Stimulation of Replication Template-Switching by DNA-Protein Crosslinks


Abstract
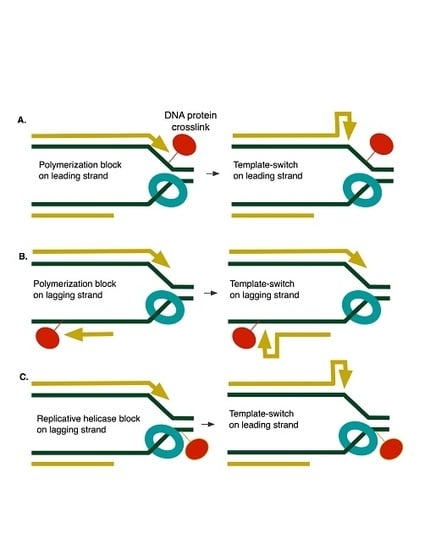
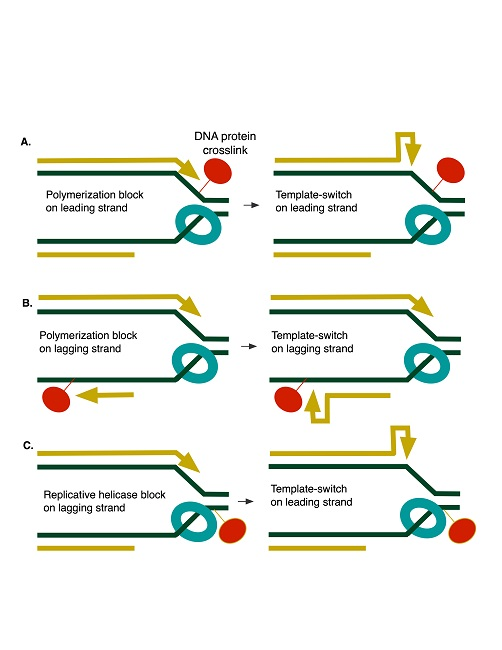
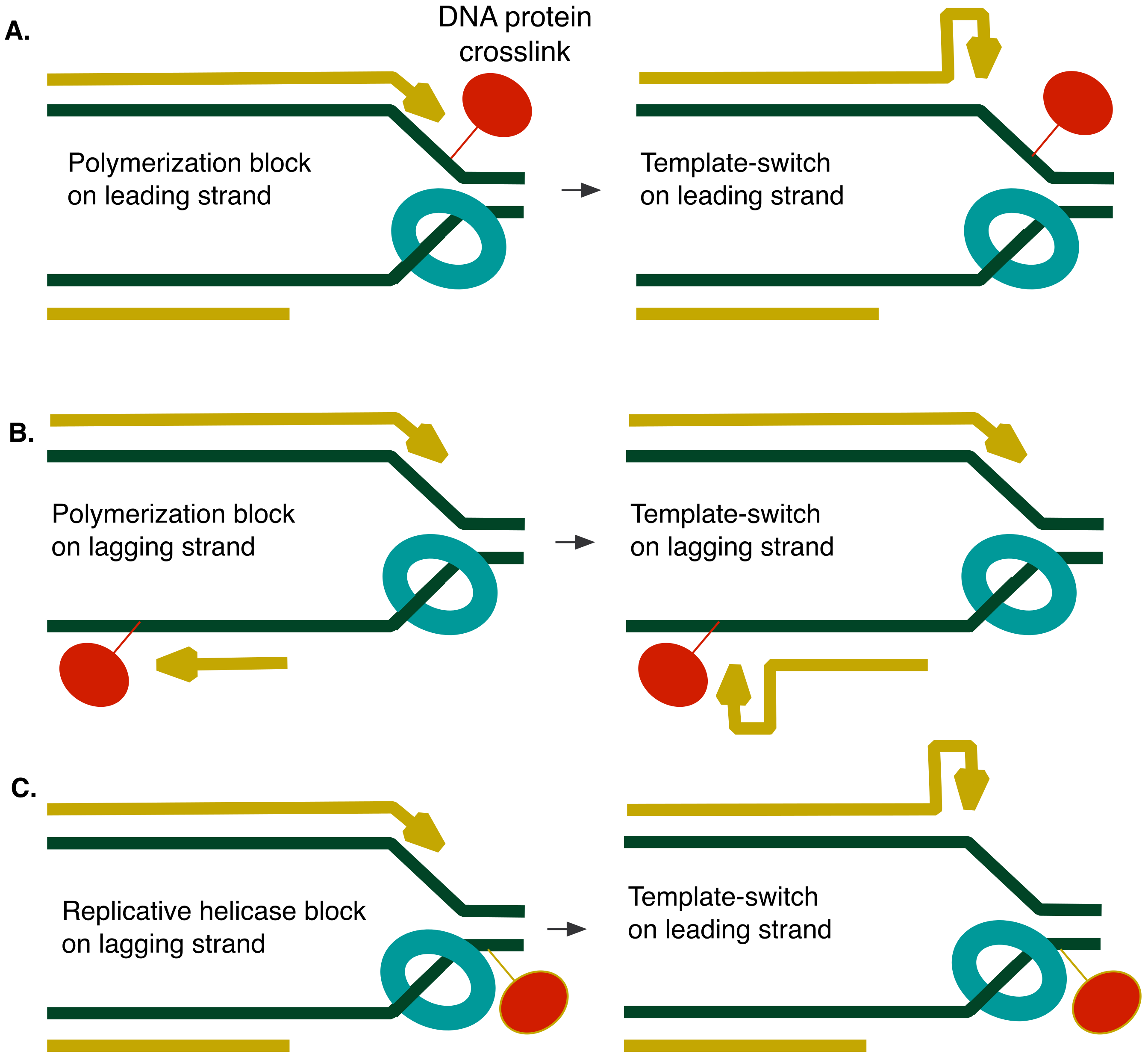
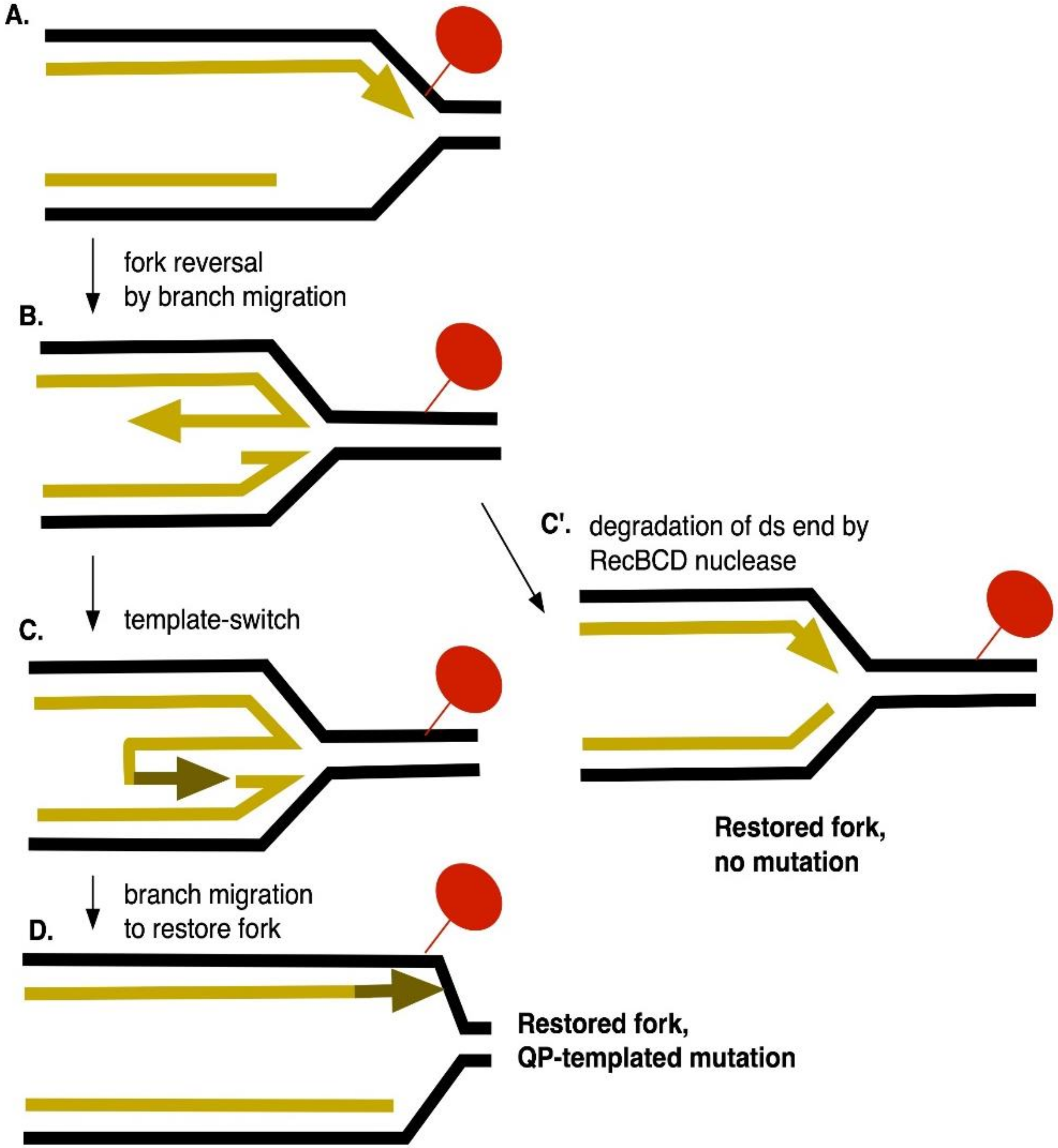
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains, Growth Conditions, and Media
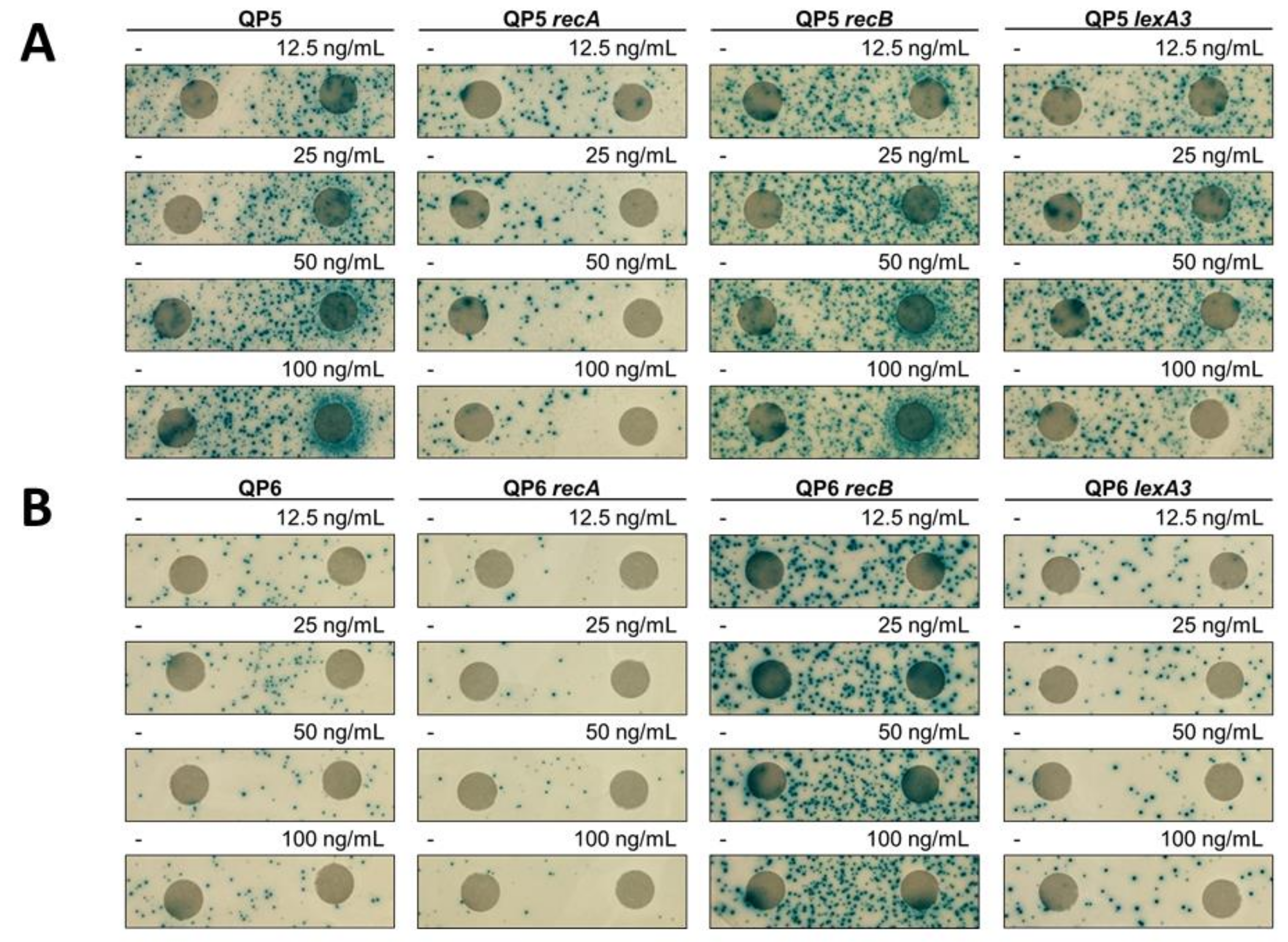
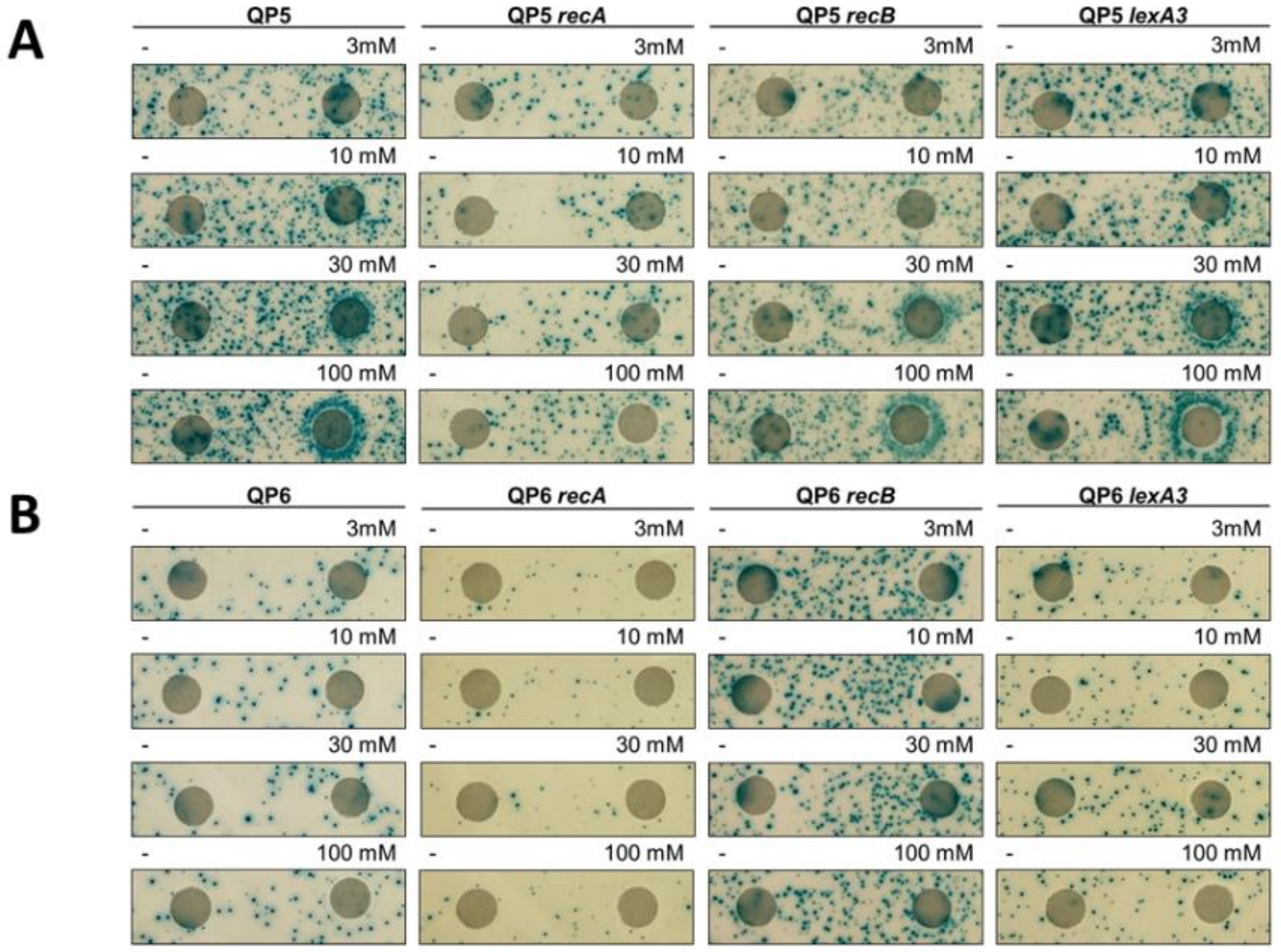
2.2. Disk Diffusion Assays
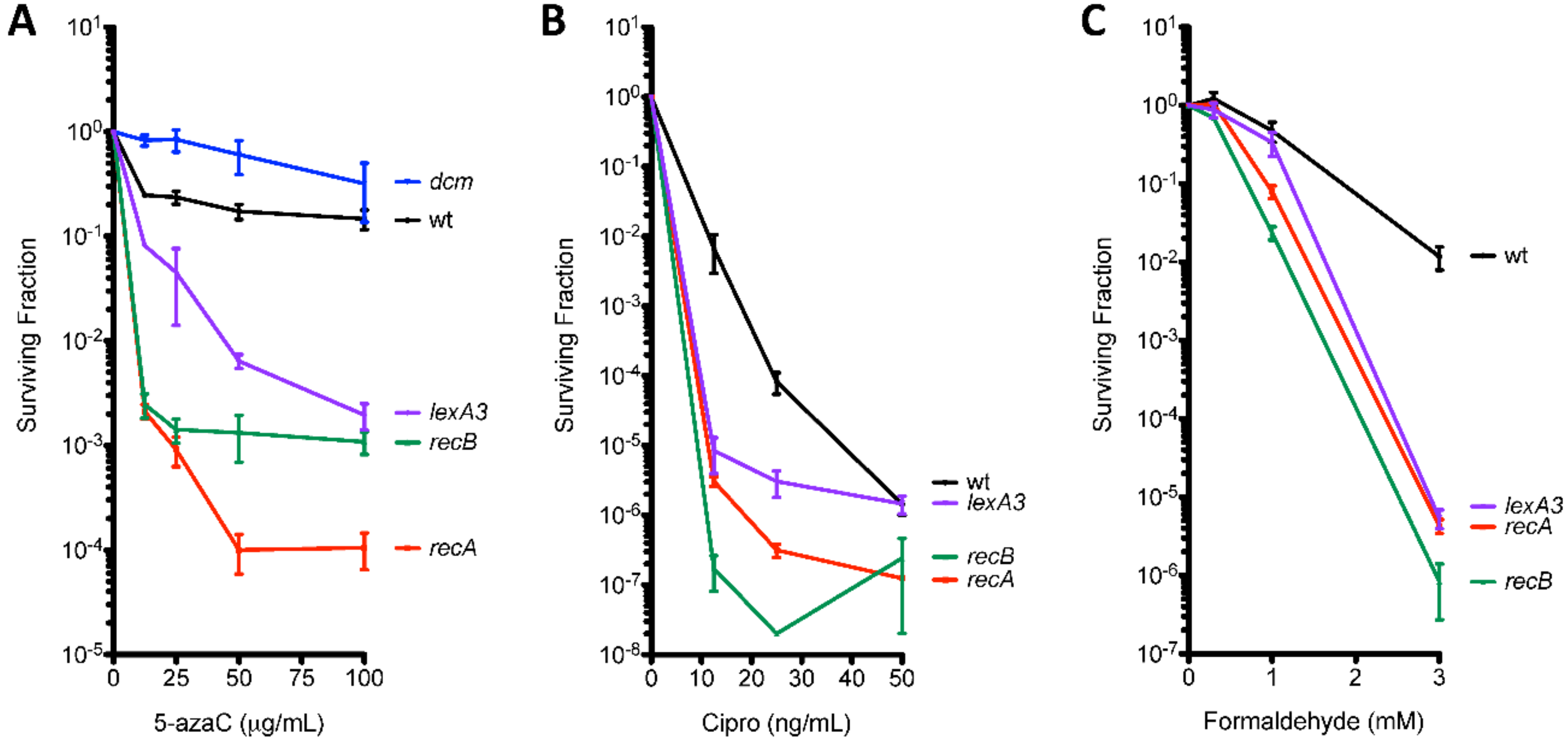
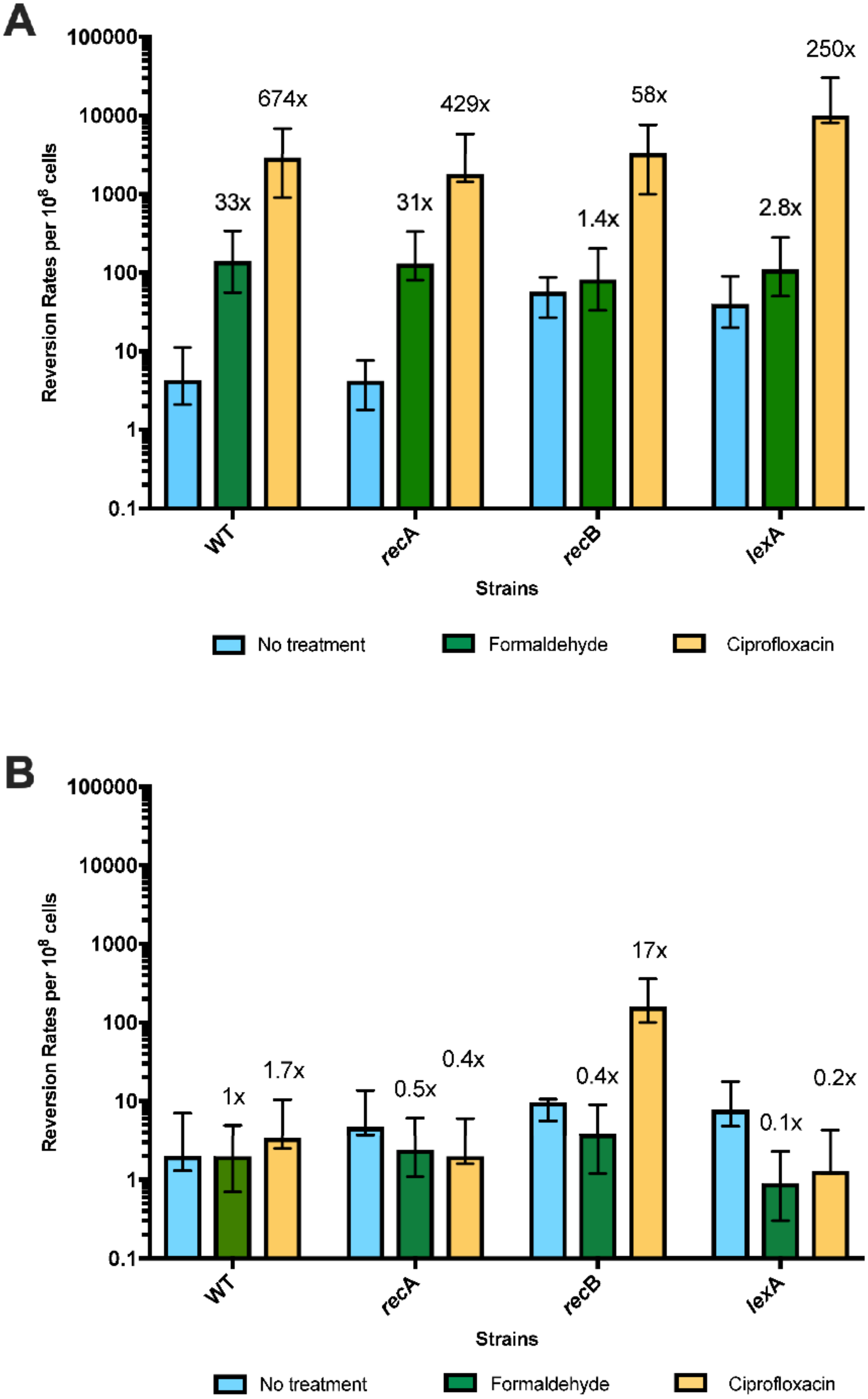
2.3. Survival Assays
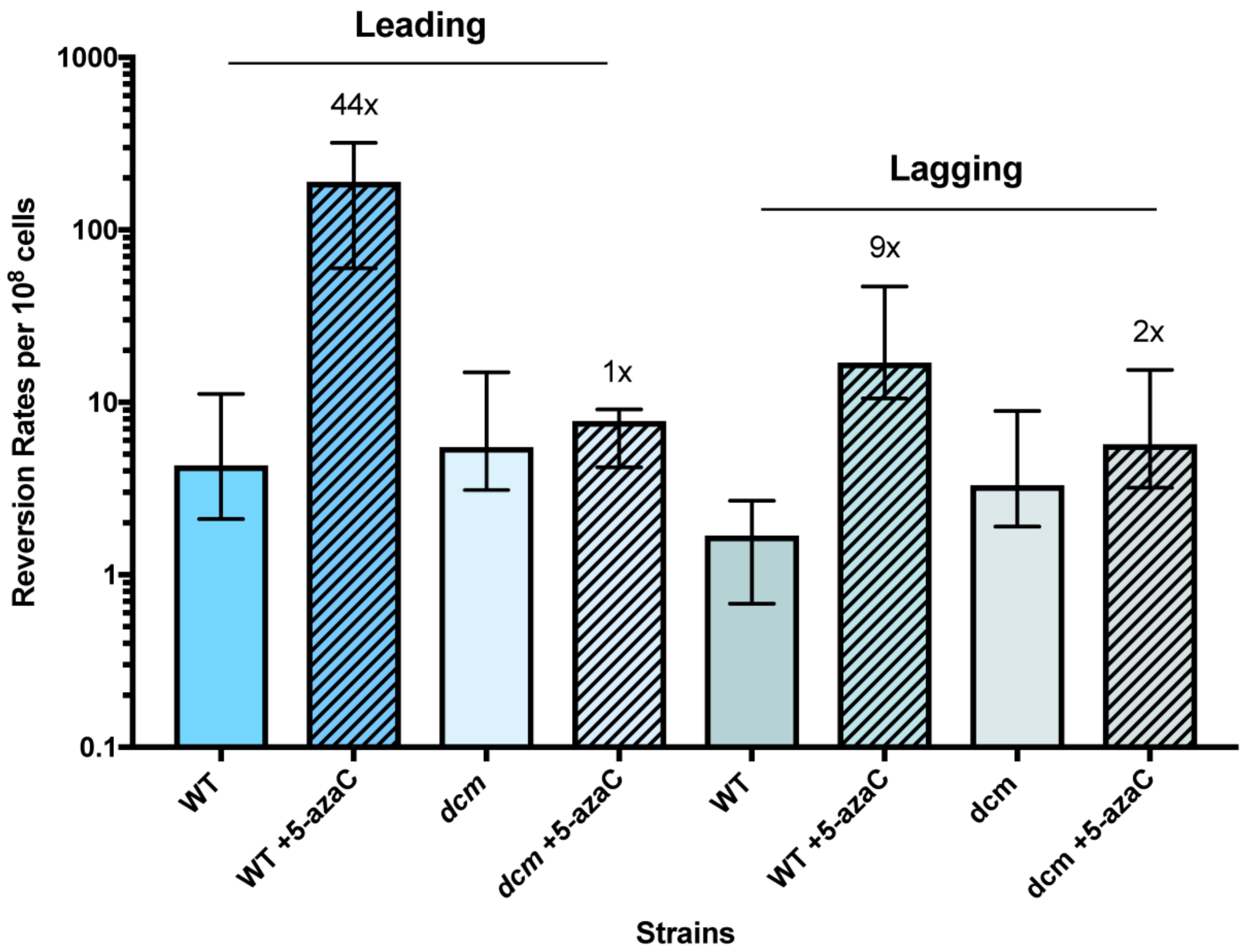
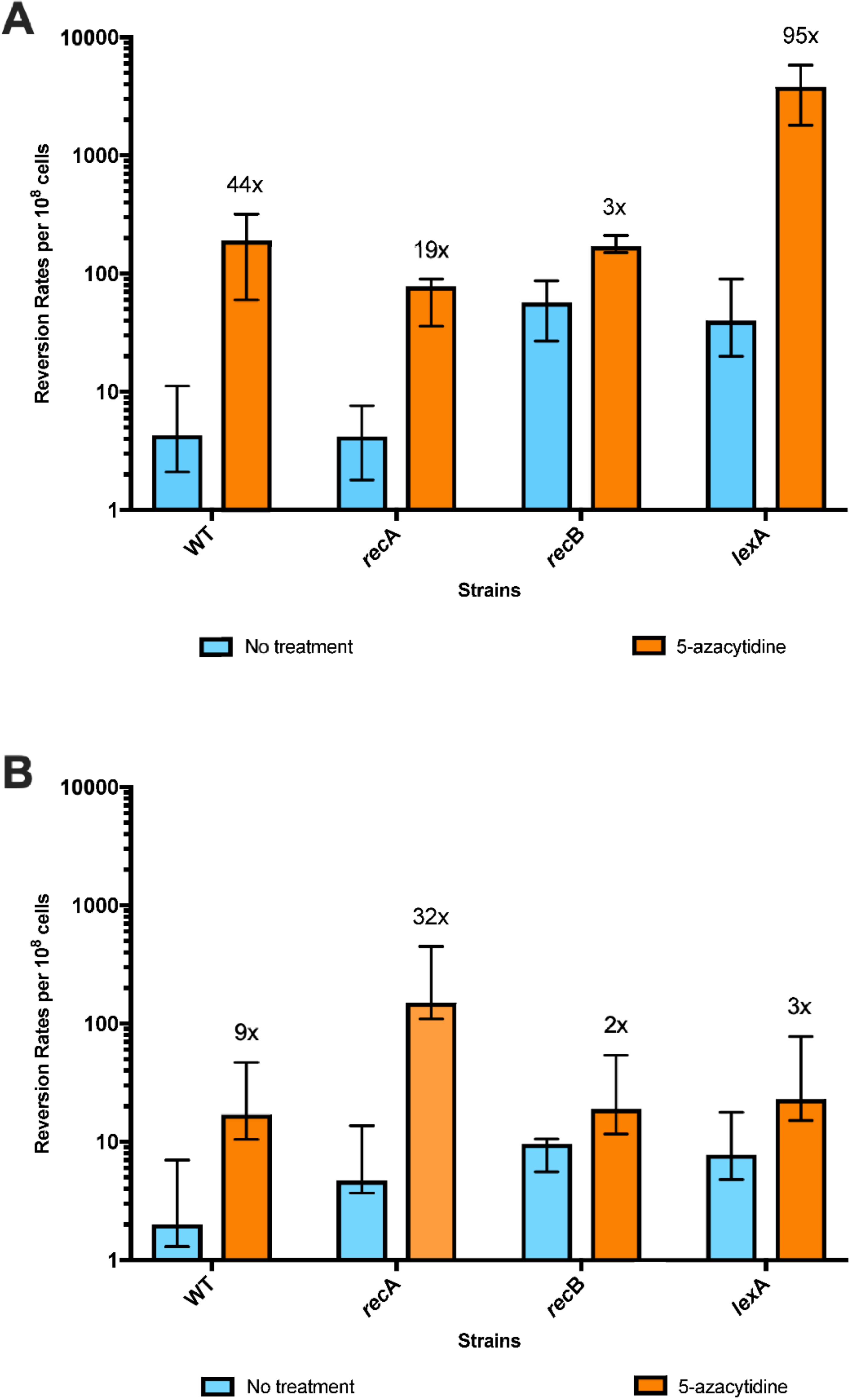
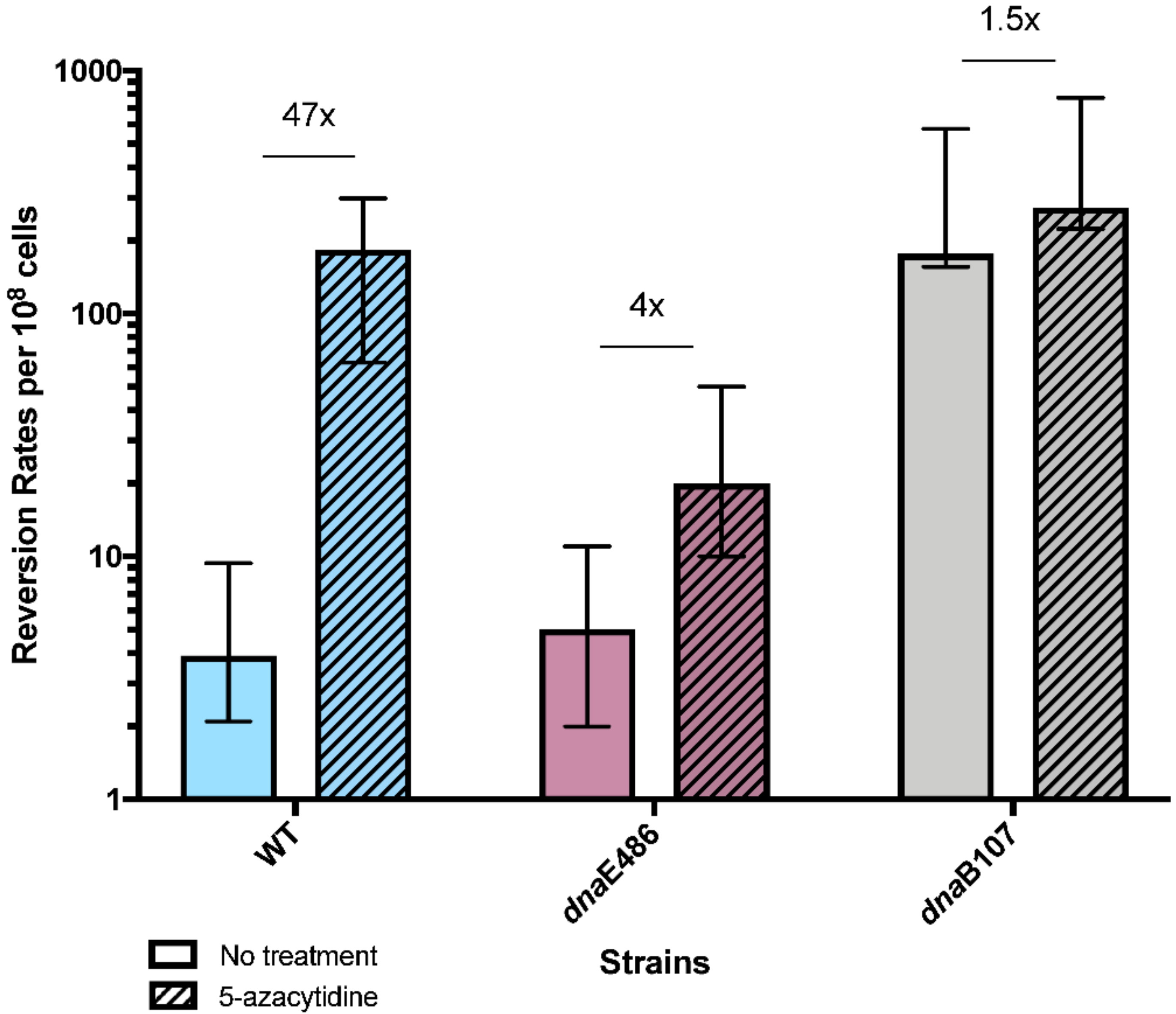
2.4. Mutation Assays
3. Results
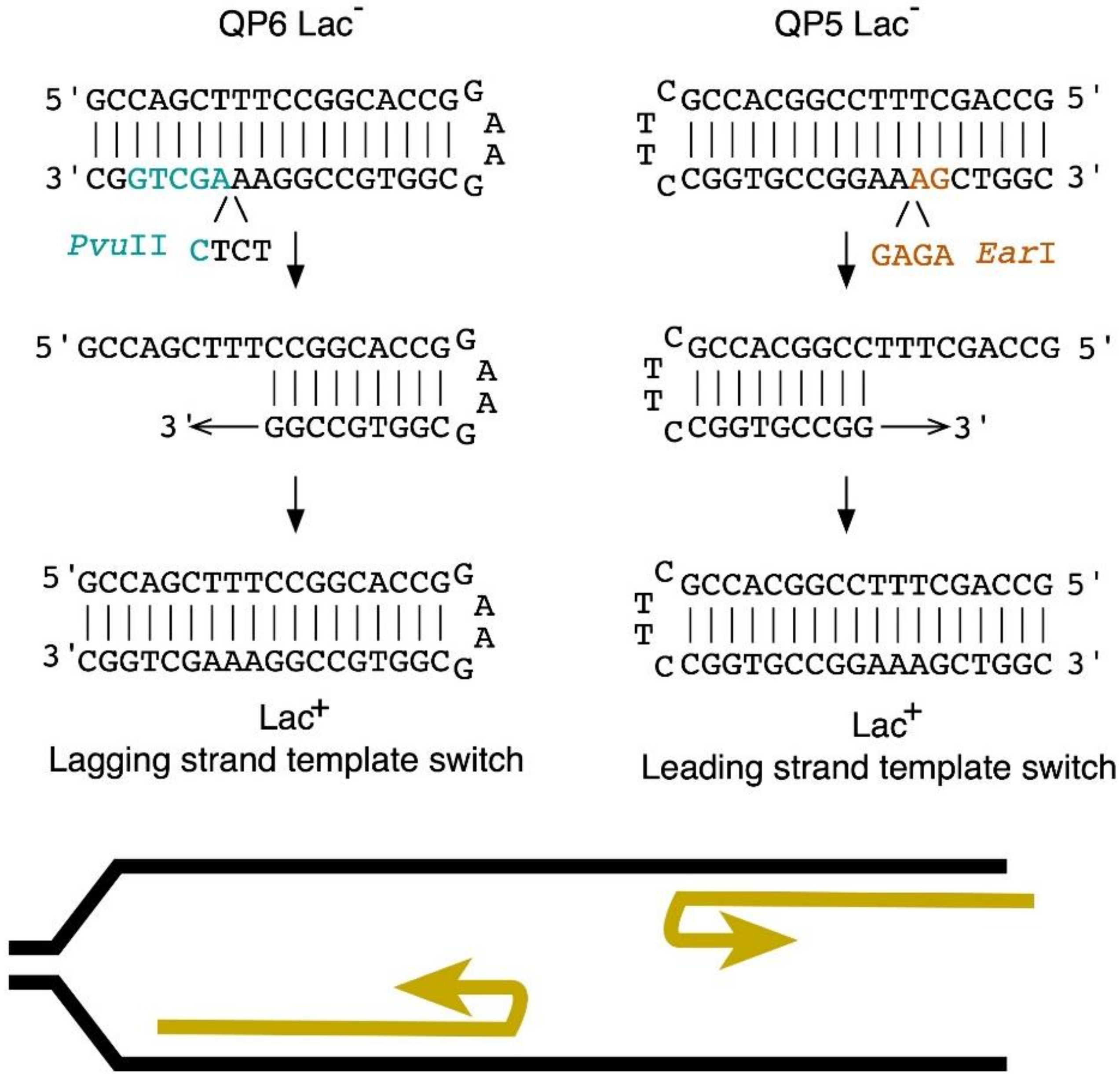
3.1. 5-Azacytidine Induces DNA Cytosine Methylase-Dependent Mutagenesis at Quasipalindromes
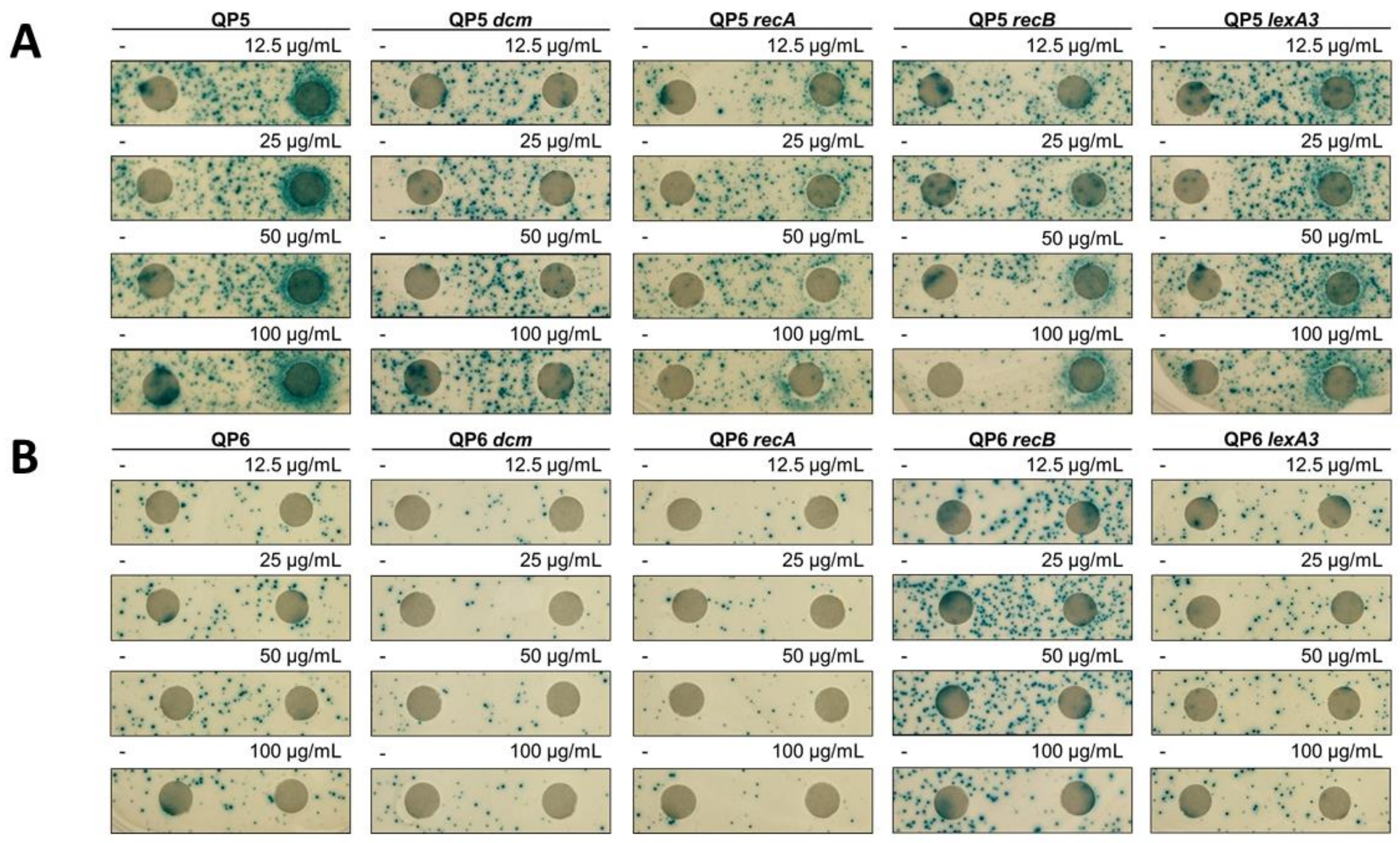
3.2. 5-Azacytidine-Induced Mutagenesis Is Largely RecA-Independent and Does Not Require DSB Repair
3.3. The SOS Response Is Antimutagenic for Template-Switch Mutations
3.4. Replication Mutants and Template-Switching
3.5. Template-Switching Is Induced by Other DPC-Inducing Agents
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cox, M.M.; Goodman, M.F.; Kreuzer, K.N.; Sherratt, D.J.; Sandler, S.J.; Marians, K.J. The importance of repairing stalled replication forks. Nature 2000, 404, 37–41. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef]
- Barker, S.; Weinfeld, M.; Murray, D. DNA-protein crosslinks: Their induction, repair, and biological consequences. Mutat. Res. 2005, 589, 111–135. [Google Scholar] [CrossRef] [PubMed]
