Comparison of the Whole-Plastome Sequence between the Bonin Islands Endemic Rubus boninensis and Its Close Relative, Rubus trifidus (Rosaceae), in the Southern Korean Peninsula



Abstract
:1. Introduction
2. Materials and Methods
2.1. Plastome Sequencing and Annotation
2.2. Comparative Plastome Analysis
2.3. Tandem Repeat and Microsatellite Analysis
2.4. Phylogenetic Analysis
3. Results and Discussion
3.1. Genome Size and Features
3.2. Analysis of Microsatellites
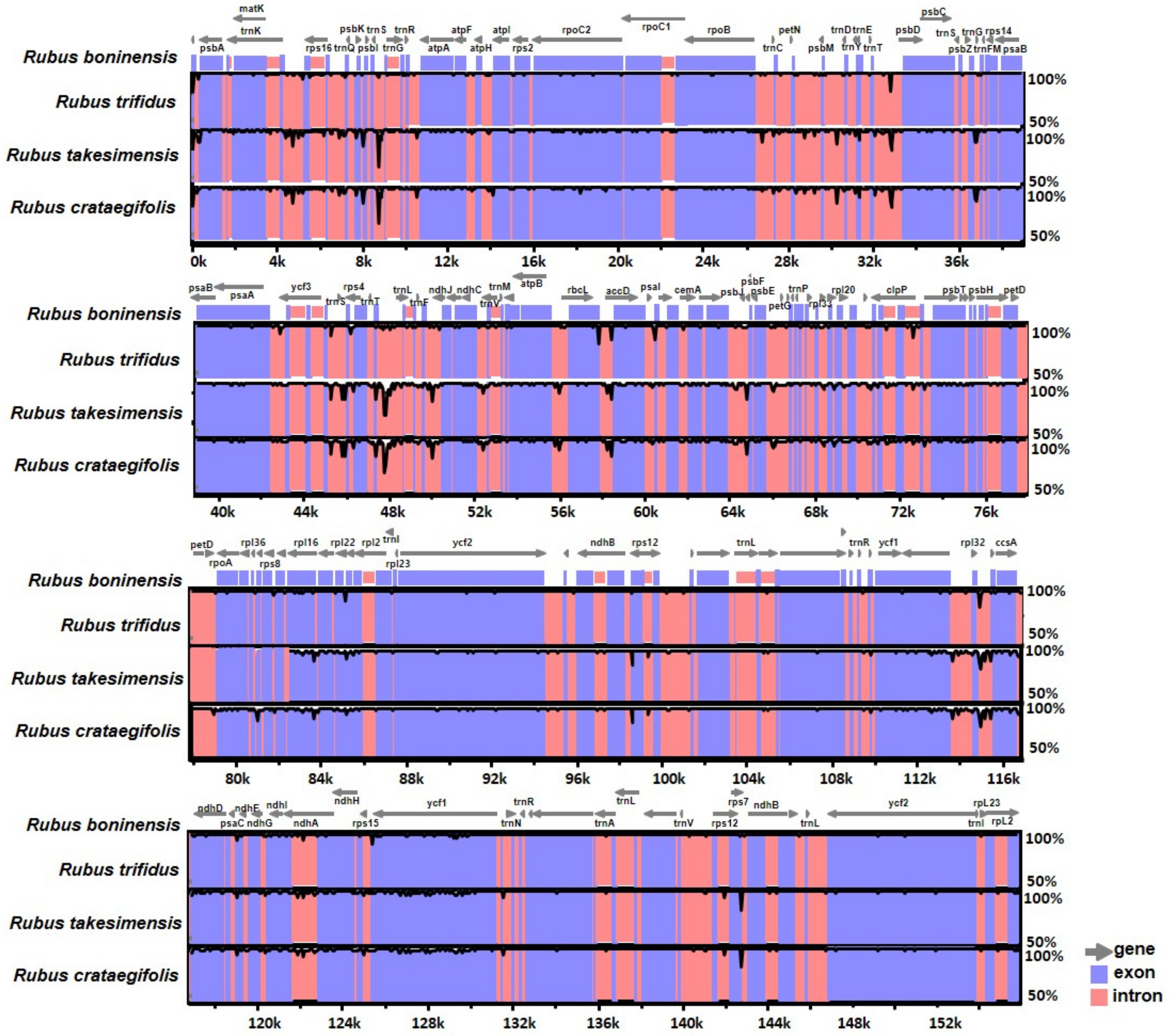
3.3. Comparative Analysis of Genome Structure
3.4. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Asami, S. Topography and geology in the Bonin Islands. In The Nature of the Bonin Islands; Tutama, T., Asami, S., Eds.; Hirokawa Shoten: Tokyo, Japan, 1970; pp. 91–108. (In Japanese) [Google Scholar]
- Toyoda, T. Flora of Bonin Islands (Enlarged & Revised); Aboc-sha: Kamakura, Japan, 2003; pp. 340–346. (In Japanese) [Google Scholar]
- Kaizuka, S. Geology and geomorphology of the Bonin Islands. Bull. Ogasawara Res. 1977, 1, 29–34. [Google Scholar]
- Imaizumi, T.; Tamura, T. Geomorphology of the Chichijima and Hahajima islands. Bull. Ogasawara Res. 1984, 8, 3–11. [Google Scholar]
- Ito, M. Origin and evolution of endemic plants of the Bonin (Ogasawara) Islands. Res. Pop. Ecol. 1988, 40, 205–212. [Google Scholar] [CrossRef]
- Chiba, S. Accelerated evolution of land snails Mandarina in the oceanic Bonin Islands: Evidence from mitochondrial DNA sequences. Evolution 1999, 53, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Chiba, S. Ecological diversity and speciation in land snails of the genus Mandarina from the Bonin Islands. Popul. Ecol. 2002, 44, 179–187. [Google Scholar] [CrossRef]
- Kobayashi, S. A list of the vascular plants occurring in the Ogasawara (Bonin) Islands. Ogasawara Res. 1978, 1, 1–33. [Google Scholar]
- Yamazaki, T. The floristic position of the Bonin Islands plants. In Illustrated Book of the Bonin Islands Plants; Toyoda, T., Ed.; Aboc-sha: Kamakura, Japan, 1981; pp. 303–308. (In Japanese) [Google Scholar]
- Shimizu, Y. Forest structures, composition, and distribution on a Pacific island, with reference to ecological release and speciation. Pac. Sci. 1991, 45, 28–49. [Google Scholar]
- Takayama, K.; Ohi-toma, T.; Kudoh, H.; Kato, H. Origin and diversification of Hibiscus glaber, species endemic to the oceanic Bonin Islands, revealed by chloroplast DNA polymorphism. Mol. Ecol. 2005, 14, 1059–1071. [Google Scholar] [CrossRef]
- Takayama, K.; Tsutsumi, C.; Kawaguchi, D.; Kato, H.; Yukawa, T. Rediscovery of Liparis hostifolia in Minami-Iwo-To Island of the Bonin (Ogasawara) Islands, Japan and molecular identification using the herbarium specimen collected more than 100 years ago. Acta Phytotax. Geobot. 2019, in press. [Google Scholar]
