De Novo Sequencing and Hybrid Assembly of the Biofuel Crop Jatropha curcas L.: Identification of Quantitative Trait Loci for Geminivirus Resistance

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. DNA Sequence Data Generation
2.3. Sequence Data Trimming, Filtering, Error Correction, and Assembly
2.4. Gene Prediction
2.5. Gene Annotation
2.6. Transcriptome Sequencing and Data Generation
2.7. Ab Initio Gene Model Validation
2.8. Identification of Simple Sequence Repeats
2.9. Genetic Diversity Analysis
2.10. Linkage Mapping and Quantitative Trait Locus Analysis for Jatropha Mosaic Virus Resistance
2.11. Identification and Annotation of Repetitive Sequences
2.12. Comparative Genomic Analysis
2.13. Data Availability
3. Results
3.1. Whole Genome Sequencing Statistics
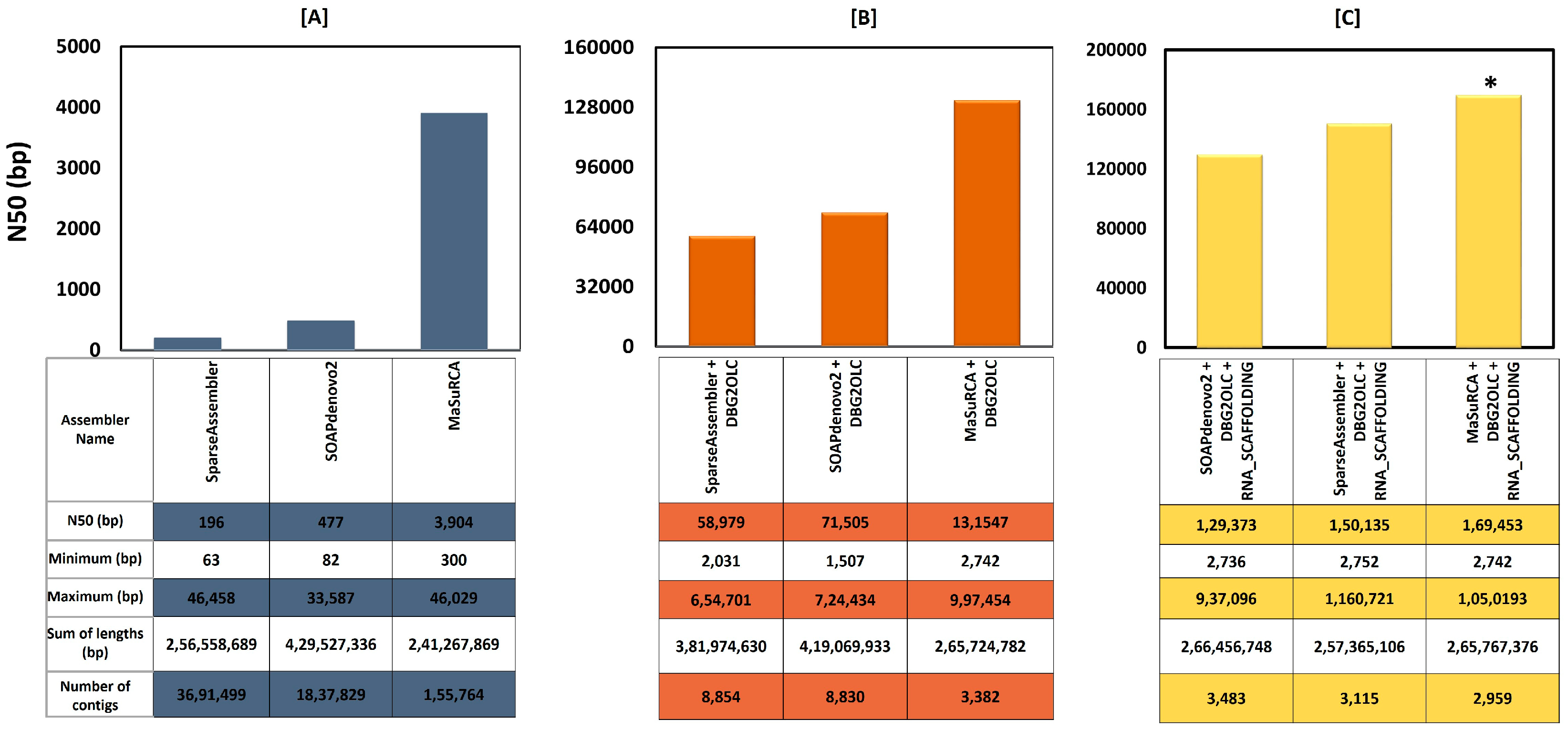
3.2. Sequence Data Trimming, Filtering, Error Correction, and Assembly
3.3. Prediction of Genes from Assembled Genome
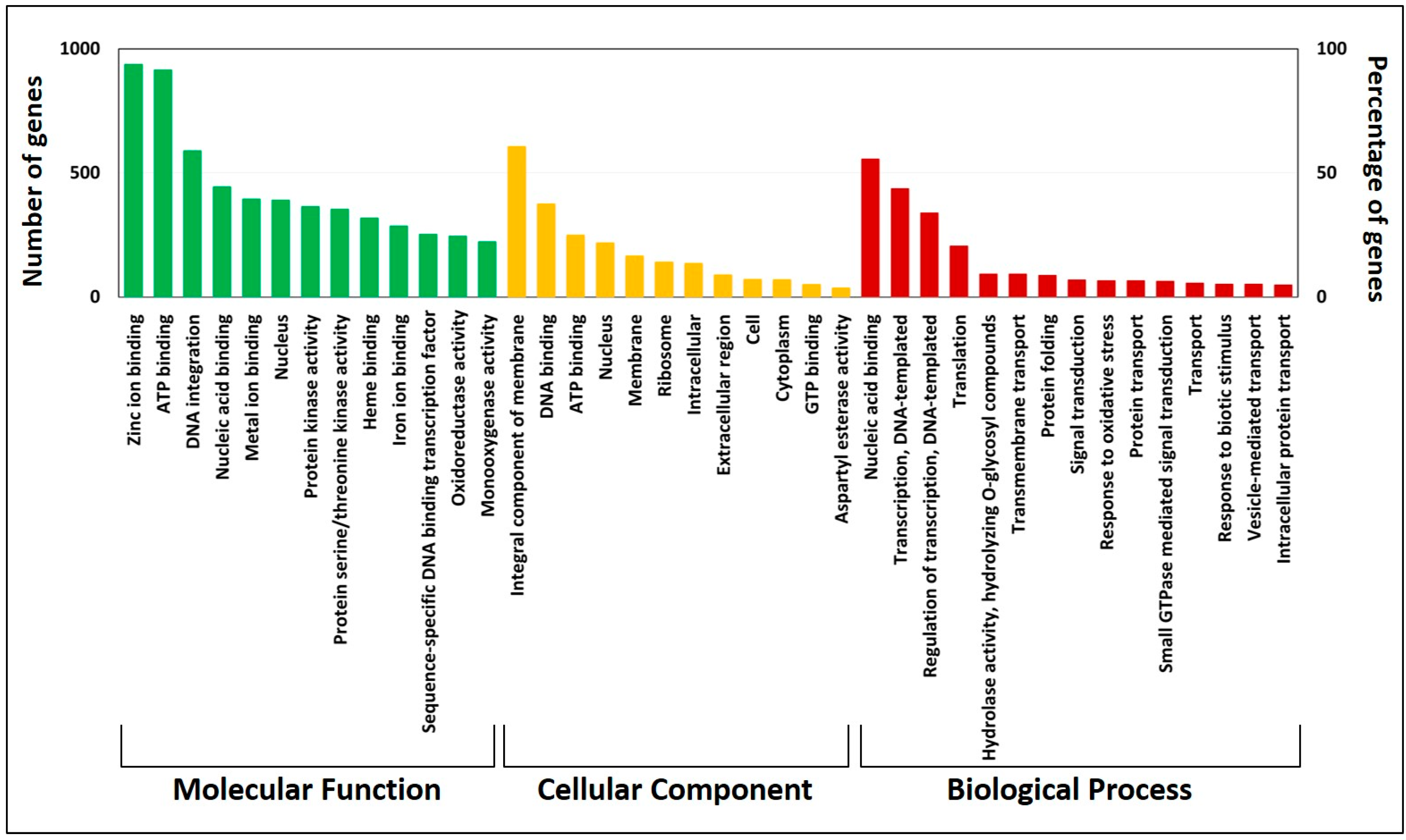
3.4. Gene Annotation and Kyoto Encyclopedia of Genes and Genomes Pathway Analysis
3.5. Transcriptome Sequencing and Gene Validation
3.6. Characterization of Genes Specific to the Drought Condition
3.7. Microsatellite Primer Designing
3.8. Genetic Diversity Analysis