- Ide, H.; Shoulkamy, M.I.; Nakano, T.; Miyamoto-Matsubara, M.; Salem, A.M. Repair and biochemical effects of DNA-protein crosslinks. Mutat. Res. 2011, 711, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Swenberg, J.A.; Lu, K.; Moeller, B.C.; Gao, L.; Upton, P.B.; Nakamura, J.; Starr, T.B. Endogenous versus exogenous DNA adducts: Their role in carcinogenesis, epidemiology, and risk assessment. Toxicol. Sci. 2011, 120 (Suppl. 1), S130–S145. [Google Scholar] [CrossRef]
- Stingele, J.; Bellelli, R.; Boulton, S.J. Mechanisms of DNA-protein crosslink repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Vaz, B.; Popovic, M.; Ramadan, K. DNA-Protein Crosslink Proteolysis Repair. Trends Biochem. Sci. 2017, 42, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Fielden, J.; Ruggiano, A.; Popovic, M.; Ramadan, K. DNA protein crosslink proteolysis repair: From yeast to premature ageing and cancer in humans. DNA Repair 2018. [Google Scholar] [CrossRef] [PubMed]
- Oleinick, N.L.; Chiu, S.M.; Ramakrishnan, N.; Xue, L.Y. The formation, identification, and significance of DNA-protein cross-links in mammalian cells. Br. J. Cancer Suppl. 1987, 8, 135–140. [Google Scholar] [PubMed]
- Craft, T.R.; Bermudez, E.; Skopek, T.R. Formaldehyde mutagenesis and formation of DNA-protein crosslinks in human lymphoblasts in vitro. Mutat. Res. 1987, 176, 147–155. [Google Scholar] [CrossRef]
- Izzotti, A.; Cartiglia, C.; Taningher, M.; De Flora, S.; Balansky, R. Age-related increases of 8-hydroxy-2′-deoxyguanosine and DNA-protein crosslinks in mouse organs. Mutat. Res. 1999, 446, 215–223. [Google Scholar] [CrossRef]
