Ubiquitin Ligases Involved in the Regulation of Wnt, TGF-β, and Notch Signaling Pathways and Their Roles in Mouse Development and Homeostasis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
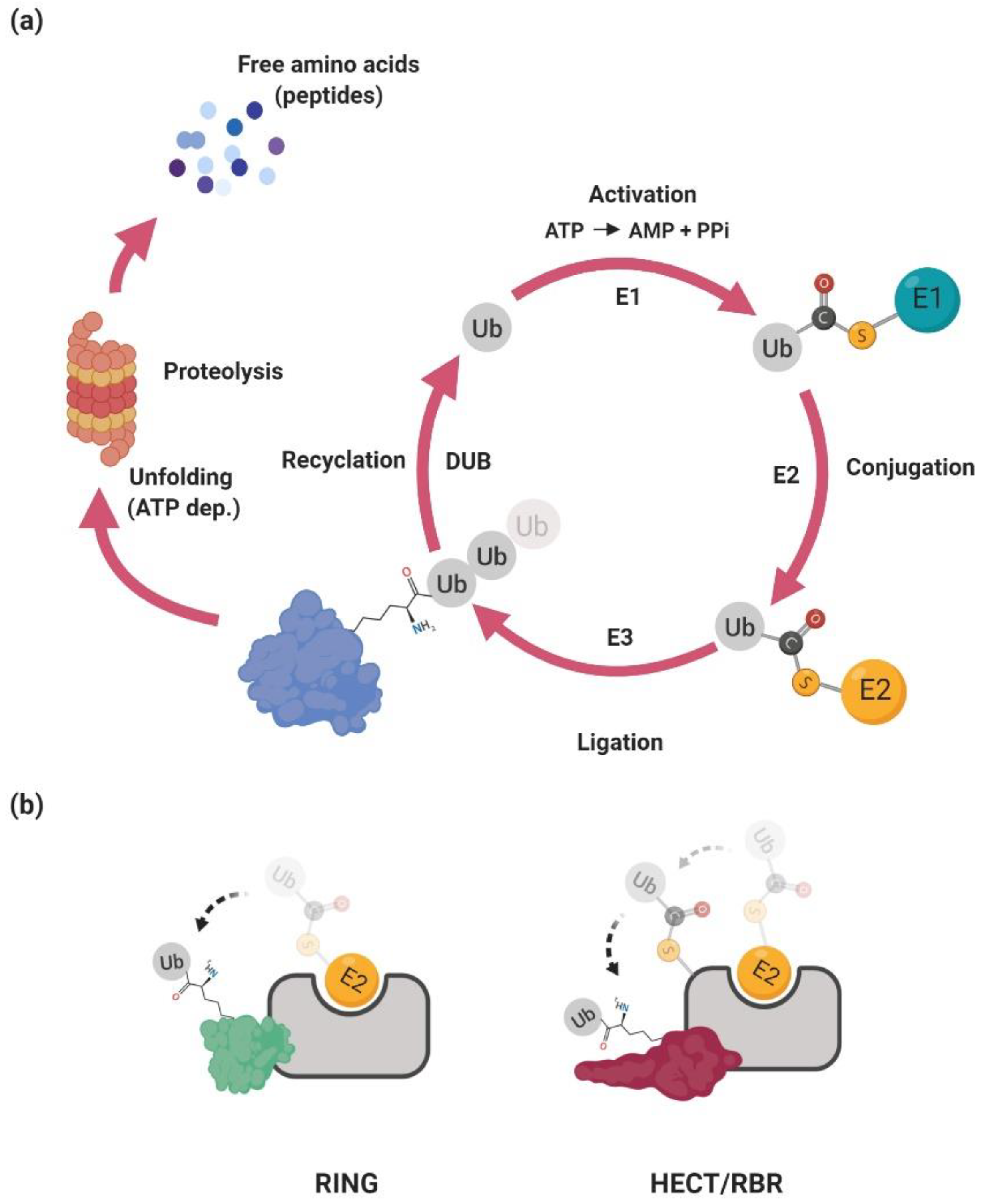
2. The Ubiquitin–Proteasome System
2.1. Ubiquitination
2.2. Ubiquitin Ligases
2.2.1. RING-Type Ubiquitin Ligases
2.2.2. U-box-Type Ubiquitin Ligases
2.2.3. HECT-Type Ubiquitin Ligases
2.2.4. RBR-Type Ubiquitin Ligases
3. Wnt Signaling Pathway and Its Regulation by Ubiquitin Ligases
3.1. Mouse Models of Ubiquitin Ligases Involved in the Wnt Signaling Pathway
3.1.1. β-Transducin Repeat-Containing Protein (β-TrCP)
3.1.2. Zinc and RING Finger 3/ RING Finger 43 (ZNRF3/RNF43)
3.1.3. RING Finger 146 (RNF146)
3.1.4. RING Finger 61 (RNF61)
3.1.5. Seven in Absentia Homolog (SIAH)
3.1.6. E3 Isolated by Differential Display (EDD)
3.1.7. Neural Precursor Cell Expressed, Developmentally Downregulated 4 (Nedd4)
3.1.8. Neural Precursor Cell Expressed, Developmentally Downregulated 4-Like (NEDD4L)
3.1.9. ITCH
3.1.10. Casitas B-Lineage Lymphoma (CBL)
4. TGF-β Signaling Pathway
4.1. TGF-β Signaling Pathway and its Regulation by Ubiquitin Ligases
4.2. Mouse Models of Ubiquitin Ligases Involved in the TGF-β Signaling Pathway
4.2.1. Smad Ubiquitination Regulatory Factor (SMURF)
4.2.2. RING Finger 111/Arkadia
4.2.3. S-Phase Kinase-Associated Protein 2 (SKP2)
4.2.4. MYC Binding Protein 2 (MYCBP2)
4.2.5. Tripartite Motif Containing 33 (TRIM33)
4.2.6. RING Finger 12 (RNF12)
4.2.7. WW Domain Containing E3 Ubiquitin Protein Ligase (WWP)
5. Notch Signaling Pathway
5.1. Notch Signaling Pathway and its Regulation by Ubiquitin Ligases
5.2. Mouse Models of Ubiquitin Ligases Involved in the Notch Signaling Pathway
5.2.1. F-box and WD Repeat Domain Containing 7 (Fbxw7)
5.2.2. Mindbomb and Neuralized (Mib and Neur)
5.2.3. Deltex-1 (Dtx1)
5.2.4. RING finger 8 (RNF8)
5.2.5. Mouse double minute 2 (MDM2)
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef]
- Kinzler, K.W.; Nilbert, M.C.; Su, L.K.; Vogelstein, B.; Bryan, T.M.; Levy, D.B.; Smith, K.J.; Preisinger, A.C.; Hedge, P.; McKechnie, D.; et al. Identification of FAP locus genes from chromosome 5q21. Science 1991, 253, 661–665. [Google Scholar] [CrossRef]
- Ellisen, L.W.; Bird, J.; West, D.C.; Soreng, A.L.; Reynolds, T.C.; Smith, S.D.; Sklar, J. TAN-1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell 1991, 66, 649–661. [Google Scholar] [CrossRef]
- Yakicier, M.C.; Irmak, M.B.; Romano, A.; Kew, M.; Ozturk, M. Smad2 and Smad4 gene mutations in hepatocellular carcinoma. Oncogene 1999, 18, 4879–4883. [Google Scholar] [CrossRef]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef]
- Skaar, J.R.; Pagan, J.K.; Pagano, M. SCF ubiquitin ligase-targeted therapies. Nat. Rev. Drug Discov. 2014, 13, 889–903. [Google Scholar] [CrossRef]
- Kumari, N.; Jaynes, P.W.; Saei, A.; Iyengar, P.V.; Richard, J.L.C.; Eichhorn, P.J.A. The roles of ubiquitin modifying enzymes in neoplastic disease. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 456–483. [Google Scholar] [CrossRef]
- Nalepa, G.; Rolfe, M.; Harper, J.W. Drug discovery in the ubiquitin-proteasome system. Nat. Rev. Drug Discov. 2006, 5, 596–613. [Google Scholar] [CrossRef]
- Buetow, L.; Huang, D.T. Structural insights into the catalysis and regulation of E3 ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2016, 17, 626–642. [Google Scholar] [CrossRef]
- Hershko, A.; Heller, H.; Elias, S.; Ciechanover, A. Components of ubiquitin-protein ligase system. Resolution, affinity purification, and role in protein breakdown. J. Biol. Chem. 1983, 258, 8206–8214. [Google Scholar]
- Deol, K.K.; Lorenz, S.; Strieter, E.R. Enzymatic Logic of Ubiquitin Chain Assembly. Front. Physiol. 2019, 10, 835. [Google Scholar] [CrossRef]
- Stewart, M.D.; Ritterhoff, T.; Klevit, R.E.; Brzovic, P.S. E2 enzymes: More than just middle men. Cell Res. 2016, 26, 423–440. [Google Scholar] [CrossRef]
- Rittinger, K.; Ikeda, F. Linear ubiquitin chains: Enzymes, mechanisms and biology. Open Biol. 2017, 7. [Google Scholar] [CrossRef]
- Akutsu, M.; Dikic, I.; Bremm, A. Ubiquitin chain diversity at a glance. J. Cell Sci. 2016, 129, 875–880. [Google Scholar] [CrossRef]
- Nakayama, K.I.; Nakayama, K. Ubiquitin ligases: Cell-cycle control and cancer. Nat. Rev. Cancer 2006, 6, 369–381. [Google Scholar] [CrossRef]
- Zheng, N.; Shabek, N. Ubiquitin Ligases: Structure, Function, and Regulation. Annu. Rev. Biochem. 2017, 86, 129–157. [Google Scholar] [CrossRef]
- Lilley, C.E.; Chaurushiya, M.S.; Boutell, C.; Landry, S.; Suh, J.; Panier, S.; Everett, R.D.; Stewart, G.S.; Durocher, D.; Weitzman, M.D. A viral E3 ligase targets RNF8 and RNF168 to control histone ubiquitination and DNA damage responses. EMBO J. 2010, 29, 943–955. [Google Scholar] [CrossRef]
