Highly Resolved Phylogenetic Relationships within Order Acipenseriformes According to Novel Nuclear Markers


 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Datasets
2.2. Quality Control and De Novo Transcriptome Assembly
2.3. Comparison of Transcripts and Extraction of Protein Sequences
2.4. Identification of Orthologous Single-Copy Genes
2.5. Exon Search and Primer Design
2.6. Taxon Sampling, DNA Extraction, and Experimental Testing
2.7. Sequence Assembly and Phylogenetic Analyses
2.8. Hypothesis Testing
2.9. Estimating Divergence Time
3. Results
3.1. Quality Control and Assembly
3.2. Identification of Putative Proteins
3.3. Characteristics of NPC Markers
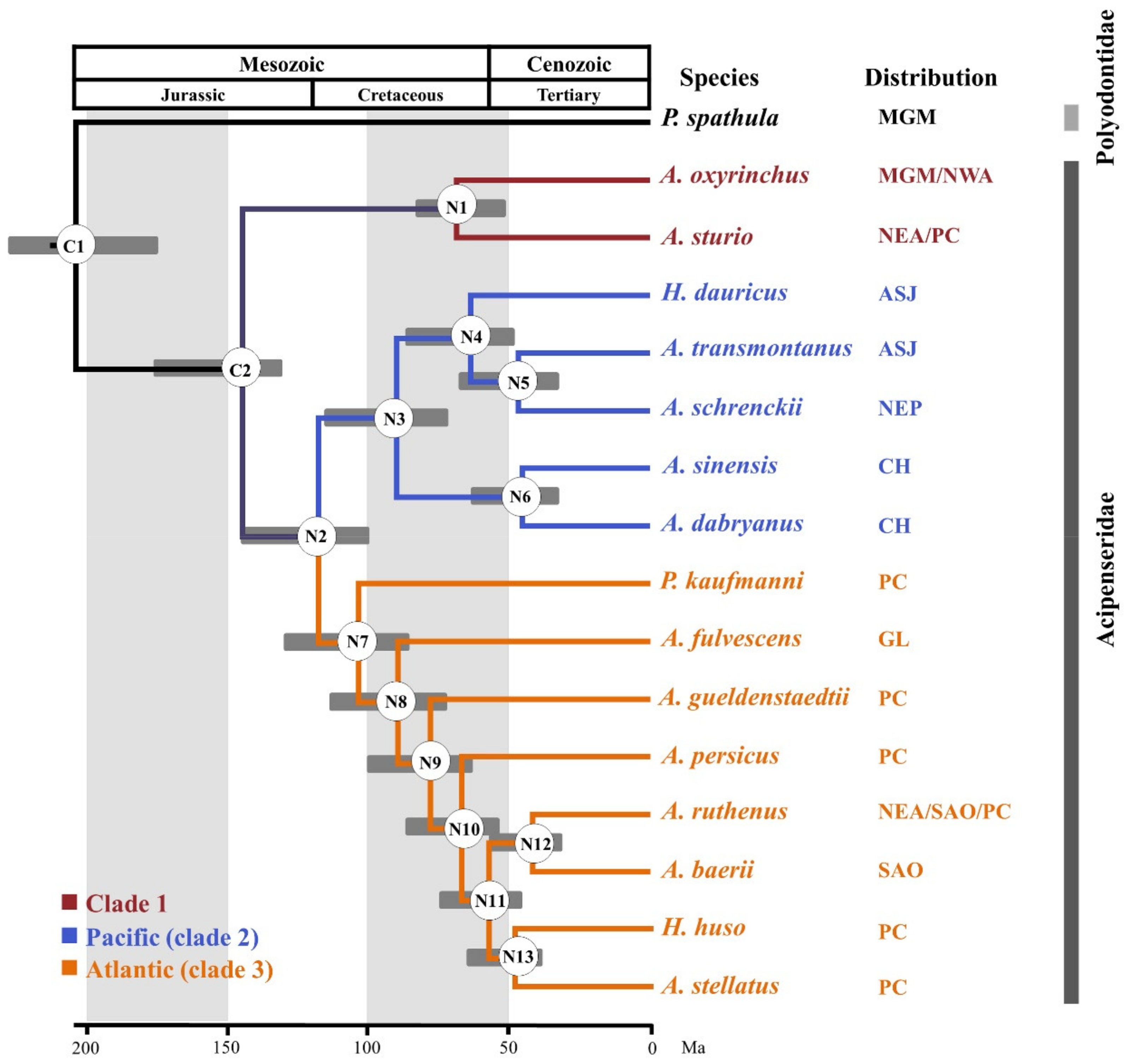
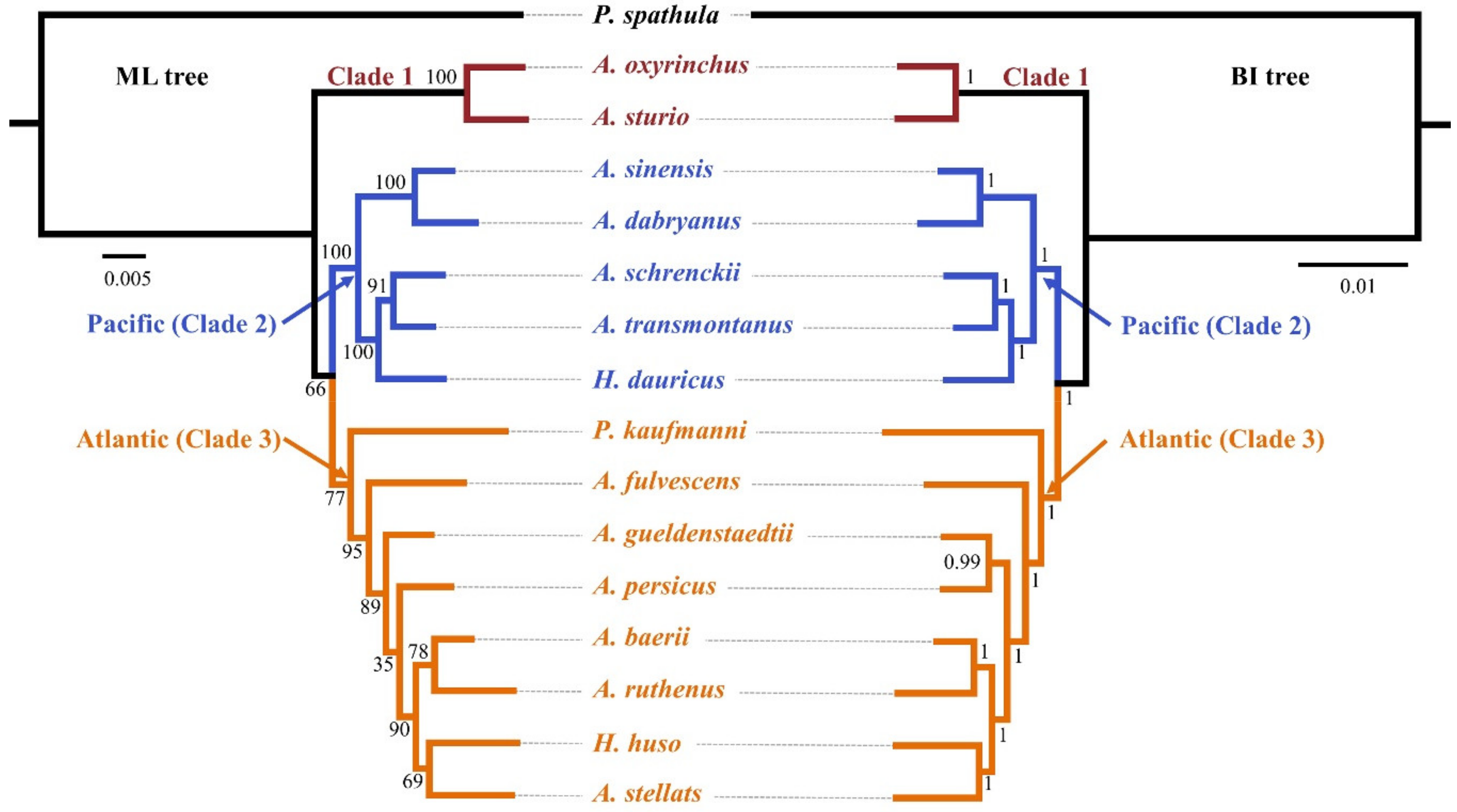
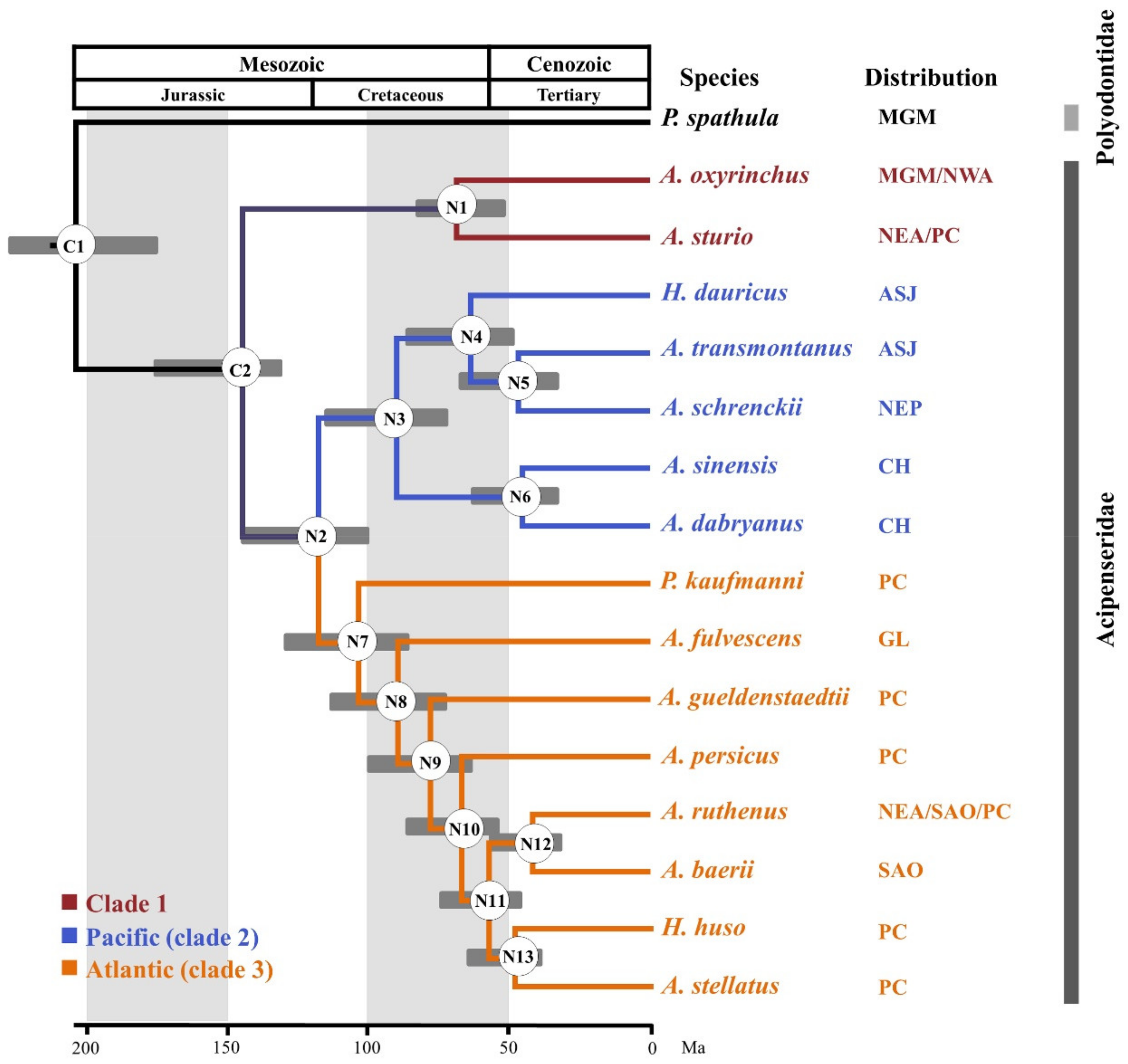
3.4. Reconstructing Phylogenetic Relationships in the Acipenseriformes
3.5. Hypothesis Testing
3.6. Divergence Times
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ludwig, A. Identification of Acipenseriformes species in trade. J. Appl. Ichthyol. 2008, 24, 2–19. [Google Scholar] [CrossRef]
- Simon, C.; Frati, B.F.; Stewart, J.B.; Beckenbach, A.T. Incorporating molecular evolution into phylogenetic analysis, and a new compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA. Annu. Rev. Ecol. Evol. Syst. 2006, 37, 545–579. [Google Scholar] [CrossRef]
- Harrison, R.G. Animal mitochondrial DNA as a genetic marker in population and evolutionary biology. Trends Ecol. Evol. 1989, 4, 6–11. [Google Scholar] [CrossRef]
- Townsend, T.; Alegre, R.; Kelley, S.; Wiens, J.; Reeder, T.W. Rapid development of multiple nuclear loci for phylogenetic analysis using genomic resources: An example from squamate reptiles. Mol. Phylogenet. Evol. 2008, 47, 129–142. [Google Scholar] [CrossRef]
- Li, Z.; De La Torre, A.R.; Sterck, L.; Cánovas, F.M.; Avila, C.; Merino, I.; Cabezas, J.A.; Cervera, M.T.; Ingvarsson, P.K.; Van de Peer, Y. Single-copy genes as molecular markers for phylogenomic studies in seed plants. Genome Biol. Evol. 2017, 9, 1130–1147. [Google Scholar] [CrossRef] [PubMed]