- Zahn, R.K.; Zahn-Daimler, G.; Ax, S.; Hosokawa, M.; Takeda, T. Assessment of DNA-protein crosslinks in the course of aging in two mouse strains by use of a modified alkaline filter elution applied to whole tissue samples. Mech. Ageing Dev. 1999, 108, 99–112. [Google Scholar] [CrossRef]
- Wu, F.Y.; Lee, Y.J.; Chen, D.R.; Kuo, H.W. Association of DNA-protein crosslinks and breast cancer. Mutat. Res. 2002, 501, 69–78. [Google Scholar] [CrossRef]
- Garaycoechea, J.I.; Crossan, G.P.; Langevin, F.; Daly, M.; Arends, M.J.; Patel, K.J. Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature 2012, 489, 571–575. [Google Scholar] [CrossRef] [PubMed]
- Kuo, H.K.; Griffith, J.D.; Kreuzer, K.N. 5-Azacytidine induced methyltransferase-DNA adducts block DNA replication in vivo. Cancer Res. 2007, 67, 8248–8254. [Google Scholar] [CrossRef]
- Lovett, S.T. Encoded errors: Mutations and rearrangements mediated by misalignment at repetitive DNA sequences. Mol. Microbiol. 2004, 52, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Lovett, S.T. Template-switching during replication fork repair in bacteria. DNA Repair 2017, 56, 118–128. [Google Scholar] [CrossRef]
- Saveson, C.J.; Lovett, S.T. Enhanced deletion formation by aberrant DNA replication in Escherichia coli. Genetics 1997, 146, 457–470. [Google Scholar] [PubMed]
- Morag, A.S.; Saveson, C.J.; Lovett, S.T. Expansion of DNA repeats in Escherichia coli: Effects of recombination and replication functions. J. Mol. Biol. 1999, 289, 21–27. [Google Scholar] [CrossRef]