- Huen, M.S.; Grant, R.; Manke, I.; Minn, K.; Yu, X.; Yaffe, M.B.; Chen, J. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell 2007, 131, 901–914. [Google Scholar] [CrossRef]
- Skaar, J.R.; Pagan, J.K.; Pagano, M. Mechanisms and function of substrate recruitment by F-box proteins. Nat. Rev. Mol. Cell Biol. 2013, 14, 369–381. [Google Scholar] [CrossRef]
- Honda, R.; Tanaka, H.; Yasuda, H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997, 420, 25–27. [Google Scholar] [CrossRef]
- de Toledo, S.M.; Azzam, E.I.; Dahlberg, W.K.; Gooding, T.B.; Little, J.B. ATM complexes with HDM2 and promotes its rapid phosphorylation in a p53-independent manner in normal and tumor human cells exposed to ionizing radiation. Oncogene 2000, 19, 6185–6193. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Khosravi, R.; Maya, R.; Gottlieb, T.; Oren, M.; Shiloh, Y.; Shkedy, D. Rapid ATM-dependent phosphorylation of MDM2 precedes p53 accumulation in response to DNA damage. Proc. Natl. Acad. Sci. USA 1999, 96, 14973–14977. [Google Scholar] [CrossRef] [PubMed]
- Frescas, D.; Pagano, M. Deregulated proteolysis by the F-box proteins SKP2 and beta-TrCP: Tipping the scales of cancer. Nat. Rev. Cancer 2008, 8, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Bulatov, E.; Ciulli, A. Targeting Cullin-RING E3 ubiquitin ligases for drug discovery: Structure, assembly and small-molecule modulation. Biochem. J. 2015, 467, 365–386. [Google Scholar] [CrossRef]
- Aravind, L.; Koonin, E.V. The U box is a modified RING finger—A common domain in ubiquitination. Curr. Biol. 2000, 10, R132–R134. [Google Scholar] [CrossRef]
- Wang, T.; Wang, W.; Wang, Q.; Xie, R.; Landay, A.; Chen, D. The E3 ubiquitin ligase CHIP in normal cell function and in disease conditions. Ann. N. Y. Acad. Sci. 2019. [Google Scholar] [CrossRef]
- Rotin, D.; Kumar, S. Physiological functions of the HECT family of ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2009, 10, 398–409. [Google Scholar] [CrossRef]
- Podos, S.D.; Hanson, K.K.; Wang, Y.C.; Ferguson, E.L. The DSmurf ubiquitin-protein ligase restricts BMP signaling spatially and temporally during Drosophila embryogenesis. Dev. Cell 2001, 1, 567–578. [Google Scholar] [CrossRef]
- Staub, O.; Dho, S.; Henry, P.; Correa, J.; Ishikawa, T.; McGlade, J.; Rotin, D. WW domains of Nedd4 bind to the proline-rich PY motifs in the epithelial Na+ channel deleted in Liddle’s syndrome. EMBO J. 1996, 15, 2371–2380. [Google Scholar] [CrossRef]
- Ebisawa, T.; Fukuchi, M.; Murakami, G.; Chiba, T.; Tanaka, K.; Imamura, T.; Miyazono, K. Smurf1 interacts with transforming growth factor-beta type I receptor through Smad7 and induces receptor degradation. J. Biol. Chem. 2001, 276, 12477–12480. [Google Scholar] [CrossRef]
- Spratt, D.E.; Walden, H.; Shaw, G.S. RBR E3 ubiquitin ligases: New structures, new insights, new questions. Biochem. J. 2014, 458, 421–437. [Google Scholar] [CrossRef] [PubMed]
- Marin, I.; Ferrus, A. Comparative genomics of the RBR family, including the Parkinson’s disease-related gene parkin and the genes of the ariadne subfamily. Mol. Biol. Evol. 2002, 19, 2039–2050. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, S.R.; Kishi, C.; Ishihara, N.; Mizushima, N. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J. Biol. Chem. 2011, 286, 19630–19640. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Nagano, Y.; Taylor, J.P.; Lim, K.L.; Yao, T.P. Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J. Cell Biol. 2010, 189, 671–679. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Nusse, R.; Clevers, H. Wnt/beta-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef]
- Tolwinski, N.S.; Wehrli, M.; Rives, A.; Erdeniz, N.; DiNardo, S.; Wieschaus, E. Wg/Wnt signal can be transmitted through arrow/LRP5,6 and Axin independently of Zw3/Gsk3beta activity. Dev. Cell 2003, 4, 407–418. [Google Scholar] [CrossRef]
- Hao, H.X.; Xie, Y.; Zhang, Y.; Charlat, O.; Oster, E.; Avello, M.; Lei, H.; Mickanin, C.; Liu, D.; Ruffner, H.; et al. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature 2012, 485, 195–200. [Google Scholar] [CrossRef]
- Koo, B.K.; Spit, M.; Jordens, I.; Low, T.Y.; Stange, D.E.; van de Wetering, M.; van Es, J.H.; Mohammed, S.; Heck, A.J.; Maurice, M.M.; et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature 2012, 488, 665–669. [Google Scholar] [CrossRef]
- Jiang, X.; Charlat, O.; Zamponi, R.; Yang, Y.; Cong, F. Dishevelled promotes Wnt receptor degradation through recruitment of ZNRF3/RNF43 E3 ubiquitin ligases. Mol. Cell 2015, 58, 522–533. [Google Scholar] [CrossRef]
- Peters, J.M.; McKay, R.M.; McKay, J.P.; Graff, J.M. Casein kinase I transduces Wnt signals. Nature 1999, 401, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, S.; Kishida, S.; Yamamoto, H.; Murai, H.; Koyama, S.; Kikuchi, A. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. EMBO J. 1998, 17, 1371–1384. [Google Scholar] [CrossRef] [PubMed]