- Nakano, S.; Matsumoto, A.; Ohta, Y.; Nakamura, H.; Furukawa, R. K-Ar ages of volcanic rocks from Kita-Iwo-To and Minami-Iwo-To Islands. In Proceedings of the Japan Geoscience Union Annual Meeting, Chiba, Japan, 16–21 May 2009; pp. V160–P012. [Google Scholar]
- Fujita, T.; Takayama, K.; Shumiya, T.; Kato, H. Vascular Flora of Minami-Iwo-To Island. Ogasawara Res. 2008, 33, 49–62. [Google Scholar]
- Ohba, H. The Higher Plant Flora of Minami-Iwojima Island (Ins. San Augustino), the Volcano Group of Islands, Japan; Wildlife Research Center: Minami-Iwojima no Shizen, Japan, 1983; pp. 61–143, (In Japanese with English summary). [Google Scholar]
- Stuessy, T.F.; Jakubowsky, G.; Gomez, R.S.; Pfosser, M.; Schlüter, P.M.; Fer, T.; Sun, B.-Y.; Kato, H. Anagenetic evolution in island plants. J. Biogeogr. 2006, 33, 1259–1265. [Google Scholar] [CrossRef]
- Tuyama, T. Plantae Boninenses Novae vel Criticae. IX. Bot. Mag. 1937, 51, 131. [Google Scholar]
- Ohba, H.; Boufford, D.E.; Iwatsuki, K. Flora of Japan. IIb; Kodansha: Tokyo, Japan, 2001; p. 155. [Google Scholar]
- Koidzumi, G. Conspectus rosacearum japonicarum. J. Coll. Sci. Imp. Univ. Tokyo 1913, 34, 113. [Google Scholar]
- Yang, J.Y.; Pak, J.-H.; Maki, M.; Kim, S.-C. Multiple origins and the population genetic structure of Rubus takesimensis (Rosaceae) on Ulleung Island: Implication for the genetic consequences of anagenetic speciation. PLoS ONE 2019, 14, e0222707. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Yang, J.Y.; Jung, K.-S.; Pak, J.-H.; Maki, M.; Kim, S.-C. Chloroplast DNA assessment of anagenetic speciation in Rubus takesimensis (Rosaceae) on Ulleung Island, Korea. J. Plant Biol. 2017, 60, 163–174. [Google Scholar] [CrossRef]
- Yang, J.Y.; Pak, J.-H.; Kim, S.-C. The complete plastome sequence of Rubus takesimensis endemic to Ulleung Island, Korea: Insights into molecular evolution of anagenetically derived species in Rubus (Rosaceae). GENE 2018, 668, 221–228. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Lohse, M.; Drechsel, O.; Bock, R. Organellar genome DRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2009, 25, 1451–1452. [Google Scholar]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res 2014, 32, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Brudno, M.; Malde, S.; Poliakov, A.; Do, C.B.; Couronne, O.; Dubchak, I.; Batzoglou, S. Glocal alignment: Finding rearrangements during alignment. Bioinformatics 2003, 19, i54–i62. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software v7: Improvements in performance and usability. Mol. Biol. Evol 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP v6: DNA sequence polymorphism analysis of large datasets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis v7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Sharp, P.M.; Li, W.H. An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol. 1986, 24, 28–38. [Google Scholar] [CrossRef]