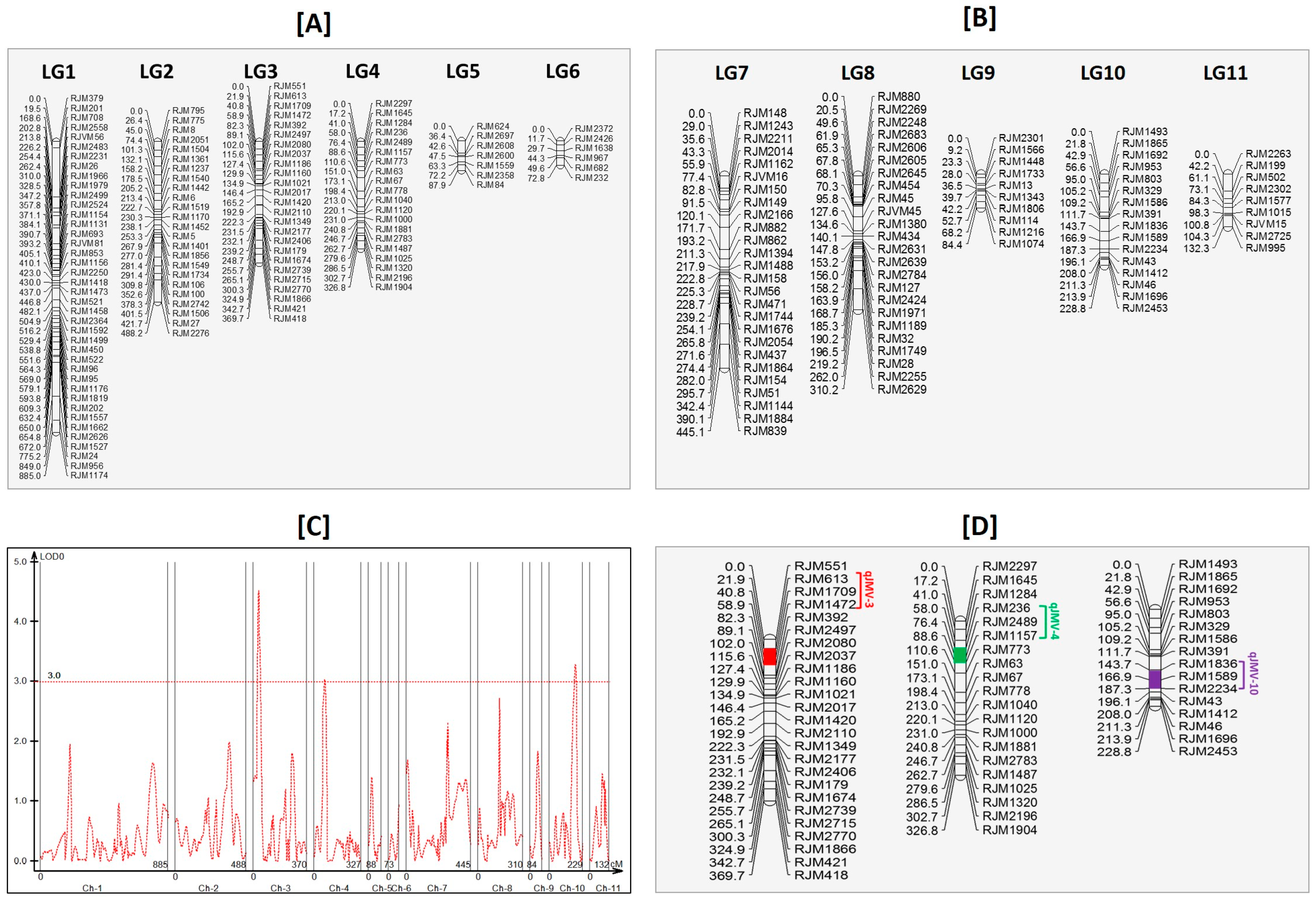
3.9. Linkage Mapping and Quantitative Trait Loci Analysis
3.10. Repetitive Sequences in Whole Genome Sequencing
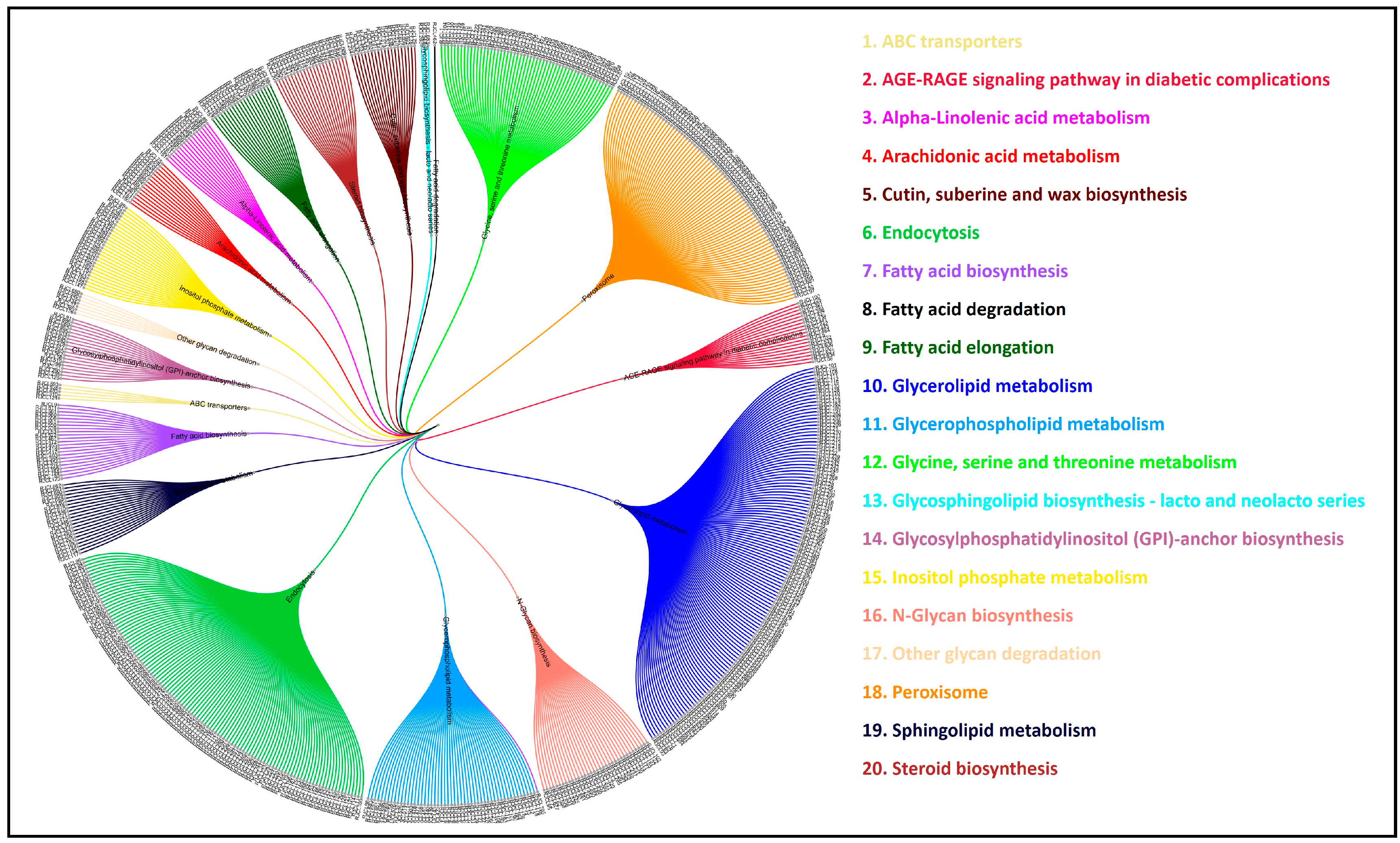
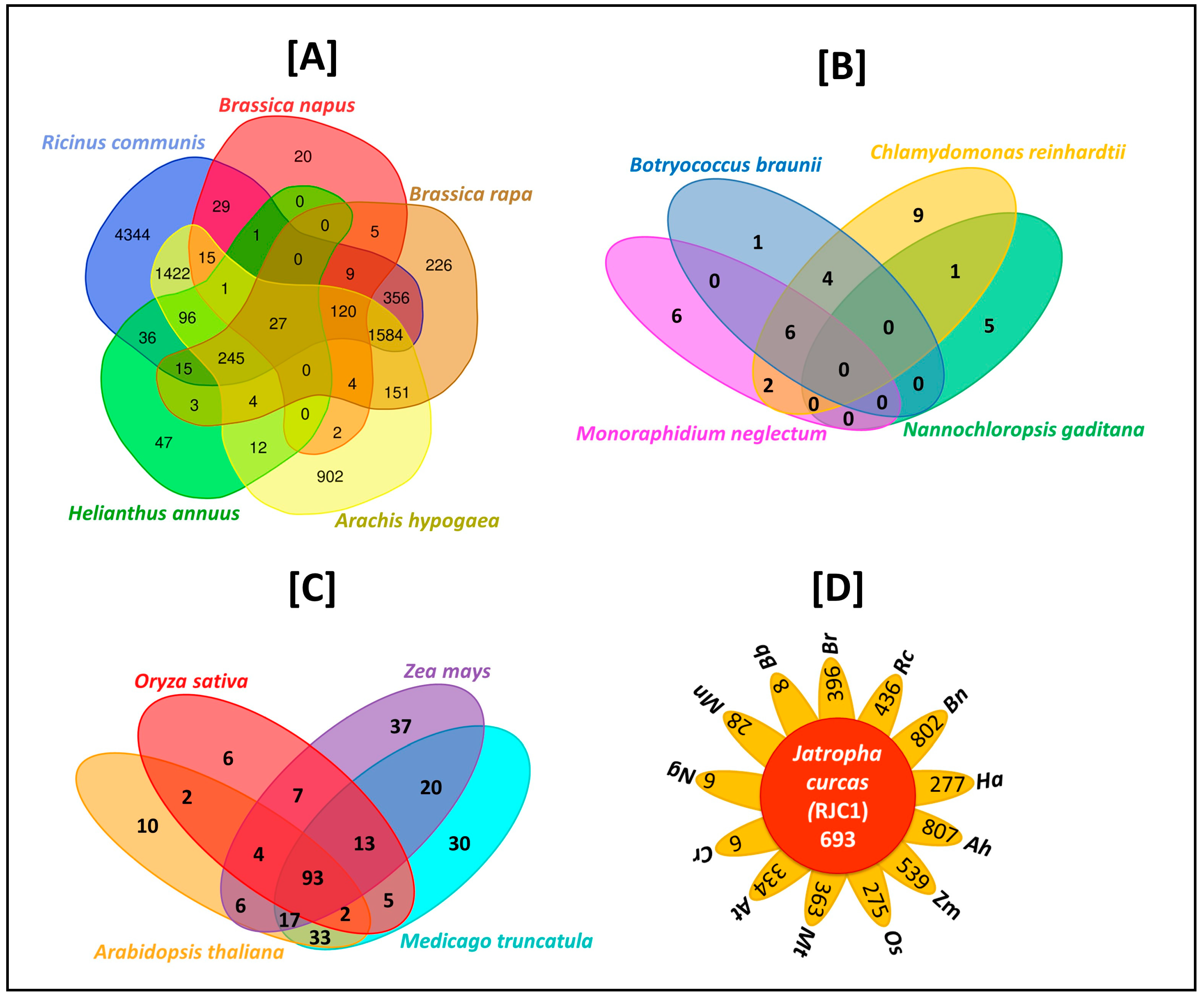
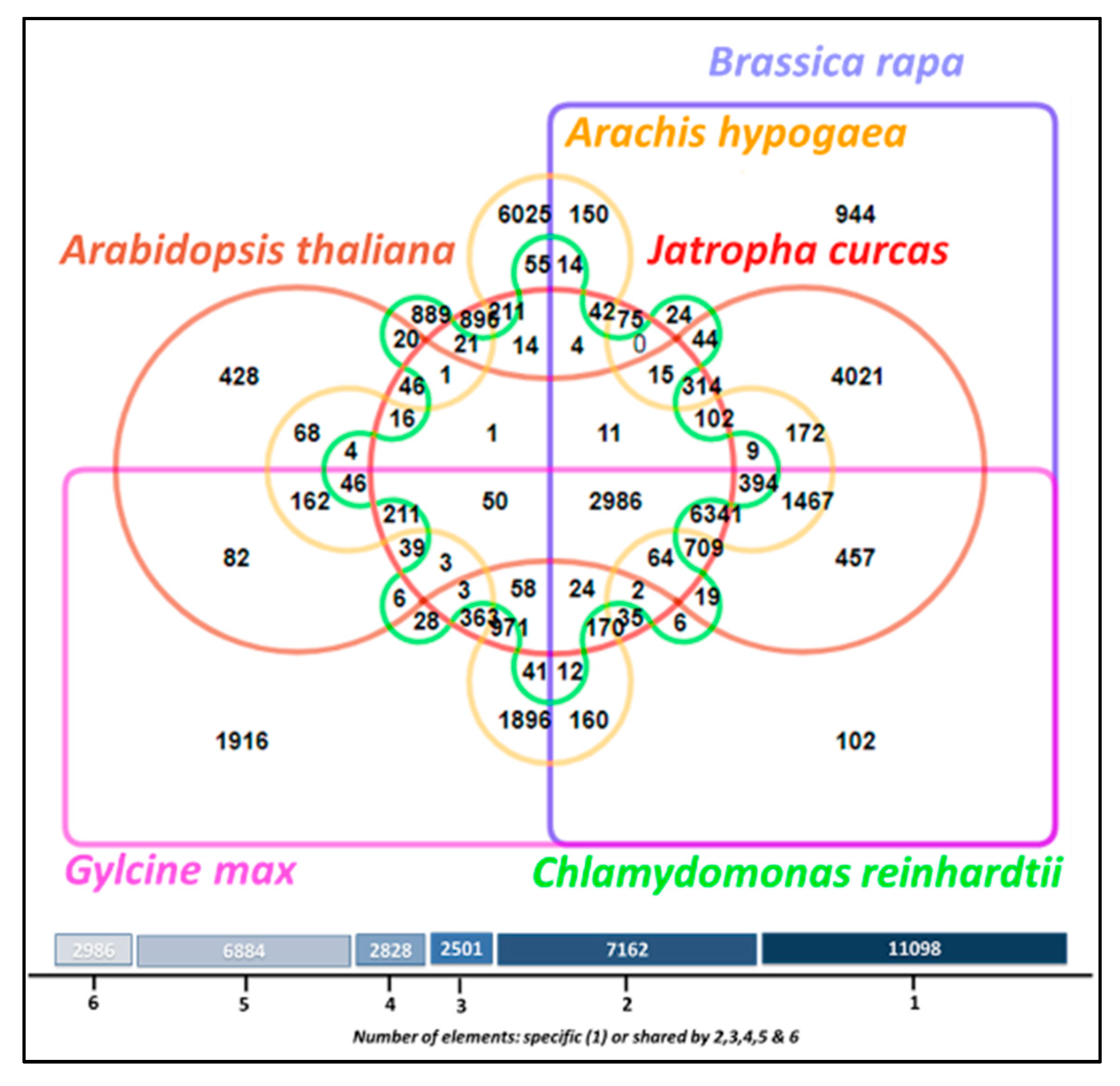
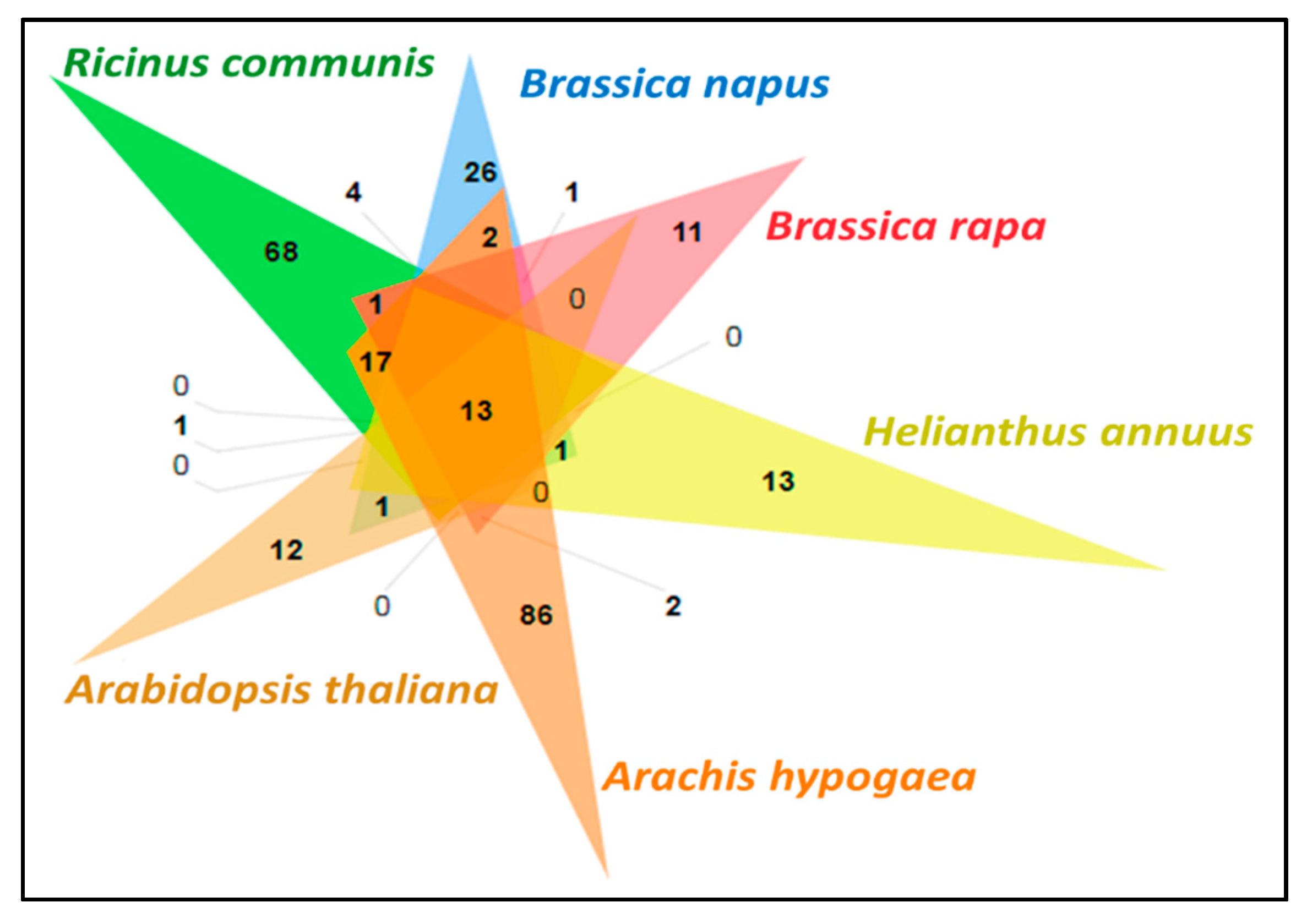
3.11. Comparative Analysis of Lipid Metabolism Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hastilestari, B.R.; Mudersbach, M.; Tomala, F.; Vogt, H.; Biskupek-Korell, B.; Van Damme, P.; Guretzki, S.; Papenbrock, J. Euphorbia tirucalli L.-comprehensive characterization of a drought tolerant plant with a potential as biofuel source. PLoS ONE 2013, 8, e63501. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Hirakawa, H.; Isobe, S.; Fukai, E.; Watanabe, A.; Kato, M.; Kawashima, K.; Minami, C.; Muraki, A.; Nakazaki, N.; et al. Sequence analysis of the genome of an oil-bearing tree, Jatropha curcas L. DNA Res. 2011, 18, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Da Fonseca Lira-Medeiros, C.; Grativol, C.; Cavalcanti Gomes Ferreira, P. Relationship of the genetic diversity of Jatropha curcas in Brazil and worldwide. In Jatropha, Challenges for a New Energy Crop: Volume 2: Genetic Improvement and Biotechnology; Springer: New York, NY, USA, 2013; pp. 251–262. ISBN 9781461449157. [Google Scholar]