- Doble, B.W.; Patel, S.; Wood, G.A.; Kockeritz, L.K.; Woodgett, J.R. Functional redundancy of GSK-3alpha and GSK-3beta in Wnt/beta-catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev. Cell 2007, 12, 957–971. [Google Scholar] [CrossRef] [PubMed]
- Morin, P.J.; Sparks, A.B.; Korinek, V.; Barker, N.; Clevers, H.; Vogelstein, B.; Kinzler, K.W. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 1997, 275, 1787–1790. [Google Scholar] [CrossRef]
- Korinek, V.; Barker, N.; Morin, P.J.; van Wichen, D.; de Weger, R.; Kinzler, K.W.; Vogelstein, B.; Clevers, H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science 1997, 275, 1784–1787. [Google Scholar] [CrossRef]
- Marikawa, Y.; Elinson, R.P. beta-TrCP is a negative regulator of Wnt/beta-catenin signaling pathway and dorsal axis formation in Xenopus embryos. Mech. Dev. 1998, 77, 75–80. [Google Scholar] [CrossRef]
- Hart, M.; Concordet, J.P.; Lassot, I.; Albert, I.; del los Santos, R.; Durand, H.; Perret, C.; Rubinfeld, B.; Margottin, F.; Benarous, R.; et al. The F-box protein beta-TrCP associates with phosphorylated beta-catenin and regulates its activity in the cell. Curr. Biol. 1999, 9, 207–210. [Google Scholar] [CrossRef]
- Zeng, X.; Huang, H.; Tamai, K.; Zhang, X.; Harada, Y.; Yokota, C.; Almeida, K.; Wang, J.; Doble, B.; Woodgett, J.; et al. Initiation of Wnt signaling: Control of Wnt coreceptor Lrp6 phosphorylation/activation via frizzled, dishevelled and axin functions. Development 2008, 135, 367–375. [Google Scholar] [CrossRef]
- Pan, W.; Choi, S.C.; Wang, H.; Qin, Y.; Volpicelli-Daley, L.; Swan, L.; Lucast, L.; Khoo, C.; Zhang, X.; Li, L.; et al. Wnt3a-mediated formation of phosphatidylinositol 4,5-bisphosphate regulates LRP6 phosphorylation. Science 2008, 321, 1350–1353. [Google Scholar] [CrossRef]
- Li, V.S.; Ng, S.S.; Boersema, P.J.; Low, T.Y.; Karthaus, W.R.; Gerlach, J.P.; Mohammed, S.; Heck, A.J.; Maurice, M.M.; Mahmoudi, T.; et al. Wnt signaling through inhibition of beta-catenin degradation in an intact Axin1 complex. Cell 2012, 149, 1245–1256. [Google Scholar] [CrossRef]
- Ji, L.; Jiang, B.; Jiang, X.; Charlat, O.; Chen, A.; Mickanin, C.; Bauer, A.; Xu, W.; Yan, X.; Cong, F. The SIAH E3 ubiquitin ligases promote Wnt/beta-catenin signaling through mediating Wnt-induced Axin degradation. Genes Dev. 2017, 31, 904–915. [Google Scholar] [CrossRef] [PubMed]
- Callow, M.G.; Tran, H.; Phu, L.; Lau, T.; Lee, J.; Sandoval, W.N.; Liu, P.S.; Bheddah, S.; Tao, J.; Lill, J.R.; et al. Ubiquitin ligase RNF146 regulates tankyrase and Axin to promote Wnt signaling. PLoS ONE 2011, 6, e22595. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, S.; Mickanin, C.; Feng, Y.; Charlat, O.; Michaud, G.A.; Schirle, M.; Shi, X.; Hild, M.; Bauer, A.; et al. RNF146 is a poly(ADP-ribose)-directed E3 ligase that regulates axin degradation and Wnt signalling. Nat. Cell Biol. 2011, 13, 623–629. [Google Scholar] [CrossRef] [PubMed]
- DaRosa, P.A.; Klevit, R.E.; Xu, W. Structural basis for tankyrase-RNF146 interaction reveals noncanonical tankyrase-binding motifs. Protein Sci. A Publ. Protein Soc. 2018, 27, 1057–1067. [Google Scholar] [CrossRef]
- Matsumoto, Y.; La Rose, J.; Lim, M.; Adissu, H.A.; Law, N.; Mao, X.; Cong, F.; Mera, P.; Karsenty, G.; Goltzman, D.; et al. Ubiquitin ligase RNF146 coordinates bone dynamics and energy metabolism. J. Clin. Investig. 2017, 127, 2612–2625. [Google Scholar] [CrossRef]
- Zhu, X.; Xing, R.; Tan, R.; Dai, R.; Tao, Q. The RNF146 E3 ubiquitin ligase is required for the control of Wnt signaling and body pattern formation in Xenopus. Mech. Dev. 2017, 147, 28–36. [Google Scholar] [CrossRef]
- Wang, Z.; Tacchelly-Benites, O.; Noble, G.P.; Johnson, M.K.; Gagne, J.P.; Poirier, G.G.; Ahmed, Y. A Context-Dependent Role for the RNF146 Ubiquitin Ligase in Wingless/Wnt Signaling in Drosophila. Genetics 2019, 211, 913–923. [Google Scholar] [CrossRef]
- Fei, C.; He, X.; Xie, S.; Miao, H.; Zhou, Z.; Li, L. Smurf1-mediated axin ubiquitination requires Smurf1 C2 domain and is cell cycle-dependent. J. Biol. Chem. 2014, 289, 14170–14177. [Google Scholar] [CrossRef]
- Fei, C.; Li, Z.; Li, C.; Chen, Y.; Chen, Z.; He, X.; Mao, L.; Wang, X.; Zeng, R.; Li, L. Smurf1-mediated Lys29-linked nonproteolytic polyubiquitination of axin negatively regulates Wnt/beta-catenin signaling. Mol. Cell. Biol. 2013, 33, 4095–4105. [Google Scholar] [CrossRef]
- Kim, S.; Jho, E.H. The protein stability of Axin, a negative regulator of Wnt signaling, is regulated by Smad ubiquitination regulatory factor 2 (Smurf2). J. Biol. Chem. 2010, 285, 36420–36426. [Google Scholar] [CrossRef]