- Kozak, M. Comparison of initiation of protein synthesis in procaryotes, eucaryotes, and organelles. Microbiol. Rev. 1983, 47, 1–45. [Google Scholar]
- Thiel, T.; Michalek, W.; Varshney, R.K.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Daniell, H.; Wardack, K.J.; Kanagaraj, A.; Lee, S.-B.; Saski, C.; Jansen, R.K. The complete nucleotide sequence of the cassava (Manihot esculenta) chloroplast genome and the evolution of atpF in Malpighiales: RNA editing and multiple losses of a group II intron. Theor. Appl. Genet. 2008, 116, 723–737. [Google Scholar] [CrossRef]
- Howarth, D.G.; Gardner, D.E.; Morden, C.W. Phylogeny of Rubus subgenus Idaeobatus (Rosaceae) and its implications toward colonization of the Hawaiian Islands. Syst. Bot. 1997, 22, 433–441. [Google Scholar] [CrossRef]
- Kim, K.J.; Jansen, R.K. ndhF sequence evolution and the major clades in the sunflower family. Proc. Natl. Acad. Sci. USA 1995, 92, 10379–10383. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.H.; Kim, S.-C. Comparative analysis of the complete chloroplast genome sequences of three closely related East-Asian wild Roses (Rosa sect. Synstylae; Rosaceae). GENES 2019, 10, 23. [Google Scholar] [CrossRef] [PubMed]
- Morton, B.R. Selection on the codon bias of chloroplast and cyanelle genes in different plant and algal lineages. J. Mol. Evol. 1998, 46, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Zhou, T.; Ma, J.; Sun, X.; Lu, Z. The relationship between synonymous codon usage and protein structure in Escherichia coli and Homo sapiens. Biosystems 2004, 73, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.; Deng, P.; Feng, K.; Liu, P.; Du, X.; You, F.M.; Song, W. Comparative analysis of codon usage patterns in chloroplast genomes of the Asteraceae family. Plant Mol. Biol. Rep. 2014, 32, 828–840. [Google Scholar] [CrossRef]
- Ravi, V.; Khurana, J.P.; Tyagi, A.K.; Khurana, P. An update chloroplast genomes. Plant Syst. Evol. 2008, 271, 101–122. [Google Scholar] [CrossRef]
- Cheng, H.; Li, J.; Zhang, H.; Cai, B.; Gao, Z.; Qiao, Y.; Mi, L. The complete chloroplast genome sequence of strawberry (Fragaria× ananassa Duch.) and comparison with related species of Rosaceae. PeerJ. 2017, 5, e3919. [Google Scholar] [CrossRef] [PubMed]
- Jian, H.-Y.; Zhang, Y.-H.; Yan, H.-J.; Qiu, X.-Q.; Wang, Q.-G.; Li, S.-B.; Zhang, S.-D. The complete chloroplast genome of a key ancestor of modern roses, Rosa chinensis var. spontanea, and a comparison with congeneric species. Molecules 2018, 23, 389. [Google Scholar]