- Teasdale, L.C.; Köhler, F.; Murray, K.D.; O’Hara, T.; Moussalli, A. Identification and qualification of 500 nuclear, single-copy, orthologous genes for the Eupulmonata (Gastropoda) using transcriptome sequencing and exon capture. Mol. Ecol. Resour. 2016, 16, 1107–1123. [Google Scholar] [CrossRef] [PubMed]
- Rothfels, C.J.; Larsson, A.; Li, F.W.; Sigel, E.M.; Huiet, L.; Burge, D.O.; Ruhsam, M.; Graham, S.W.; Stevenson, D.W.; Wong, G.K.; et al. Transcriptome-mining for single-copy nuclear markers in ferns. PLoS ONE 2013, 8, e76957. [Google Scholar] [CrossRef]
- Peng, Z.; Elango, N.; Wildman, D.E.; Yi, S.V. Primate phylogenomics: Developing numerous nuclear non-coding, non-repetitive markers for ecological and phylogenetic applications and analysis of evolutionary rate variation. BMC Genom. 2009, 10, 247. [Google Scholar] [CrossRef]
- Li, C.; Ortí, G.; Gong, Z.; Lu, G. A practical approach to phylogenomics: The phylogeny of ray-finned fish (Actinopterygii) as a case study. BMC Evol. Biol. 2007, 7, 44. [Google Scholar] [CrossRef]
- Duarte, J.M.; Wall, P.K.; Edger, P.P.; Landherr, L.L.; Hong, M.; Pires, P.K.; Leebens-Mack, J.; Depamphilis, C.W. Identification of shared single copy nuclear genes in Arabidopsis, Populus, Vitis and Oryza and their phylogenetic utility across various taxonomic levels. BMC Evol. Biol. 2010, 10, 61. [Google Scholar] [CrossRef]
- Deng, H.; Zhang, G.Q.; Lin, M.; Wang, Y.; Liu, Z.J. Mining from transcriptomes: 315 single-copy orthologous genes concatenated for the phylogenetic analyses of Orchidaceae. Ecol. Evol. 2015, 5, 3800–3807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Che, L.H.; Zhang, S.Q.; Li, Y.; Liang, D.; Pang, H.; Ślipiński, A.; Zhang, P. Genome-wide survey of nuclear protein-coding markers for beetle phylogenetics and their application in resolving both deep and shallow-level divergences. Mol. Ecol. Resour. 2017, 17, 1342–1358. [Google Scholar] [CrossRef] [PubMed]