- Wendimu, M.A. Jatropha potential on marginal land in Ethiopia: Reality or myth? Energy Sustain. Dev. 2016, 30, 14–20. [Google Scholar] [CrossRef]
- Mohibbe Azam, M.; Waris, A.; Nahar, N.M. Prospects and potential of fatty acid methyl esters of some non-traditional seed oils for use as biodiesel in India. Biomass Bioenergy 2005, 29, 293–302. [Google Scholar] [CrossRef]
- Ginwal, H.S.; Rawat, P.S.; Srivastava, R.L. Seed source variation in growth performance and oil yield of Jatropha curcas Linn. in Central India. Silvae Genet. 2004, 53, 186–191. [Google Scholar] [CrossRef]
- Mozo, T.; Dewar, K.; Dunn, P.; Ecker, J.R.; Fischer, S.; Kloska, S.; Lehrach, H.; Marra, M.; Martienssen, R.; Meier-Ewert, S.; et al. A complete BAC-based physical map of the Arabidopsis thaliana genome. Nat. Genet. 1999, 22, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Batzoglou, S.; Jaffe, D.B.; Stanley, K.; Butler, J.; Gnerre, S.; Mauceli, E.; Berger, B.; Mesirov, J.P.; Lander, E.S. ARACHNE: A whole-genome shotgun assembler. Genome Res. 2002, 12, 177–189. [Google Scholar] [CrossRef]