- Zhang, X.; Rong, C.; Qin, L.; Mo, C.; Fan, L.; Yan, J.; Zhang, M. Complete chloroplast genome sequence of Malus hupehensis: Genome structure, comparative analysis, and phylogenetic relationships. Molecules 2018, 23, 2917. [Google Scholar] [CrossRef] [PubMed]
- Gichira, A.W.; Li, Z.; Saina, J.K.; Long, Z.; Hu, G.; Gituru, R.W.; Wang, Q.; Chen, J. The complete chloroplast genome sequence of an endemic monotypic genus Hagenia (Rosaceae): Structural comparative analysis, gene content and microsatellite detection. PeerJ. 2017, 5, e2846. [Google Scholar] [CrossRef] [PubMed]
- Powell, W.; Morgante, M.; Andre, C.; McNicol, J.W.; Machray, G.C.; Doyle, J.J.; Tingey, S.V.; Rafalski, J.A. Hypervariable microsatellites provide a general source of polymorphic DNA markers for the chloroplast genome. Curr. Biol. 1995, 5, 1023–1029. [Google Scholar] [CrossRef] [Green Version]
- Provan, J.; Soranzo, N.; Wilson, N.J.; McNicol, J.W.; Forrest, G.I.; Cottrell, J.; Powell, W. Gene-Pool variation in Caledonian and European Scots pine (Pinus sylvestris L.) revealed by chloroplast simple sequence repeats. Proc. R. Soc. Lond. Biol. Sci. 1998, 265, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Mori, N.; Ogihara, Y. Evaluation of allelic diversity at chloroplast microsatellite loci among common wheat and its ancestral species. Theor. Appl. Genet. 2001, 103, 896–904. [Google Scholar] [CrossRef]
- Vendramin, G.G.; Degen, B.; Petit, R.J.; Anzidei, M.; Madaghiele, A.; Ziegenhagen, B. High level of variation at Abies alba chloroplast microsatellite loci in Europe. Mol. Ecol. 1999, 8, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.M.; Staub, J.E.; Chen, J.F. Molecular phylogeny of Cucumis species as revealed by consensus chloroplast SSR marker length and sequence variation. Genome 2006, 49, 219–229. [Google Scholar] [CrossRef]
- Katayama, H.; Uematsu, C. Structural analysis of chloroplast DNA in Prunus (Rosaceae): Evolution, genetic diversity and unequal mutations. Theor. Appl. Genet. 2005, 111, 1430–1439. [Google Scholar] [CrossRef] [PubMed]
- Korotkova, N.; Nauheumer, L.; Ter-Voskanyan, H.; Allgaier, M.; Borsch, T. Variability Among the most rapidly evolving plastid genomic region is lineage-specific: Implication of pairwise genome comparisons in Pyrus (Rosaceae) and other angiosperms for marker choice. PLoS ONE 2014, 9, e112998. [Google Scholar] [CrossRef]
- Panga, X.; Songa, J.; Zhua, Y.; Xub, H.; Huanga, L.; Chena, S. Applying plant DNAbarcodes for Rosaceae species identification. Cladistics 2011, 27, 165–170. [Google Scholar] [CrossRef]