- Thomson, R.C.; Wang, I.J.; Johnson, J.R. Genome-enabled development of DNA markers for ecology, evolution and conservation. Mol. Ecol. 2010, 19, 2184–2195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, C.C.; Creevey, C.J.; O’Connell, M.J. Mitochondrial data are not suitable for resolving placental mammal phylogeny. Mamm. Genome 2014, 25, 636–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krieger, J.; Hett, A.K.; Fuerst, P.A.; Artyukhin, E.A.; Ludwig, A. The molecular phylogeny of the order Acipenseriformes revised. J. Appl. Ichthyol. 2010, 24, 36–45. [Google Scholar] [CrossRef]
- Rajkov, J.; Shao, Z.; Berrebi, P. Evolution of polyploidy and functional diploidization in sturgeons: Microsatellite analysis in 10 sturgeon species. J. Hered. 2014, 105, 521–531. [Google Scholar] [CrossRef]
- Grande, L.; Bemis, W.E. Osteology and phylogenetic relationships of fossil and recent paddlefishes (Polyodontidae) with comments on the interrelationships of Acipenseriformes. J. Vertebr. Paleontol. 1991, 11, 1–121. [Google Scholar] [CrossRef]
- Peng, Z.; Ludwig, A.; Wang, D.; Rui, D.; Wei, Q.; He, S. Age and biogeography of major clades in sturgeons and paddlefishes (pisces: Acipenseriformes). Mol. Phylogenet. Evol. 2007, 42, 854–862. [Google Scholar] [CrossRef]
- Fontana, F.; Tagliavini, J.; Congiu, L. Sturgeon genetics and cytogenetics: Recent advancements and perspectives. Genetica 2001, 111, 359–373. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, Y.; Zheng, X.; Chen, Y.; Deng, H.; Wang, D.; Wei, Q.; Zhang, Y.; Long, N.; Wu, Q. Molecular phylogenetic systematics of twelve species of acipenseriformes based on mtDNA ND4L–ND4 gene sequence analysis. Sci. China Life Sci. 2000, 43, 129–137. [Google Scholar] [CrossRef]
- Ludwig, A.; May, B.; Debus, L.; Jenneckens, I. Heteroplasmy in the mtdna control region of sturgeon (Acipenser, Huso and Scaphirhynchus). Genetics 2000, 156, 1933–1947. [Google Scholar] [PubMed]
- Ludwig, A.; Belfiore, N.M.; Pitra, C.; Svirsky, V.; Jenneckens, I. Genome duplication events and functional reduction of ploidy levels in sturgeon (Acipenser, Huso and Scaphirhynchus). Genetics 2001, 158, 1203–1215. [Google Scholar] [PubMed]