- Seier, T.; Padgett, D.R.; Zilberberg, G.; Sutera, V.A., Jr.; Toha, N.; Lovett, S.T. Insights into mutagenesis using Escherichia coli chromosomal lacZ strains that enable detection of a wide spectrum of mutational events. Genetics 2011, 188, 247–262. [Google Scholar] [CrossRef]
- Seier, T.; Zilberberg, G.; Zeiger, D.M.; Lovett, S.T. Azidothymidine and other chain terminators are mutagenic for template-switch-generated genetic mutations. Proc. Natl. Acad. Sci. USA 2012, 109, 6171–6174. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, B.J. Pedigrees of some mutant strains of Escherichia coli K-12. Bacteriol. Rev. 1972, 36, 525–557. [Google Scholar] [PubMed]
- Miller, J.H. A Short Course in Bacterial Genetics; Cold Spring Harbor Press: New York, NY, USA, 1992. [Google Scholar]
- Willetts, N.S.; Clark, A.J.; Low, B. Genetic location of certain mutations conferring recombination deficiency in Escherichia coli. J. Bacteriol. 1969, 97, 244–249. [Google Scholar] [PubMed]
- Laranjo, L.T.; Gross, S.J.; Zeiger, D.M.; Lovett, S.T. SSB recruitment of Exonuclease I aborts template-switching in Escherichia coli. DNA Repair 2017, 57, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Ma, W.T.; Sandri, G.H. On fluctuation analysis: A new, simple and efficient method for computing the expected number of mutants. Genetica 1992, 85, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Rosche, W.A.; Foster, P.L. Determining mutation rates in bacterial populations. Methods 2000, 20, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Ara, T.; Hasegawa, M.; Takai, Y.; Okumura, Y.; Baba, M.; Datsenko, K.; Tomita, M.; Wanner, B.; Mori, H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol. Syst. Biol. 2006, 2. [Google Scholar] [CrossRef] [PubMed]