- Margulies, M.; Egholm, M.; Altman, W.; Attiya, S.; Bader, J. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005, 437, 376–380. [Google Scholar] [CrossRef] [Green Version]
- Baker, M. De novo genome assembly: What every biologist should know. Nat. Methods 2012, 9, 333–337. [Google Scholar] [CrossRef]
- Huang, S.; Li, R.; Zhang, Z.; Li, L.; Gu, X.; Fan, W.; Lucas, W.J.; Wang, X.; Xie, B.; Ni, P.; et al. The genome of the cucumber, Cucumis sativus L. Nat. Genet. 2009, 41, 1275–1281. [Google Scholar] [CrossRef]
- Velasco, R.; Zharkikh, A.; Affourtit, J.; Dhingra, A.; Cestaro, A.; Kalyanaraman, A.; Fontana, P.; Bhatnagar, S.K.; Troggio, M.; Pruss, D.; et al. The genome of the domesticated apple (Malus × domestica Borkh.). Nat. Genet. 2010, 42, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, H.; Tsuchimoto, S.; Sakai, H.; Nakayama, S.; Fujishiro, T.; Kishida, Y.; Kohara, M.; Watanabe, A.; Yamada, M.; Aizu, T.; et al. Upgraded genomic information of Jatropha curcas L. Plant Biotechnol. 2012, 29, 123–130. [Google Scholar] [CrossRef]
- Wu, P.; Zhou, C.; Cheng, S.; Wu, Z.; Lu, W.; Han, J.; Chen, Y.; Ni, P.; Wang, Y.; Xu, X.; et al. Integrated genome sequence and linkage map of physic nut (Jatropha curcas L.), a biodiesel plant. Plant J. 2015, 81, 810–821. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.A.; Iwata, A.; Lee, S.H.; Schmutz, J.; Shoemaker, R. Sequencing crop genomes: Approaches and applications. New Phytol. 2011, 191, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.R.; Zhou, P.; Mudge, J.; Gurtowski, J.; Lee, H.; Ramaraj, T.; Walenz, B.P.; Liu, J.; Stupar, R.M.; Denny, R.; et al. Hybrid assembly with long and short reads improves discovery of gene family expansions. BMC Genomics 2017, 18, 541. [Google Scholar] [CrossRef] [PubMed]
- Utturkar, S.M.; Klingeman, D.M.; Land, M.L.; Schadt, C.W.; Doktycz, M.J.; Pelletier, D.A.; Brown, S.D. Evaluation and validation of de novo and hybrid assembly techniques to derive high-quality genome sequences. Bioinformatics 2014, 30, 2709–2716. [Google Scholar] [CrossRef] [PubMed]
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [PubMed]
- Collard, B.C.; Mackill, D.J. Marker-assisted selection: An approach for precision plant breeding in the twenty-first century. Philos. Trans. R. Soc. B Biol. Sci. 2008, 363, 557–572. [Google Scholar] [CrossRef]
- Malmstrøm, M.; Matschiner, M.; Tørresen, O.K.; Jakobsen, K.S.; Jentoft, S. Data descriptor: Whole genome sequencing data and de novo draft assemblies for 66 teleost species. Sci. Data 2017, 4, 160132. [Google Scholar] [CrossRef]