- Krieger, J.; Fuerst, P.A. Evidence for a slowed rate of molecular evolution in the order Acipenseriformes. Mol. Biol. Evol. 2002, 19, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Birstein, V.J.; Doukakis, P.; Desalle, R. Molecular phylogeny of Acipenseridae: Nonmonophyly of scaphirhynchinae. Copeia 2002, 2002, 287–301. [Google Scholar] [CrossRef]
- Birstein, V.J.; Desalle, R. Molecular phylogeny of Acipenserinae. Mol. Phylogenet. Evol. 1998, 9, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Bemis, W.E.; Kynard, B. Sturgeon rivers: An introduction to Acipenseriform biogeography and life history. Environ. Biol. Fishes 1997, 48, 167–183. [Google Scholar] [CrossRef]
- Bemis, W.E.; Findeis, E.K.; Grande, L. An overview of Acipenseriformes. Environ. Biol. Fishes 1997, 48, 25–71. [Google Scholar] [CrossRef]
- Artyukhin, E.N. Morphological phylogeny of the order Acipenseriformes. J. Appl. Ichthyol. 2010, 22, 66–69. [Google Scholar] [CrossRef]
- Findeis, E.K. Osteology and phylogenetic interrelationships of sturgeons (Acipenseridae). Environ. Biol. Fishes 1997, 48, 73–126. [Google Scholar] [CrossRef]
- Kaitetzidou, E.; Ludwig, A.; Gessner, J.; Sarropoulou, E. Expression patterns of atlantic sturgeon (Acipenser oxyrinchus) during embryonic development. G3-Genes Genoms Genet. 2017, 7, 533–542. [Google Scholar] [CrossRef]
- Yue, H.; Li, C.; Du, H.; Zhang, S.; Wei, Q. Sequencing and de novo assembly of the gonadal transcriptome of the endangered Chinese sturgeon (Acipenser sinensis). PLoS ONE 2015, 10, e0127332. [Google Scholar] [CrossRef]
- Song, W.; Jiang, K.; Zhang, F.; Lin, Y.; Ma, L. Transcriptome sequencing, de novo assembly and differential gene expression analysis of the early development of Acipenser baeri. PLoS ONE 2015, 10, e0137450. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.Y.; Liang, D.; Zhang, P. Selecting question-specific genes to reduce incongruence in phylogenomics: A case study of jawed vertebrate backbone phylogeny. Syst. Biol. 2015, 64, 1104–1120. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Zhang, X.; Li, L.; Jiang, H.; Chen, J. High-throughput sequencing of microrna transcriptome and expression assay in the sturgeon, Acipenser schrenckii. PLoS ONE 2014, 9, e115251. [Google Scholar] [CrossRef]