- Santi, D.V.; Norment, A.; Garrett, C.E. Covalent bond formation between a DNA-cytosine methyltransferase and DNA containing 5-azacytosine. Proc. Natl. Acad. Sci. USA 1984, 81, 6993–6997. [Google Scholar] [CrossRef]
- Friedman, S. The irreversible binding of azacytosine-containing DNA fragments to bacterial DNA(cytosine-5)methyltransferases. J. Biol. Chem. 1985, 260, 5698–5705. [Google Scholar]
- Bhagwat, A.S.; Roberts, R.J. Genetic analysis of the 5-azacytidine sensitivity of Escherichia coli K-12. J. Bacteriol. 1987, 169, 1537–1546. [Google Scholar] [CrossRef]
- Lal, D.; Som, S.; Friedman, S. Survival and mutagenic effects of 5-azacytidine in Escherichia coli. Mutat. Res. 1988, 193, 229–236. [Google Scholar] [CrossRef]
- Butala, M.; Busby, S.J.W.; Lee, D.J. DNA sampling: A method for probing protein binding at specific loci on bacterial chromosomes. Nucleic Acids Res. 2009, 37, e37. [Google Scholar] [CrossRef] [PubMed]
- Salem, A.M.; Nakano, T.; Takuwa, M.; Matoba, N.; Tsuboi, T.; Terato, H.; Yamamoto, K.; Yamada, M.; Nohmi, T.; Ide, H. Genetic analysis of repair and damage tolerance mechanisms for DNA-protein cross-links in Escherichia coli. J. Bacteriol. 2009, 191, 5657–5668. [Google Scholar] [CrossRef] [PubMed]
- Krasich, R.; Wu, S.Y.; Kuo, H.K.; Kreuzer, K.N. Functions that protect Escherichia coli from DNA-protein crosslinks. DNA Repair 2015, 28, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Simmons, L.A.; Foti, J.J.; Cohen, S.E.; Walker, G.C. The SOS Regulatory Network. EcoSal Plus 2008, 3. [Google Scholar] [CrossRef] [PubMed]