- Li, W.; Lu, Y.; Xie, X.; Li, B.; Han, Y.; Sun, T.; Xian, Y.; Yang, H.; Liu, K. Development of chloroplast genomic resources for Pyrus hopeiensis (Rosaceae). Conserv. Genet. Resour. 2018, 10, 511–513. [Google Scholar] [CrossRef]
- Alice, L.A.; Campbell, C.S. Phylogeny of Rubus (Rosaceae) based on nuclear ribosomal DNA internal transcribed spacer region sequences. Am. J. Bot. 1999, 86, 81–97. [Google Scholar] [CrossRef] [PubMed]
- Morton, B.R. The role of context-dependent mutations in generating compositional and codon usage bias in grass chloroplast DNA. J. Mol. Evol. 2003, 56, 616–629. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.Y.; Pak, H.-H. Phylogeny of Korean Rubus (Rosaceae) based on ITS (nrDNA) and trnL/F intergenic region (cpDNA). J. Plant Biol. 2006, 49, 44–54. [Google Scholar] [CrossRef]
- Yang, J.Y.; Yoon, H.-S.; Pak, J.-H. Phylogeny of Korean Rubus (Rosaceae) based on the second intron of the LEAFY gene. Can. J. Plant Sci. 2012, 92, 461–472. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, Q.; Chen, T.; Tang, H.; Liu, L.; Wang, X. Phylogenetic insights into Chinese Rubus (Rosaceae) from multiple chloroplast and nuclear DNAs. Front. Plant Sci. 2016, 7, 968. [Google Scholar] [CrossRef] [PubMed]
- Potter, D.; Eriksson, T.; Evans, R.C.; Oh, S.; Smedmark, J.E.E.; Morgan, D.R.; Kerr, M.; Robertson, K.R.; Arsenault, M.; Dickinson, T.A.; et al. Phylogeny and classification of Rosaceae. Pl. Syst. Evol. 2007, 266, 5–43. [Google Scholar] [CrossRef]
- Xiang, Y.; Huang, C.-H.; Hu, Y.; Wen, J.; Li, S.; Yi, T.; Chen, H.; Xiang, J.; Ma, H. Evolution of Rosaceae fruit types based on nuclear phylogeny in the context of geological times and genome duplication. Mol. Biol. Evol. 2016, 34, 262–281. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxa | R. boninensis | R. trifidus |
|---|---|---|
| Accession Number | MH734123 | MK465682 |
| Total cpDNA size (bp)/GC content (%) | 155,807/37.1 | 155,823/37.1 |
| LSC size (bp)/GC content (%) | 85,438/34.9 | 85,466/35.0 |
| IR size (bp)/GC content (%) | 25,793/42.8 | 25,799/42.8 |
| SSC size (bp)/GC content (%) | 18,783/31.0 | 18,759/31.0 |
| Number of genes | 131 | 131 |
| Number of protein-coding genes | 84 | 84 |
| Number of tRNA genes | 37 | 37 |
| Number of rRNA genes | 8 | 8 |
| Number of duplicated genes | 17 | 17 |
| Codon | Amino Acid | Count BON/TRF | RSCU BON/TRF | tRNA BON/TRF | Codon | Amino Acid | Count BON/TRF | RSCU BON/TRF | tRNA BON/TRF |
|---|---|---|---|---|---|---|---|---|---|
| UUU | F | 986/985 | 1.31 | UCU | S | 561/554 | 1.68/1.67 | ||
| UUC | F | 514 | 0.69 | trnF-GAA | UCC | S | 343 | 1.03 | trnS-GGA |
| UUA | L | 882/887 | 1.92/1.93 | trnL-UAA | UCA | S | 382/380 | 1.15 | - / trnS-UGA |