- Vidotto, M.; Grapputo, A.; Boscari, E.; Barbisan, F.; Coppe, A.; Grandi, G.; Kumar, A.; Congiu, L. Transcriptome sequencing and de novo annotation of the critically endangered adriatic sturgeon. BMC Genom. 2013, 14, 407. [Google Scholar] [CrossRef] [PubMed]
- Braasch, I.; Gehrke, A.R.; Smith, J.J.; Kawasaki, K.; Manousaki, T.; Pasquier, J.; Amores, A.; Desvignes, T.; Batzel, P.; Catchen, J.; et al. The spotted gar genome illuminates vertebrate evolution and facilitates human-teleost comparisons. Nat. Genet. 2016, 48, 427–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Bolger, A.M.; Marc, L.; Bjoern, U. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Haas, B.J.; Alexie, P.; Moran, Y.; Manfred, G.; Blood, P.D.; Joshua, B.; Matthew Brian, C.; David, E.; Bo, L.; Matthias, L. De novo transcript sequence reconstruction from rna-seq using the trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. Orthomcl: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef]
- Shen, X.-X.; Liang, D.; Wen, J.-Z.; Zhang, P. Multiple genome alignments facilitate development of NPCL markers: A case study of tetrapod phylogeny focusing on the position of turtles. Mol. Biol. Evol. 2011, 28, 3237–3252. [Google Scholar] [CrossRef]
- Lu, G.; Moriyama, E.N. Vector NTI, a balanced all-in-one sequence analysis suite. Brief. Bioinform. 2004, 5, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. Mega7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, G.; Lohman, D.J.; Meier, R. Sequencematrix: Concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 2011, 27, 171–180. [Google Scholar] [CrossRef]
- Xia, X. Dambe 5: A comprehensive software package for data analysis in molecular biology and evolution. Mol. Biol. Evol. 2013, 30, 1720–1728. [Google Scholar] [CrossRef]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. Partitionfinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. Raxml version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. Mrbayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarisation in bayesian phylogenetics using tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Zhang, C.; Sayyari, E.; Mirarab, S. Astral-III: Increased scalability and impacts of contracting low support branches. In Proceedings of the RECOMB International Workshop on Comparative Genomics, Barcelona, Spain, 4–6 October 2017; pp. 53–75. [Google Scholar]