- Pootakham, W.; Sonthirod, C.; Naktang, C.; Ruang-Areerate, P.; Yoocha, T.; Sangsrakru, D.; Theerawattanasuk, K.; Rattanawong, R.; Lekawipat, N.; Tangphatsornruang, S. De novo hybrid assembly of the rubber tree genome reveals evidence of paleotetraploidy in Hevea species. Sci. Rep. 2017, 7, 41457. [Google Scholar] [CrossRef]
- Vining, K.J.; Johnson, S.R.; Ahkami, A.; Lange, I.; Parrish, A.N.; Trapp, S.C.; Croteau, R.B.; Straub, S.C.K.; Pandelova, I.; Lange, B.M. Draft genome sequence of Mentha longifolia and development of resources for mint cultivar improvement. Mol. Plant 2017, 10, 323–339. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Singh, R. Natural occurrence of leaf curl mosaic virus of Jatropha in Uttar Pradesh of India. World J. Pharm. Pharm. Sci. 2015, 4, 1631–1638. [Google Scholar]
- Kashina, B.D.; Alegbejo, M.D.; Banwo, O.O.; Nielsen, S.L.; Nicolaisen, M. Molecular identification of a new begomovirus associated with mosaic disease of Jatropha curcas L. in Nigeria. Arch. Virol. 2013, 158, 511–514. [Google Scholar] [CrossRef] [PubMed]
- Nextera® Mate Pair Library Prep Reference Guide; 15035209 v02; Illumina: San Diego, CA, USA, 2016; pp. 1–28.