| UUG | L | 550/552 | 1.2 | trnL-CAA | UCG | S | 200/204 | 0.6 | |
| CUU | L | 592/590 | 1.29 | CCU | P | 408/409 | 1.5 | ||
| CUC | L | 185/138 | 0.4 | CCC | P | 212/213 | 0.78 | ||
| CUA | L | 356/355 | 0.78/0.77 | trnL-UAG | CCA | P | 304/300 | 1.12/1.1 | trnP-UGG |
| CUG | L | 185/186 | 0.4/0.41 | CCG | P | 164 | 0.6 | ||
| AUU | I | 1118/1114 | 1.48 | ACU | T | 523/525 | 1.57 | ||
| AUC | I | 435 | 0.58 | trnI-GAU | ACC | T | 254 | 0.76/0.78 | trnT-GGU |
| AUA | I | 711/703 | 0.94 | ACA | T | 406/407 | 1.22 | trnT-UGU | |
| AUG | M | 619/620 | 1 | trnI-CAU, trnfM-CAU/trnfM-CAU | ACG | T | 148 | 0.44 | |
| GUU | V | 518 | 1.45 | GCU | A | 630 | 1.8/1.81 | ||
| GUC | V | 169/167 | 0.47 | trnV-GAC | GCC | A | 219 | 0.63 | |
| GUA | V | 543 | 1.52 | trnV-UAC | GCA | A | 387/384 | 1.11 | trnA-UGC |
| GUG | V | 202/201 | 0.56 | GCG | A | 162 | 0.46 | ||
| UAU | Y | 768/767 | 1.59 | UGU | C | 229 | 1.53 | ||
| UAC | Y | 196 | 0.41 | trnY-GUA | UGC | C | 71 | 0.47 | trnC-GCA |
| UAA | * | 48 | 1.71/1.73 | UGA | * | 16 | 0.57/0.58 | ||
| UAG | * | 20 | 0.71/0.69 | -/trnI-CAT | UGG | W | 454/452 | 1 | trnW-CCA |
| CAU | H | 486/487 | 1.53 | CGU | R | 343/344 | 1.3/1.31 | trnR-ACG | |
| CAC | H | 151/150 | 0.47 | trnH-GUG | CGC | R | 104 | 0.39 | |
| CAA | Q | 711/713 | 1.54/1.55 | trnK-UUG - | CGA | R | 357/358 | 1.36 | |
| CAG | Q | 210/209 | 0.46/0.45 | CGG | R | 116/115 | 0.44 | ||
| AAU | N | 971/968 | 1.52/1.51 | AGU | S | 390/387 | 1.17/1.16 | ||
| AAC | N | 310/311 | 0.48/0.49 | trnN-GUU | AGC | S | 125/126 | 0.37/0.38 | trnS-GCU |
| AAA | K | 1083/1070 | 1.5/1.49 | trnK-UUU | AGA | R | 482 | 1.83 | trnR-UCU |
| AAG | K | 362 | 0.5/0.51 | AGG | R | 178 | 0.68 | ||
| GAU | D | 873/874 | 1.62 | GGU | G | 581/582 | 1.3 | ||
| GAC | D | 203/202 | 0.38 | trnD-GUC | GGC | G | 194/193 | 0.43 | trnS-GCC |
| GAA | E | 1020/1022 | 1.48/1.49 | trnE-UUC | GGA | G | 701/699 | 1.57 | trnG-UCC |
| GAG | E | 358/354 | 0.52/0.51 | GGG | G | 313/312 | 0.7 |
| Codon | Amino Acid | Count CRA/TAK | RSCU CRA/TAK | tRNA CRA/TAK | Codon | Amino Acid | Count CRA/TAK | RSCU CRA/TAK | tRNA CRA/TAK |
|---|---|---|---|---|---|---|---|---|---|
| UUU | F | 980/979 | 1.31 | UCU | S | 557 | 1.68 | ||
| UUC | F | 514 | 0.69 | trnF-GAA | UCC | S | 345 | 1.04 | trnS-GGA |
| UUA | L | 886/887 | 1.93 | trnL-UAA | UCA | S | 383 | 1.15 | trnS-UGA |
| UUG | L | 557/555 | 1.21 | trnL-CAA | UCG | S | 198 | 0.6 | |
| CUU | L | 593/592 | 1.29 | CCU | P | 412 | 1.52 | ||
| CUC | L | 181 | 0.39 | CCC | P | 211 | 0.78 | ||
| CUA | L | 363/361 | 0.79 | trnL-UAG | CCA | P | 303 | 1.11 | trnP-UGG |
| CUG | L | 181/182 | 0.39/0.4 | CCG | P | 161 | 0.59 | ||
| AUU | I | 1105/1105 | 1.48 | ACU | T | 525/523 | 1.57 | ||