- Sayyari, E.; Mirarab, S. Fast coalescent-based computation of local branch support from quartet frequencies. Mol. Biol. Evol. 2016, 33, 1654–1668. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, H.A.; Strimmer, K.; Vingron, M.; von Haeseler, A. Tree-puzzle: Maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics 2002, 18, 502–504. [Google Scholar] [CrossRef] [PubMed]
- Shimodaira, H.; Hasegawa, M. Consel: For assessing the confidence of phylogenetic tree selection. Bioinformatics 2001, 17, 1246–1247. [Google Scholar] [CrossRef] [PubMed]
- Shimodaira, H. An approximately unbiased test of phylogenetic tree selection. Syst. Biol. 2002, 51, 492–508. [Google Scholar] [CrossRef] [PubMed]
- Kishino, H.; Hasegawa, M. Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in hominoidea. J. Mol. Evol. 1989, 29, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Shimodaira, H.; Hasegawa, M. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol. Biol. Evol. 1999, 16, 1114–1116. [Google Scholar] [CrossRef]
- Dos Reis, M.; Yang, Z. Approximate likelihood calculation on a phylogeny for bayesian estimation of divergence times. Mol. Biol. Evol. 2011, 28, 2161–2172. [Google Scholar] [CrossRef] [PubMed]
- Jin, F. Late mesozoic Acipenseriformes (osteichthyes: Actinopterygii) in central asia and their biogrograochical implications. In Sixth Symposium on Mesozoic Terrestrial Ecosystems and Biota: Short Papers; China Ocean Press: Beijing, China, 1995; pp. 15–21. [Google Scholar]
- Benton, R. Fossil butte national monument kemmerer, wyoming: Twenty years of visitor service and research. Rocks Miner. 1993, 68, 180–182. [Google Scholar] [CrossRef]