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 3 December 2018).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Xu, H.; Luo, X.; Qian, J.; Pang, X.; Song, J.; Qian, G.; Chen, J.; Chen, S. FastUniq: A fast de novo duplicates removal tool for paired short reads. PLoS ONE 2012, 7, e52249. [Google Scholar] [CrossRef]
- Chikhi, R.; Medvedev, P. Informed and automated k-mer size selection for genome assembly. Bioinformatics 2014, 30, 31–37. [Google Scholar] [CrossRef]
- Zimin, A.V.; Marçais, G.; Puiu, D.; Roberts, M.; Salzberg, S.L.; Yorke, J.A. The MaSuRCA genome assembler. Bioinformatics 2013, 29, 2669–2677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, C.; Ma, Z.S.; Cannon, C.H.; Pop, M.; Yu, D.W. SparseAssembler: De novo Assembly with the Sparse de Bruijn Graph. arXiv, 2011; arXiv:11062603. [Google Scholar]
- Ye, C.; Ma, Z.; Cannon, C.H.; Pop, M.; Yu, D.W. Exploiting sparseness in de novo genome assembly. BMC Bioinform. 2012, 13, S1. [Google Scholar] [CrossRef] [Green Version]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; Wang, J. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef]
- Ye, C.; Hill, C.M.; Wu, S.; Ruan, J.; Ma, Z. DBG2OLC: Efficient assembly of large genomes using long erroneous reads of the third generation sequencing technologies. Sci. Rep. 2016, 6, 31900. [Google Scholar] [CrossRef]
- Xue, W.; Li, J.T.; Zhu, Y.P.; Hou, G.Y.; Kong, X.F.; Kuang, Y.Y.; Sun, X.W. L_RNA_scaffolder: Scaffolding genomes with transcripts. BMC Genomics 2013, 14, 604. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Parra, G.; Bradnam, K.; Korf, I. CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 2007, 23, 1061–1067. [Google Scholar] [CrossRef]
- Stanke, M.; Morgenstern, B. AUGUSTUS: A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005, 33, W465–W467. [Google Scholar] [CrossRef]
- Hoff, K.J.; Lange, S.; Lomsadze, A.; Borodovsky, M.; Stanke, M. BRAKER1: Unsupervised RNA-Seq-Based Genome Annotation with GeneMark-ET and AUGUSTUS: Table 1. Bioinformatics 2016, 32, 767–769. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. TRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1996, 25, 955–964. [Google Scholar] [CrossRef]
- Wikipedia, F. RefSeq RefSeq categories. Nucleic Acids Res. 2015, 33, 5–7. [Google Scholar] [CrossRef]
- Langmead, B. Aligning short sequencing reads with Bowtie. Curr. Protoc. Bioinforma. 2010, 32, 11.7.1–11.7.14. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Quinlan, A.R. BEDTools: The Swiss-Army tool for genome feature analysis. Curr. Protoc. Bioinform. 2014, 11.12.1–11.12.34. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [PubMed]
- Rohlf, F.J. NTSYSpc, Version 2.02g, Exter Software; Applied Bio-Systematics Inc.: Setauket, NY, USA, 1998. [Google Scholar]
- Van Ooijen, J.W.; Voorrips, R.E. JoinMap 3.0: Software for the Calculation of Genetic Linkage Maps; Plant Research International: Wageningen, The Netherlands, 2001. [Google Scholar]
- Basten, C.J.; Weir, B.S.; Zeng, Z.B. QTL Cartographer: A Reference Manual and Tutorial for QTL Mapping; North Carolina State Univ.: Raleigh, NC, USA, 1997. [Google Scholar]
- Price, A.L.; Jones, N.C.; Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 2005, 21, i351–i358. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. 2010. Available online: http://www.repeatmasker.org (accessed on 3 December 2018).