| AUC | I | 442 | 0.59 | trnI-GAU | ACC | T | 251 | 0.75 | trnT-GGU |
| AUA | I | 693 | 0.93 | ACA | T | 406/408 | 1.22 | trnT-UGU | |
| AUG | M | 617 | 1 | trnI-CAU, trnfM-CAU, trnM-CAU | ACG | T | 152/151 | 0.46/0.45 | |
| GUU | V | 519 | 1.45 | GCU | A | 634 | 1.82 | ||
| GUC | V | 172 | 0.48 | trnV-GAC | GCC | A | 218 | 0.63 | |
| GUA | V | 536/538 | 1.5/1.51 | trnV-UAC | GCA | A | 377/376 | 1.08 | trnA-UGC |
| GUG | V | 201/200 | 0.56 | GCG | A | 163/164 | 0.47 | ||
| UAU | Y | 766 | 1.59 | UGU | C | 229 | 1.53 | ||
| UAC | Y | 195 | 0.41 | trnY-GUA | UGC | C | 70 | 0.47 | trnC-GCA |
| UAA | * | 47 | 1.68 | UGA | * | 14 | 0.5 | ||
| UAG | * | 23 | 0.82 | UGG | W | 451 | 1 | trnW-CCA | |
| CAU | H | 479/477 | 1.52/1.51 | CGU | R | 341 | 1.29 | trnR-ACG | |
| CAC | H | 152/154 | 0.48/0.49 | trnH-GUG | CGC | R | 105/104 | 0.49/0.39 | |
| CAA | Q | 714 | 1.54 | trnQ-UUG | CGA | R | 362/360 | 1.36 | |
| CAG | Q | 211 | 0.46 | CGG | R | 116/117 | 0.44 | ||
| AAU | N | 969 | 1.52 | AGU | S | 388/387 | 1.17/1.16 | ||
| AAC | N | 307 | 0.48 | trnN-GUU | AGC | S | 124/125 | 0.37 | trnS-GCU |
| AAA | K | 1065/1066 | 1.5 | trnK-UUU | AGA | R | 486/488 | 1.83/1.84 | trnR-UCU |
| AAG | K | 353/352 | 0.5 | AGG | R | 182 | 0.69 | ||
| GAU | D | 867 | 1.62 | GGU | G | 576/577 | 1.29 | ||
| GAC | D | 204/205 | 0.38 | trnD-GUC | GGC | G | 198/197 | 0.44 | trnS-GCC |
| GAA | E | 1017 | 1.48 | trnE-UUC | GGA | G | 700/697 | 1.57 | trnG-UCC |
| GAG | E | 358 | 0.52 | GGG | G | 311/313 | 0.7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Takayama, K.; Pak, J.-H.; Kim, S.-C. Comparison of the Whole-Plastome Sequence between the Bonin Islands Endemic Rubus boninensis and Its Close Relative, Rubus trifidus (Rosaceae), in the Southern Korean Peninsula. Genes 2019, 10, 774. https://doi.org/10.3390/genes10100774
Yang J, Takayama K, Pak J-H, Kim S-C. Comparison of the Whole-Plastome Sequence between the Bonin Islands Endemic Rubus boninensis and Its Close Relative, Rubus trifidus (Rosaceae), in the Southern Korean Peninsula. Genes. 2019; 10(10):774. https://doi.org/10.3390/genes10100774
Chicago/Turabian StyleYang, JiYoung, Koji Takayama, Jae-Hong Pak, and Seung-Chul Kim. 2019. "Comparison of the Whole-Plastome Sequence between the Bonin Islands Endemic Rubus boninensis and Its Close Relative, Rubus trifidus (Rosaceae), in the Southern Korean Peninsula" Genes 10, no. 10: 774. https://doi.org/10.3390/genes10100774
APA StyleYang, J., Takayama, K., Pak, J. -H., & Kim, S. -C. (2019). Comparison of the Whole-Plastome Sequence between the Bonin Islands Endemic Rubus boninensis and Its Close Relative, Rubus trifidus (Rosaceae), in the Southern Korean Peninsula. Genes, 10(10), 774. https://doi.org/10.3390/genes10100774