- Inoue, J.G.; Miya, M.; Venkatesh, B.; Nishida, M. The mitochondrial genome of indonesian coelacanth Latimeria menadoensis (sarcopterygii: Coelacanthiformes) and divergence time estimation between the two coelacanths. Gene 2005, 349, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Van de Peer, Y.; Mizrachi, E.; Marchal, K. The evolutionary significance of polyploidy. Nat. Rev. Genet. 2017, 18, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Zhang, N.; Zhang, Q.; Endress, P.K.; Huang, J.; Ma, H. Resolution of deep eudicot phylogeny and their temporal diversification using nuclear genes from transcriptomic and genomic datasets. New Phytol. 2017, 214, 1338–1354. [Google Scholar] [CrossRef]
- Spinks, P.Q.; Thomson, R.C.; Lovely, G.A.; Shaffer, H.B. Assessing what is needed to resolve a molecular phylogeny: Simulations and empirical data from emydid turtles. BMC Evol. Biol. 2009, 9, 56. [Google Scholar] [CrossRef] [PubMed]
- Rokas, A.; Williams, B.L.; King, N.; Carroll, S.B. Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature 2003, 425, 798–804. [Google Scholar] [CrossRef] [PubMed]
- Birstein, V.J.; Bemis, W.E. How many species are there within the genus Acipenser? Environ. Biol. Fishes 1997, 48, 157–163. [Google Scholar] [CrossRef]
- Chen, M.Y.; Mao, R.L.; Liang, D.; Kuro-o, M.; Zeng, X.M.; Zhang, P. A reinvestigation of phylogeny and divergence times of hynobiidae (Amphibia, Caudata) based on 29 nuclear genes. Mol. Phylogenet. Evol. 2015, 83, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.G.; Smith, D.G.; Funnell, B.M. Atlas of Mesozoic and Cenozoic Coastlines; Cambridge University Press: Cambridge, UK, 2004; ISBN 052-145-155-8. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
| Species | Common Name | Collection Locality | Distribution 1 |
|---|---|---|---|
| Acipenser | |||
| A. baerii | Siberian sturgeon | Germany | SAO |
| A. dabryanus | Yangtze sturgeon | Yibin, Sichuang, China | CH |
| A. fulvescens | Lake sturgeon | Wolfgangsee, Wisconsin, America | GL |
| A. gueldenstaedtii | Danube sturgeon | Germany | PC |
| A. oxyrinchus | Atlantic sturgeon | Canada | MGM/NWA |
| A. persicus | Persian sturgeon | Iran | PC |
| A. ruthenus | Sterlet sturgeon | The Danube, Germany | NEA/SAO/PC |