- Lee, H.K.; Lee, E.W.; Seo, J.; Jeong, M.; Lee, S.H.; Kim, S.Y.; Jho, E.H.; Choi, C.H.; Chung, J.Y.; Song, J. Ubiquitylation and degradation of adenomatous polyposis coli by MKRN1 enhances Wnt/beta-catenin signaling. Oncogene 2018, 37, 4273–4286. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Zhang, Y.; Xu, C.; Tao, Q.H.; Chen, Y.G. HECT domain-containing E3 ubiquitin ligase NEDD4L negatively regulates Wnt signaling by targeting dishevelled for proteasomal degradation. J. Biol. Chem. 2013, 288, 8289–8298. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Li, M.; Wang, J.; Nie, F.; Li, L. The E3 ubiquitin ligase ITCH negatively regulates canonical Wnt signaling by targeting dishevelled protein. Mol. Cell. Biol. 2012, 32, 3903–3912. [Google Scholar] [CrossRef] [PubMed]
- Nethe, M.; de Kreuk, B.J.; Tauriello, D.V.; Anthony, E.C.; Snoek, B.; Stumpel, T.; Salinas, P.C.; Maurice, M.M.; Geerts, D.; Deelder, A.M.; et al. Rac1 acts in conjunction with Nedd4 and dishevelled-1 to promote maturation of cell-cell contacts. J. Cell Sci. 2012, 125, 3430–3442. [Google Scholar] [CrossRef]
- Zhang, Y.; Ding, Y.; Chen, Y.G.; Tao, Q. NEDD4L regulates convergent extension movements in Xenopus embryos via Disheveled-mediated non-canonical Wnt signaling. Dev. Biol. 2014, 392, 15–25. [Google Scholar] [CrossRef]
- Tsukiyama, T.; Fukui, A.; Terai, S.; Fujioka, Y.; Shinada, K.; Takahashi, H.; Yamaguchi, T.P.; Ohba, Y.; Hatakeyama, S. Molecular Role of RNF43 in Canonical and Noncanonical Wnt Signaling. Mol. Cell. Biol. 2015, 35, 2007–2023. [Google Scholar] [CrossRef]
- Angers, S.; Thorpe, C.J.; Biechele, T.L.; Goldenberg, S.J.; Zheng, N.; MacCoss, M.J.; Moon, R.T. The KLHL12-Cullin-3 ubiquitin ligase negatively regulates the Wnt-beta-catenin pathway by targeting Dishevelled for degradation. Nat. Cell Biol. 2006, 8, 348–357. [Google Scholar] [CrossRef]
- Lee, H.; Cheong, S.M.; Han, W.; Koo, Y.; Jo, S.B.; Cho, G.S.; Yang, J.S.; Kim, S.; Han, J.K. Head formation requires Dishevelled degradation that is mediated by March2 in concert with Dapper1. Development 2018, 145. [Google Scholar] [CrossRef]
- Zhou, M.I.; Wang, H.; Foy, R.L.; Ross, J.J.; Cohen, H.T. Tumor suppressor von Hippel-Lindau (VHL) stabilization of Jade-1 protein occurs through plant homeodomains and is VHL mutation dependent. Cancer Res. 2004, 64, 1278–1286. [Google Scholar] [CrossRef]
- Shafique, S.; Rashid, S. Structural basis for renal cancer by the dynamics of pVHL-dependent JADE1 stabilization and beta-catenin regulation. Prog. Biophys. Mol. Biol. 2019, 145, 65–77. [Google Scholar] [CrossRef]
- Chitalia, V.C.; Foy, R.L.; Bachschmid, M.M.; Zeng, L.; Panchenko, M.V.; Zhou, M.I.; Bharti, A.; Seldin, D.C.; Lecker, S.H.; Dominguez, I.; et al. Jade-1 inhibits Wnt signalling by ubiquitylating beta-catenin and mediates Wnt pathway inhibition by pVHL. Nat. Cell Biol. 2008, 10, 1208–1216. [Google Scholar] [CrossRef] [PubMed]
- Shivanna, S.; Harrold, I.; Shashar, M.; Meyer, R.; Kiang, C.; Francis, J.; Zhao, Q.; Feng, H.; Edelman, E.R.; Rahimi, N.; et al. The c-Cbl ubiquitin ligase regulates nuclear beta-catenin and angiogenesis by its tyrosine phosphorylation mediated through the Wnt signaling pathway. J. Biol. Chem. 2015, 290, 12537–12546. [Google Scholar] [CrossRef] [PubMed]
- Chitalia, V.; Shivanna, S.; Martorell, J.; Meyer, R.; Edelman, E.; Rahimi, N. c-Cbl, a ubiquitin E3 ligase that targets active beta-catenin: A novel layer of Wnt signaling regulation. J. Biol. Chem. 2013, 288, 23505–23517. [Google Scholar] [CrossRef] [PubMed]
- Flack, J.E.; Mieszczanek, J.; Novcic, N.; Bienz, M. Wnt-Dependent Inactivation of the Groucho/TLE Co-repressor by the HECT E3 Ubiquitin Ligase Hyd/UBR5. Mol. Cell 2017, 67, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Hay-Koren, A.; Caspi, M.; Zilberberg, A.; Rosin-Arbesfeld, R. The EDD E3 ubiquitin ligase ubiquitinates and up-regulates beta-catenin. Mol. Biol. Cell 2011, 22, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, Y.; Wong, C.C.; Zhang, J.; Dong, Y.; Li, X.; Kang, W.; Chan, F.K.L.; Sung, J.J.Y.; Yu, J. RNF6 Promotes Colorectal Cancer by Activating the Wnt/beta-Catenin Pathway via Ubiquitination of TLE3. Cancer Res. 2018, 78, 1958–1971. [Google Scholar] [CrossRef] [PubMed]
- Glaeser, K.; Urban, M.; Fenech, E.; Voloshanenko, O.; Kranz, D.; Lari, F.; Christianson, J.C.; Boutros, M. ERAD-dependent control of the Wnt secretory factor Evi. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Perrody, E.; Abrami, L.; Feldman, M.; Kunz, B.; Urbe, S.; van der Goot, F.G. Ubiquitin-dependent folding of the Wnt signaling coreceptor LRP6. Elife 2016, 5. [Google Scholar] [CrossRef]
- Feldman, M.; van der Goot, F.G. Novel ubiquitin-dependent quality control in the endoplasmic reticulum. Trends Cell Biol. 2009, 19, 357–363. [Google Scholar] [CrossRef]