- Kuban, W.; Banach-Orlowska, M.; Bialoskorska, M.; Lipowska, A.; Schaaper, R.M.; Jonczyk, P.; Fijalkowska, I.J. Mutator phenotype resulting from DNA polymerase IV overproduction in Escherichia coli: Preferential mutagenesis on the lagging strand. J. Bacteriol. 2005, 187, 6862–6866. [Google Scholar] [CrossRef] [PubMed]
- Lovett, S.T.; Hurley, R.L.; Sutera, V.A., Jr.; Aubuchon, R.H.; Lebedeva, M.A. Crossing over between regions of limited homology in Escherichia coli. RecA-dependent and RecA-independent pathways. Genetics 2002, 160, 851–859. [Google Scholar]
- Seigneur, M.; Ehrlich, S.D.; Michel, B. RuvABC-dependent double-strand breaks in dnaBts mutants require recA. Mol. Microbiol. 2000, 38, 565–574. [Google Scholar] [CrossRef]
- Stewart, J.W.; Sherman, F. Yeast frameshift mutants identified by sequence changes in iso-1-cytochrome C. In Molecular and Environmental Aspects of Mutagenesis; Miller, M.W., Ed.; Charles C. Thomas Pub. Inc.: Springfield, IL, USA, 1974; pp. 102–127. [Google Scholar]
- Viswanathan, M.; Lacirignola, J.J.; Hurley, R.L.; Lovett, S.T. A novel mutational hotspot in a natural quasipalindrome in Escherichia coli. J. Mol. Biol. 2000, 302, 553–564. [Google Scholar] [CrossRef]
- Ripley, L.S. Model for the participation of quasi-palindromic DNA sequences in frameshift mutation. Proc. Natl. Acad. Sci. USA 1982, 79, 4128–4132. [Google Scholar] [CrossRef]
- Dutra, B.E.; Lovett, S.T. Cis and trans-acting effects on a mutational hotspot involving a replication template switch. J. Mol. Biol. 2006, 356, 300–311. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Eichelmann, M.C.; Ramirez-Santos, J. Methylated cytosine at Dcm (CCATGG) sites in Escherichia coli: Possible function and evolutionary implications. J. Mol. Evol. 1993, 37, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Cupples, C.G.; Miller, J.H. A set of lacZ strains in Escherichia coli that allow rapid detection of each of the six base substitutions. Proc. Natl. Acad. Sci. USA 1989, 86, 5345–5349. [Google Scholar] [CrossRef] [PubMed]
- Jackson-Grusby, L.; Laird, P.W.; Magge, S.N.; Moeller, B.J.; Jaenisch, R. Mutagenicity of 5-aza-2′-deoxycytidine is mediated by the mammalian DNA methyltransferase. Proc. Natl. Acad. Sci. USA 1997, 94, 4681–4685. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef]
- Hooper, D.C.; Wolfson, J.S.; Ng, E.Y.; Swartz, M.N. Mechanisms of action of and resistance to ciprofloxacin. Am. J. Med. 1987, 82, 12–20. [Google Scholar]
- Kaguni, J.M. The Macromolecular machines that duplicate the Escherichia coli chromosome as targets for drug discovery. Antibiotics 2018, 7. [Google Scholar] [CrossRef]
- O’Donnell, M.; Langston, L.; Stillman, B. Principles and concepts of DNA replication in bacteria, archaea, and eukarya. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef]
- Karon, M.; Benedict, W.F. Chromatid breakage: Differential effect of inhibitors of DNA synthesis during G 2 phase. Science 1972, 178, 62. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Genotype | Origin or Reference |
|---|---|---|
| MG1655 | F- rph-1 | |
| STL7180 | recA::cat | Lab collection |
| STL12071 | lexA3 malF::Tn10 kan | Lab collection |
| STL15144 | lacZ(QP5) lexA3 malF::Tn10kan mphC281::Tn10 | This work |
| STL15654 | lacZ(QP6) mphC281::Tn10 | Lab collection |