| A. schrenckii | Amur sturgeon | Wanzhou, Chongqing, China | ASJ |
| A. sinensis | Chinese sturgeon | Yangtze river fisheries research institute, Hubei, China | CH |
| A. stellatus | Starry sturgeon | The Danube, Romania | PC |
| A. sturio | Sturgeon | Gironde River, France | NEA/PC |
| A. transmontanus | White sturgeon | Amur river, China | NEP |
| Huso | |||
| H. dauricus | Kaluga | Amur river, China | ASJ |
| H. huso | Beluga | The Danube, Romania | PC |
| Pseudoscaphirhynchus | |||
| P. kaufmanni | Amu Darya sturgeon | Amu Darya, Turkmenistan | PC |
| Polyodon | |||
| P. spathula | Paddlefish | Wanzhou, Chongqing, China | MGM |
| Rank | Topology Tested | log L | AU | KH | SH |
|---|---|---|---|---|---|
| 1 | Best tree (ML tree) | −47,716.74 | 1.000 | 1.000 | 1.000 |
| 2 | (((((ASTE,PK),AR)),HH),(((AT,ASC),(ASI,AD)),HD) 1 | −47,944.33 | 6 × 10−8 | 0 | 0.011 |
| 3 | ((ASTE,PK),((AR,HH),HD),AF) 2 | −48,422.00 | 2 × 10−25 | 0 | 0 |
| 4 | ((Acipenser, PK),(HH,HD)) 3 | −48,869.58 | 0.008 | 0 | 0 |
| 5 | (((((HH,HD),(AR,ASC)),ASTE),(ASTU,AO)),PK) 4 | −48,915.92 | 2 × 10−62 | 0 | 0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, D.; Li, Y.; Zhao, Q.; Zhao, L.; Ludwig, A.; Peng, Z. Highly Resolved Phylogenetic Relationships within Order Acipenseriformes According to Novel Nuclear Markers. Genes 2019, 10, 38. https://doi.org/10.3390/genes10010038
Luo D, Li Y, Zhao Q, Zhao L, Ludwig A, Peng Z. Highly Resolved Phylogenetic Relationships within Order Acipenseriformes According to Novel Nuclear Markers. Genes. 2019; 10(1):38. https://doi.org/10.3390/genes10010038
Chicago/Turabian StyleLuo, Dehuai, Yanping Li, Qingyuan Zhao, Lianpeng Zhao, Arne Ludwig, and Zuogang Peng. 2019. "Highly Resolved Phylogenetic Relationships within Order Acipenseriformes According to Novel Nuclear Markers" Genes 10, no. 1: 38. https://doi.org/10.3390/genes10010038
APA StyleLuo, D., Li, Y., Zhao, Q., Zhao, L., Ludwig, A., & Peng, Z. (2019). Highly Resolved Phylogenetic Relationships within Order Acipenseriformes According to Novel Nuclear Markers. Genes, 10(1), 38. https://doi.org/10.3390/genes10010038