- Abrami, L.; Kunz, B.; Iacovache, I.; van der Goot, F.G. Palmitoylation and ubiquitination regulate exit of the Wnt signaling protein LRP6 from the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2008, 105, 5384–5389. [Google Scholar] [CrossRef]
- Joshi, V.; Amanullah, A.; Upadhyay, A.; Mishra, R.; Kumar, A.; Mishra, A. A Decade of Boon or Burden: What Has the CHIP Ever Done for Cellular Protein Quality Control Mechanism Implicated in Neurodegeneration and Aging? Front. Mol. Neurosci. 2016, 9, 93. [Google Scholar] [CrossRef] [PubMed]
- Bhuripanyo, K.; Wang, Y.; Liu, X.; Zhou, L.; Liu, R.; Duong, D.; Zhao, B.; Bi, Y.; Zhou, H.; Chen, G.; et al. Identifying the substrate proteins of U-box E3s E4B and CHIP by orthogonal ubiquitin transfer. Sci. Adv. 2018, 4, e1701393. [Google Scholar] [CrossRef] [PubMed]
- Rudloff, S.; Kemler, R. Differential requirements for beta-catenin during mouse development. Development 2012, 139, 3711–3721. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Guo, N.; Nathans, J. The role of Frizzled3 and Frizzled6 in neural tube closure and in the planar polarity of inner-ear sensory hair cells. J. Neurosci. 2006, 26, 2147–2156. [Google Scholar] [CrossRef] [PubMed]
- Korinek, V.; Barker, N.; Moerer, P.; van Donselaar, E.; Huls, G.; Peters, P.J.; Clevers, H. Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nat. Genet. 1998, 19, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.; Hatakeyama, S.; Maruyama, S.; Kikuchi, A.; Onoe, K.; Good, R.A.; Nakayama, K.I. Impaired degradation of inhibitory subunit of NF-kappa B (I kappa B) and beta-catenin as a result of targeted disruption of the beta-TrCP1 gene. Proc. Natl. Acad. Sci. USA 2003, 100, 8752–8757. [Google Scholar] [CrossRef] [PubMed]
- Narimatsu, M.; Bose, R.; Pye, M.; Zhang, L.; Miller, B.; Ching, P.; Sakuma, R.; Luga, V.; Roncari, L.; Attisano, L.; et al. Regulation of planar cell polarity by Smurf ubiquitin ligases. Cell 2009, 137, 295–307. [Google Scholar] [CrossRef]
- Lee, M.S.; Han, H.J.; Han, S.Y.; Kim, I.Y.; Chae, S.; Lee, C.S.; Kim, S.E.; Yoon, S.G.; Park, J.W.; Kim, J.H.; et al. Loss of the E3 ubiquitin ligase MKRN1 represses diet-induced metabolic syndrome through AMPK activation. Nat. Commun. 2018, 9, 3404. [Google Scholar] [CrossRef]
- Lu, C.; Thoeni, C.; Connor, A.; Kawabe, H.; Gallinger, S.; Rotin, D. Intestinal knockout of Nedd4 enhances growth of Apc(min) tumors. Oncogene 2016, 35, 5839–5849. [Google Scholar] [CrossRef]
- Grima, B.; Lamouroux, A.; Chelot, E.; Papin, C.; Limbourg-Bouchon, B.; Rouyer, F. The F-box protein slimb controls the levels of clock proteins period and timeless. Nature 2002, 420, 178–182. [Google Scholar] [CrossRef]
- Nakagawa, T.; Zhang, T.; Kushi, R.; Nakano, S.; Endo, T.; Nakagawa, M.; Yanagihara, N.; Zarkower, D.; Nakayama, K. Regulation of mitosis-meiosis transition by the ubiquitin ligase beta-TrCP in male germ cells. Development 2017, 144, 4137–4147. [Google Scholar] [CrossRef] [PubMed]
- Guardavaccaro, D.; Kudo, Y.; Boulaire, J.; Barchi, M.; Busino, L.; Donzelli, M.; Margottin-Goguet, F.; Jackson, P.K.; Yamasaki, L.; Pagano, M. Control of meiotic and mitotic progression by the F box protein beta-Trcp1 in vivo. Dev. Cell 2003, 4, 799–812. [Google Scholar] [CrossRef]
- Ohsaki, K.; Oishi, K.; Kozono, Y.; Nakayama, K.; Nakayama, K.I.; Ishida, N. The role of {beta}-TrCP1 and {beta}-TrCP2 in circadian rhythm generation by mediating degradation of clock protein PER2. J. Biochem. 2008, 144, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Kanarek, N.; Horwitz, E.; Mayan, I.; Leshets, M.; Cojocaru, G.; Davis, M.; Tsuberi, B.Z.; Pikarsky, E.; Pagano, M.; Ben-Neriah, Y. Spermatogenesis rescue in a mouse deficient for the ubiquitin ligase SCF{beta}-TrCP by single substrate depletion. Genes Dev. 2010, 24, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Kudo, Y.; Guardavaccaro, D.; Santamaria, P.G.; Koyama-Nasu, R.; Latres, E.; Bronson, R.; Yamasaki, L.; Pagano, M. Role of F-box protein betaTrcp1 in mammary gland development and tumorigenesis. Mol. Cell. Biol. 2004, 24, 8184–8194. [Google Scholar] [CrossRef] [PubMed]
- Baguma-Nibasheka, M.; Kablar, B. Abnormal retinal development in the Btrc null mouse. Dev. Dyn. 2009, 238, 2680–2687. [Google Scholar] [CrossRef]
- Kanarek, N.; Grivennikov, S.I.; Leshets, M.; Lasry, A.; Alkalay, I.; Horwitz, E.; Shaul, Y.D.; Stachler, M.; Voronov, E.; Apte, R.N.; et al. Critical role for IL-1beta in DNA damage-induced mucositis. Proc. Natl. Acad. Sci. USA 2014, 111, E702–E711. [Google Scholar] [CrossRef]
- Nakagawa, T.; Araki, T.; Nakagawa, M.; Hirao, A.; Unno, M.; Nakayama, K. S6 Kinase- and beta-TrCP2-Dependent Degradation of p19Arf Is Required for Cell Proliferation. Mol. Cell. Biol. 2015, 35, 3517–3527. [Google Scholar] [CrossRef]
