1 (IAP) [28,29,30,31,32,33,34,35,36,45,46] | Active: Trxl1-CVVC |
| TUSC3 (N33) [28,29,37,38,39,40,41,42,43,44,45] | Active: Trxl1-CSVC | |
| Protein Folding (Chaperones) | ||
| BiP * | PDI (PDA1, P4HB) [49] | Active: Trxl1,4-CGHC; Inactive: Trxl2,3 |
| P5 [49] | Active: Trxl1,2-CGHC | |
| ERdj5 (JPDI) [49,50] | Active: Trxl1-CPPC, Trxl2-CGPC, Trxl3-CHPC, Trxl4-CSHC | |
| CNX | ERp57 [51,52,53,54,55] | Active: Trxl1,4-CGHC; Inactive: Trxl2,3 |
| CRT | ERp57 [53,54,55] | |
| ERp72 [56] | Active: Trxl1,2,5-CGHC; Inactive: Trxl3,4 | |
| PDIR [56] | Active: Trxl2-CSMC, Trxl3-CGHC, Trxl4-CPHC; Inactive: Trxl1 | |
| N-Glycan Processing (Glucose addition) | ||
| UGGT | Inactive: Trxl1 (aa 45–220), Trxl2 (aa 414–656), Trxl3 (aa 667–880), Trxl (aa 275–410 and 897–950) [57,58] | |
| SELENOF (Sep15) [59] | Active: Trxl1-CGU | |
| N-Glycan Processing (Mannose trimming) | ||
| EDEM1 | P4HB (PDA1, PDI) [60,61,62,63,64] | |
| TXNDC11 [63,65] | Active: Trxl1-CELC, Trxl5-CGFC; Inactive: Trxl2,3,4 | |
| EDEM2 | PDI (PDA1, P4HB) [60,61,62,63,64] | |
| TXNDC11 [63,65,66] | ||
| EDEM3 | TXNDC11 [65] | |
| ERp46 (TXNDC5) [67] | Active: Trxl1,2,3-CGHC | |
| ER-Golgi trafficking and oligomerization | ||
| ERGIC-53 | ERp44 (TXNDC4) [68,69,70,71,72,73,74] | Active: Trxl1-CRFS; Inactive: Trxl2,3 |
| ERAD (Retrotranslocation) | ||
| OS-9 and SEL1L [75,76,77,78] | ERp90 (TXNDC16) | Inactive: Trxl1-CX8C, Trxl2-CX9C, Trxl3-CX6C, Trxl4,5 |
| ERFAD (FOXRED2) ** | NADPH-dependent reductase | |
| EDEM1 | ERdj5 (JPDI) [79,80,81] | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patel, C.; Saad, H.; Shenkman, M.; Lederkremer, G.Z. Oxidoreductases in Glycoprotein Glycosylation, Folding, and ERAD. Cells 2020, 9, 2138. https://doi.org/10.3390/cells9092138
Patel C, Saad H, Shenkman M, Lederkremer GZ. Oxidoreductases in Glycoprotein Glycosylation, Folding, and ERAD. Cells. 2020; 9(9):2138. https://doi.org/10.3390/cells9092138
Chicago/Turabian StylePatel, Chaitanya, Haddas Saad, Marina Shenkman, and Gerardo Z. Lederkremer. 2020. "Oxidoreductases in Glycoprotein Glycosylation, Folding, and ERAD" Cells 9, no. 9: 2138. https://doi.org/10.3390/cells9092138