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Coleman-Derr, D.; Chen, G.; Gu, Y.Q. OrthoVenn: A web server for genome wide comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 2015, 43, W78–W84. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Jayakumar, V.; Nitasaka, E.; Toyoda, A.; Noguchi, H.; Itoh, T.; Shin, I.T.; Minakuchi, Y.; Koda, Y.; Nagano, A.J.; et al. Genome sequence and analysis of the Japanese morning glory Ipomoea nil. Nat. Commun. 2016, 7, 13295. [Google Scholar] [CrossRef] [PubMed]
- Apweiler, R. UniProt: The Universal Protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef] [PubMed]
- UniProt Consortium, T.U. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar] [CrossRef] [PubMed]
- Goyer, A. Thiamine in plants: Aspects of its metabolism and functions. Phytochemistry 2010, 71, 1615–1624. [Google Scholar] [CrossRef]
- Yazaki, K.; Arimura, G.; Ohnishi, T. ‘Hidden’ terpenoids in plants: Their biosynthesis, localization and ecological roles. Plant Cell Physiol. 2017, 58, 1615–1621. [Google Scholar] [CrossRef]
- Jalmi, S.K.; Sinha, A.K. ROS mediated MAPK signaling in abiotic and biotic stress- striking similarities and differences. Front. Plant Sci. 2015, 6, 769. [Google Scholar] [CrossRef]
- Saha, B.; Borovskii, G.; Panda, S.K. Alternative oxidase and plant stress tolerance. Plant Signal. Behav. 2016, 11, e1256530. [Google Scholar] [CrossRef]
- Bilgin, D.D.; Zavala, J.A.; Zhu, J.; Clough, S.J.; Ort, D.R.; Delucia, E.H. Biotic stress globally downregulates photosynthesis genes. Plant. Cell Environ. 2010, 33, 1597–1613. [Google Scholar] [CrossRef] [Green Version]
- Alter, S.; Bader, K.C.; Spannagl, M.; Wang, Y.; Bauer, E.; Schon, C.-C.; Mayer, K.F.X. DroughtDB: An expert-curated compilation of plant drought stress genes and their homologs in nine species. Database 2015, 2015, bav046. [Google Scholar] [CrossRef] [PubMed]
- You, F.M.; Huo, N.; Gu, Y.; Luo, M.-C.; Ma, Y.; Hane, D.; Lazo, G.R.; Dvorak, J.; Anderson, O.D. BatchPrimer3: A high throughput web application for PCR and sequencing primer design. BMC Bioinform. 2008, 9, 253. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, P.; Varshney, A.; Johnson, T.S.; Jha, T.B. Jatropha curcas: A review on biotechnological status and challenges. Plant Biotechnol. Rep. 2011, 5, 197–215. [Google Scholar] [CrossRef]
- Natarajan, P.; Kanagasabapathy, D.; Gunadayalan, G.; Panchalingam, J.; Shree, N.; Sugantham, P.A.; Singh, K.K.; Madasamy, P. Gene discovery from Jatropha curcas by sequencing of ESTs from normalized and full-length enriched cDNA library from developing seeds. BMC Genomics 2010, 11, 606. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Deng, Z.; Qin, B.; Liu, X.; Men, Z. De novo assembly and characterization of bark transcriptome using Illumina sequencing and development of EST-SSR markers in rubber tree (Hevea brasiliensis Muell. Arg.). BMC Genomics 2012, 13, 192. [Google Scholar] [CrossRef] [PubMed]
- Rivarola, M.; Foster, J.T.; Chan, A.P.; Williams, A.L.; Rice, D.W.; Liu, X.; Melake-Berhan, A.; Creasy, H.H.; Puiu, D.; Rosovitz, M.J.; et al. Castor Bean Organelle genome sequencing and worldwide genetic diversity analysis. PLoS ONE 2011, 6, e21743. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Xia, Y.; Li, X.; Hou, L.; Yu, J. The Rice Genome Knowledgebase (RGKbase): An annotation database for rice comparative genomics and evolutionary biology. Nucleic Acids Res. 2012, 41, D1199–D1205. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of contigs | 2959 |
| Total bases (bp) | 265,767,376 |
| Max contig sequence (bp) | 1,050,193 |
| Average sequence (bp) | 89,817 |
| N50 length (bp) | 169,453 |
| GC content genome (%) | 33.40 |
| Protein-coding genes (AUGUSTUS 3.2.1) | 20,759 |
| Protein-coding genes (BRAKER2) | 18,890 |
| Average gene length (Kb) | 1.16 |
| GC content of genes (%) | 43.85 |
| Gene space-covered BUSCO (%) | 84.8 |
| Repeats identified (%) | 41.09 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kancharla, N.; Jalali, S.; Narasimham, J.V.; Nair, V.; Yepuri, V.; Thakkar, B.; Reddy, V.; Kuriakose, B.; Madan, N.; S, A. De Novo Sequencing and Hybrid Assembly of the Biofuel Crop Jatropha curcas L.: Identification of Quantitative Trait Loci for Geminivirus Resistance. Genes 2019, 10, 69. https://doi.org/10.3390/genes10010069
Kancharla N, Jalali S, Narasimham JV, Nair V, Yepuri V, Thakkar B, Reddy V, Kuriakose B, Madan N, S A. De Novo Sequencing and Hybrid Assembly of the Biofuel Crop Jatropha curcas L.: Identification of Quantitative Trait Loci for Geminivirus Resistance. Genes. 2019; 10(1):69. https://doi.org/10.3390/genes10010069
Chicago/Turabian StyleKancharla, Nagesh, Saakshi Jalali, J. V. Narasimham, Vinod Nair, Vijay Yepuri, Bijal Thakkar, VB Reddy, Boney Kuriakose, Neeta Madan, and Arockiasamy S. 2019. "De Novo Sequencing and Hybrid Assembly of the Biofuel Crop Jatropha curcas L.: Identification of Quantitative Trait Loci for Geminivirus Resistance" Genes 10, no. 1: 69. https://doi.org/10.3390/genes10010069