- Bond, C.E.; McKeone, D.M.; Kalimutho, M.; Bettington, M.L.; Pearson, S.A.; Dumenil, T.D.; Wockner, L.F.; Burge, M.; Leggett, B.A.; Whitehall, V.L. RNF43 and ZNRF3 are commonly altered in serrated pathway colorectal tumorigenesis. Oncotarget 2016, 7, 70589–70600. [Google Scholar] [CrossRef]
- Giannakis, M.; Hodis, E.; Jasmine Mu, X.; Yamauchi, M.; Rosenbluh, J.; Cibulskis, K.; Saksena, G.; Lawrence, M.S.; Qian, Z.R.; Nishihara, R.; et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat. Genet. 2014, 46, 1264–1266. [Google Scholar] [CrossRef]
- Koo, B.K.; van Es, J.H.; van den Born, M.; Clevers, H. Porcupine inhibitor suppresses paracrine Wnt-driven growth of Rnf43;Znrf3-mutant neoplasia. Proc. Natl. Acad. Sci. USA 2015, 112, 7548–7550. [Google Scholar] [CrossRef] [PubMed]
- Basham, K.J.; Rodriguez, S.; Turcu, A.F.; Lerario, A.M.; Logan, C.Y.; Rysztak, M.R.; Gomez-Sanchez, C.E.; Breault, D.T.; Koo, B.K.; Clevers, H.; et al. A ZNRF3-dependent Wnt/beta-catenin signaling gradient is required for adrenal homeostasis. Genes Dev. 2019, 33, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Jameson, S.A.; Lin, Y.T.; Capel, B. Testis development requires the repression of Wnt4 by Fgf signaling. Dev. Biol. 2012, 370, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.; Siggers, P.; Corrochano, S.; Warr, N.; Sagar, D.; Grimes, D.T.; Suzuki, M.; Burdine, R.D.; Cong, F.; Koo, B.K.; et al. ZNRF3 functions in mammalian sex determination by inhibiting canonical WNT signaling. Proc. Natl. Acad. Sci. USA 2018, 115, 5474–5479. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Yu, Z.; Li, N. The E3 ubiquitin ligase RNF146 promotes colorectal cancer by activating the Wnt/beta-catenin pathway via ubiquitination of Axin1. Biochem. Biophys. Res. Commun. 2018, 503, 991–997. [Google Scholar] [CrossRef]
- Wei, W.; Zeve, D.; Suh, J.M.; Wang, X.; Du, Y.; Zerwekh, J.E.; Dechow, P.C.; Graff, J.M.; Wan, Y. Biphasic and dosage-dependent regulation of osteoclastogenesis by beta-catenin. Mol. Cell. Biol. 2011, 31, 4706–4719. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Larose, J.; Kent, O.A.; Lim, M.; Changoor, A.; Zhang, L.; Storozhuk, Y.; Mao, X.; Grynpas, M.D.; Cong, F.; et al. RANKL coordinates multiple osteoclastogenic pathways by regulating expression of ubiquitin ligase RNF146. J. Clin. Investig. 2017, 127, 1303–1315. [Google Scholar] [CrossRef]
- Ueki, Y.; Tiziani, V.; Santanna, C.; Fukai, N.; Maulik, C.; Garfinkle, J.; Ninomiya, C.; doAmaral, C.; Peters, H.; Habal, M.; et al. Mutations in the gene encoding c-Abl-binding protein SH3BP2 cause cherubism. Nat. Genet. 2001, 28, 125–126. [Google Scholar] [CrossRef]
- Levaot, N.; Voytyuk, O.; Dimitriou, I.; Sircoulomb, F.; Chandrakumar, A.; Deckert, M.; Krzyzanowski, P.M.; Scotter, A.; Gu, S.; Janmohamed, S.; et al. Loss of Tankyrase-mediated destruction of 3BP2 is the underlying pathogenic mechanism of cherubism. Cell 2011, 147, 1324–1339. [Google Scholar] [CrossRef]
- Zou, Y.F.; Xie, C.W.; Yang, S.X.; Xiong, J.P. AMPK activators suppress breast cancer cell growth by inhibiting DVL3-facilitated Wnt/beta-catenin signaling pathway activity. Mol. Med. Rep. 2017, 15, 899–907. [Google Scholar] [CrossRef][Green Version]
- Lee, H.; Kang, R.; Bae, S.; Yoon, Y. AICAR, an activator of AMPK, inhibits adipogenesis via the WNT/beta-catenin pathway in 3T3-L1 adipocytes. Int. J. Mol. Med. 2011, 28, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Ouyang, H.; Zhu, T.; Lindvall, C.; Wang, Y.; Zhang, X.; Yang, Q.; Bennett, C.; Harada, Y.; Stankunas, K.; et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 2006, 126, 955–968. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Kim, D.; Kee, S.H. Metformin-activated AMPK regulates beta-catenin to reduce cell proliferation in colon carcinoma RKO cells. Oncol. Lett. 2019, 17, 2695–2702. [Google Scholar] [CrossRef] [PubMed]
- Kondo, S.; Seo, S.Y.; Yoshizaki, T.; Wakisaka, N.; Furukawa, M.; Joab, I.; Jang, K.L.; Pagano, J.S. EBV latent membrane protein 1 up-regulates hypoxia-inducible factor 1alpha through Siah1-mediated down-regulation of prolyl hydroxylases 1 and 3 in nasopharyngeal epithelial cells. Cancer Res. 2006, 66, 9870–9877. [Google Scholar] [CrossRef]
- Dickins, R.A.; Frew, I.J.; House, C.M.; O’Bryan, M.K.; Holloway, A.J.; Haviv, I.; Traficante, N.; de Kretser, D.M.; Bowtell, D.D. The ubiquitin ligase component Siah1a is required for completion of meiosis I in male mice. Mol. Cell. Biol. 2002, 22, 2294–2303. [Google Scholar] [CrossRef]
- Frew, I.J.; Sims, N.A.; Quinn, J.M.; Walkley, C.R.; Purton, L.E.; Bowtell, D.D.; Gillespie, M.T. Osteopenia in Siah1a mutant mice. J. Biol. Chem. 2004, 279, 29583–29588. [Google Scholar] [CrossRef]
- Frew, I.J.; Hammond, V.E.; Dickins, R.A.; Quinn, J.M.; Walkley, C.R.; Sims, N.A.; Schnall, R.; Della, N.G.; Holloway, A.J.; Digby, M.R.; et al. Generation and analysis of Siah2 mutant mice. Mol. Cell. Biol. 2003, 23, 9150–9161. [Google Scholar] [CrossRef]