| STL16519 | lacZ(QP6) lexA3 malF::Tn10 kan mphC281::Tn10 | This work |
| STL17685 | lacZ(QP5) mphC281::Tn10 | Lab collection |
| STL18747 | lacZ(QP5) dcm∆::FRT kan mphC281::Tn10 | This work |
| STL18749 | lacZ(QP6) dcm∆::FRT kan mphC281::Tn10 | This work |
| STL19829 | lacZ(QP5) recA::cat mphC281::Tn10 | This work |
| STL19831 | lacZ(QP6) recA::cat mphC281::Tn10 | This work |
| STL19843 | recB∆::FRT kan | This work |
| STL20891 | lacZ(QP5) dnaE486 zae-3095::Tn10kan mphC-281::Tn10 | This work |
| STL21742 | dcm∆::FRT kan | This work |
| STL21922 | lacZ(QP5) dnaB107(ts) malE::Tn10kan | This work |
| STL21926 | lacZ(QP5) recB∆::FRT kan mphC-281::Tn10 | This work |
| STL22017 | lacZ(QP6) recB∆::FRT kan mphC-281::Tn10 | This work |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laranjo, L.T.; Klaric, J.A.; Pearlman, L.R.; Lovett, S.T. Stimulation of Replication Template-Switching by DNA-Protein Crosslinks. Genes 2019, 10, 14. https://doi.org/10.3390/genes10010014
Laranjo LT, Klaric JA, Pearlman LR, Lovett ST. Stimulation of Replication Template-Switching by DNA-Protein Crosslinks. Genes. 2019; 10(1):14. https://doi.org/10.3390/genes10010014
Chicago/Turabian StyleLaranjo, Laura T., Julie A. Klaric, Leah R. Pearlman, and Susan T. Lovett. 2019. "Stimulation of Replication Template-Switching by DNA-Protein Crosslinks" Genes 10, no. 1: 14. https://doi.org/10.3390/genes10010014
APA StyleLaranjo, L. T., Klaric, J. A., Pearlman, L. R., & Lovett, S. T. (2019). Stimulation of Replication Template-Switching by DNA-Protein Crosslinks. Genes, 10(1), 14. https://doi.org/10.3390/genes10010014