- Scortegagna, M.; Kim, H.; Li, J.L.; Yao, H.; Brill, L.M.; Han, J.; Lau, E.; Bowtell, D.; Haddad, G.; Kaufman, R.J.; et al. Fine tuning of the UPR by the ubiquitin ligases Siah1/2. PLoS Genet. 2014, 10, e1004348. [Google Scholar] [CrossRef]
- Wang, D.; Wang, Y.; Kong, T.; Fan, F.; Jiang, Y. Hypoxia-induced beta-catenin downregulation involves p53-dependent activation of Siah-1. Cancer Sci. 2011, 102, 1322–1328. [Google Scholar] [CrossRef]
- Simon, M.C. Siah proteins, HIF prolyl hydroxylases, and the physiological response to hypoxia. Cell 2004, 117, 851–853. [Google Scholar] [CrossRef][Green Version]
- Gudjonsson, T.; Altmeyer, M.; Savic, V.; Toledo, L.; Dinant, C.; Grofte, M.; Bartkova, J.; Poulsen, M.; Oka, Y.; Bekker-Jensen, S.; et al. TRIP12 and UBR5 suppress spreading of chromatin ubiquitylation at damaged chromosomes. Cell 2012, 150, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Shearer, R.F.; Iconomou, M.; Watts, C.K.; Saunders, D.N. Functional Roles of the E3 Ubiquitin Ligase UBR5 in Cancer. Mol. Cancer Res. 2015, 13, 1523–1532. [Google Scholar] [CrossRef] [PubMed]
- Saunders, D.N.; Hird, S.L.; Withington, S.L.; Dunwoodie, S.L.; Henderson, M.J.; Biben, C.; Sutherland, R.L.; Ormandy, C.J.; Watts, C.K. Edd, the murine hyperplastic disc gene, is essential for yolk sac vascularization and chorioallantoic fusion. Mol. Cell. Biol. 2004, 24, 7225–7234. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Tamai, Y.; Zorn, A.M.; Yoshida, H.; Seldin, M.F.; Nishikawa, S.; Taketo, M.M. Mouse Wnt receptor gene Fzd5 is essential for yolk sac and placental angiogenesis. Development 2001, 128, 25–33. [Google Scholar]
- Kinsella, E.; Dora, N.; Mellis, D.; Lettice, L.; Deveney, P.; Hill, R.; Ditzel, M. Use of a Conditional Ubr5 Mutant Allele to Investigate the Role of an N-End Rule Ubiquitin-Protein Ligase in Hedgehog Signalling and Embryonic Limb Development. PLoS ONE 2016, 11, e0157079. [Google Scholar] [CrossRef]
- Ye, X.; Wang, L.; Shang, B.; Wang, Z.; Wei, W. NEDD4: A promising target for cancer therapy. Curr. Cancer Drug Targets 2014, 14, 549–556. [Google Scholar] [CrossRef]
- Li, J.J.; Ferry, R.J., Jr.; Diao, S.; Xue, B.; Bahouth, S.W.; Liao, F.F. Nedd4 haploinsufficient mice display moderate insulin resistance, enhanced lipolysis, and protection against high-fat diet-induced obesity. Endocrinology 2015, 156, 1283–1291. [Google Scholar] [CrossRef]
- Liu, Y.; Oppenheim, R.W.; Sugiura, Y.; Lin, W. Abnormal development of the neuromuscular junction in Nedd4-deficient mice. Dev. Biol. 2009, 330, 153–166. [Google Scholar] [CrossRef]
- Kawabe, H.; Neeb, A.; Dimova, K.; Young, S.M., Jr.; Takeda, M.; Katsurabayashi, S.; Mitkovski, M.; Malakhova, O.A.; Zhang, D.E.; Umikawa, M.; et al. Regulation of Rap2A by the ubiquitin ligase Nedd4-1 controls neurite development. Neuron 2010, 65, 358–372. [Google Scholar] [CrossRef]
- Cao, X.R.; Lill, N.L.; Boase, N.; Shi, P.P.; Croucher, D.R.; Shan, H.; Qu, J.; Sweezer, E.M.; Place, T.; Kirby, P.A.; et al. Nedd4 controls animal growth by regulating IGF-1 signaling. Sci. Signal. 2008, 1. [Google Scholar] [CrossRef]
- Fouladkou, F.; Lu, C.; Jiang, C.; Zhou, L.; She, Y.; Walls, J.R.; Kawabe, H.; Brose, N.; Henkelman, R.M.; Huang, A.; et al. The ubiquitin ligase Nedd4-1 is required for heart development and is a suppressor of thrombospondin-1. J. Biol. Chem. 2010, 285, 6770–6780. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Jeon, S.A.; Kim, B.G.; Takeda, M.; Cho, J.J.; Kim, D.I.; Kawabe, H.; Cho, J.Y. Nedd4 Deficiency in Vascular Smooth Muscle Promotes Vascular Calcification by Stabilizing pSmad1. J. Bone Min. Res. 2017, 32, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.A.; Lee, J.H.; Kim, D.W.; Cho, J.Y. E3-ubiquitin ligase NEDD4 enhances bone formation by removing TGFbeta1-induced pSMAD1 in immature osteoblast. Bone 2018, 116, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Wiszniak, S.; Harvey, N.; Schwarz, Q. Cell autonomous roles of Nedd4 in craniofacial bone formation. Dev. Biol. 2016, 410, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Wiszniak, S.; Kabbara, S.; Lumb, R.; Scherer, M.; Secker, G.; Harvey, N.; Kumar, S.; Schwarz, Q. The ubiquitin ligase Nedd4 regulates craniofacial development by promoting cranial neural crest cell survival and stem-cell like properties. Dev. Biol. 2013, 383, 186–200. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Gay, D.L.; MacLeod, M.K.; Cao, X.; Hala, T.; Sweezer, E.M.; Kappler, J.; Marrack, P.; Oliver, P.M. Nedd4 augments the adaptive immune response by promoting ubiquitin-mediated degradation of Cbl-b in activated T cells. Nat. Immunol. 2008, 9, 1356–1363. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Qiao, G.; Ying, H.; Li, Z.; Zhao, Y.; Liang, Y.; Yang, L.; Lipkowitz, S.; Penninger, J.M.; Langdon, W.Y.; et al. E3 ubiquitin ligase Cbl-b regulates Pten via Nedd4 in T cells independently of its ubiquitin ligase activity. Cell Rep. 2012, 1, 472–482. [Google Scholar] [CrossRef]
- Russo, C.J.; Melista, E.; Cui, J.; DeStefano, A.L.; Bakris, G.L.; Manolis, A.J.; Gavras, H.; Baldwin, C.T. Association of NEDD4L ubiquitin ligase with essential hypertension. Hypertension 2005, 46, 488–491. [Google Scholar] [CrossRef]
- Yanpallewar, S.; Wang, T.; Koh, D.C.; Quarta, E.; Fulgenzi, G.; Tessarollo, L. Nedd4-2 haploinsufficiency causes hyperactivity and increased sensitivity to inflammatory stimuli. Sci. Rep. 2016, 6, 32957. [Google Scholar] [CrossRef]
- Harvey, K.F.; Dinudom, A.; Cook, D.I.; Kumar, S. The Nedd4-like protein KIAA0439 is a potential regulator of the epithelial sodium channel. J. Biol. Chem. 2001, 276, 8597–8601. [Google Scholar] [CrossRef]
- Zhou, R.; Patel, S.V.; Snyder, P.M. Nedd4-2 catalyzes ubiquitination and degradation of cell surface ENaC. J. Biol. Chem. 2007, 282, 20207–20212. [Google Scholar] [CrossRef] [PubMed]
- Hanukoglu, I.; Hanukoglu, A. Epithelial sodium channel (ENaC) family: Phylogeny, structure-function, tissue distribution, and associated inherited diseases. Gene 2016, 579, 95–132. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Hummler, E.; Hill, D.L. Selective Deletion of Sodium Salt Taste during Development Leads to Expanded Terminal Fields of Gustatory Nerves in the Adult Mouse Nucleus of the Solitary Tract. J. Neurosci. Off. J. Soc. Neurosci. 2017, 37, 660–672. [Google Scholar] [CrossRef] [PubMed]
- Shi, P.P.; Cao, X.R.; Sweezer, E.M.; Kinney, T.S.; Williams, N.R.; Husted, R.F.; Nair, R.; Weiss, R.M.; Williamson, R.A.; Sigmund, C.D.; et al. Salt-sensitive hypertension and cardiac hypertrophy in mice deficient in the ubiquitin ligase Nedd4-2. Am. J. Physiol. Ren. Physiol. 2008, 295, F462–F470. [Google Scholar] [CrossRef] [PubMed]
- Boase, N.A.; Rychkov, G.Y.; Townley, S.L.; Dinudom, A.; Candi, E.; Voss, A.K.; Tsoutsman, T.; Semsarian, C.; Melino, G.; Koentgen, F.; et al. Respiratory distress and perinatal lethality in Nedd4-2-deficient mice. Nat. Commun. 2011, 2, 287. [Google Scholar] [CrossRef] [PubMed]
- Henshall, T.L.; Manning, J.A.; Alfassy, O.S.; Goel, P.; Boase, N.A.; Kawabe, H.; Kumar, S. Deletion of Nedd4-2 results in progressive kidney disease in mice. Cell Death Differ. 2017, 24, 2150–2160. [Google Scholar] [CrossRef] [PubMed]
- Yip, K.H.; Kolesnikoff, N.; Hauschild, N.; Biggs, L.; Lopez, A.F.; Galli, S.J.; Kumar, S.; Grimbaldeston, M.A. The Nedd4-2/Ndfip1 axis is a negative regulator of IgE-mediated mast cell activation. Nat. Commun. 2016, 7, 13198. [Google Scholar] [CrossRef]
- Infante, P.; Lospinoso Severini, L.; Bernardi, F.; Bufalieri, F.; Di Marcotullio, L. Targeting Hedgehog Signalling through the Ubiquitylation Process: The Multiple Roles of the HECT-E3 Ligase Itch. Cells 2019, 8. [Google Scholar] [CrossRef]
- Liu, Y.C. The E3 ubiquitin ligase Itch in T cell activation, differentiation, and tolerance. Semin. Immunol. 2007, 19, 197–205. [Google Scholar] [CrossRef]
- Gao, M.; Labuda, T.; Xia, Y.; Gallagher, E.; Fang, D.; Liu, Y.C.; Karin, M. Jun turnover is controlled through JNK-dependent phosphorylation of the E3 ligase Itch. Science 2004, 306, 271–275. [Google Scholar] [CrossRef]
- Lohr, N.J.; Molleston, J.P.; Strauss, K.A.; Torres-Martinez, W.; Sherman, E.A.; Squires, R.H.; Rider, N.L.; Chikwava, K.R.; Cummings, O.W.; Morton, D.H.; et al. Human ITCH E3 ubiquitin ligase deficiency causes syndromic multisystem autoimmune disease. Am. J. Hum. Genet. 2010, 86, 447–453. [Google Scholar] [CrossRef]
- Perry, W.L.; Hustad, C.M.; Swing, D.A.; O’Sullivan, T.N.; Jenkins, N.A.; Copeland, N.G. The itchy locus encodes a novel ubiquitin protein ligase that is disrupted in a18H mice. Nat. Genet. 1998, 18, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Hustad, C.M.; Perry, W.L.; Siracusa, L.D.; Rasberry, C.; Cobb, L.; Cattanach, B.M.; Kovatch, R.; Copeland, N.G.; Jenkins, N.A. Molecular genetic characterization of six recessive viable alleles of the mouse agouti locus. Genetics 1995, 140, 255–265. [Google Scholar] [PubMed]
- Fang, D.; Elly, C.; Gao, B.; Fang, N.; Altman, Y.; Joazeiro, C.; Hunter, T.; Copeland, N.; Jenkins, N.; Liu, Y.C. Dysregulation of T lymphocyte function in itchy mice: A role for Itch in TH2 differentiation. Nat. Immunol. 2002, 3, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.S.; Park, Y.; Elly, C.; Liu, Y.C. Itch expression by Treg cells controls Th2 inflammatory responses. J. Clin. Investig. 2013, 123, 4923–4934. [Google Scholar] [CrossRef] [PubMed]
- Xiao, N.; Eto, D.; Elly, C.; Peng, G.; Crotty, S.; Liu, Y.C. The E3 ubiquitin ligase Itch is required for the differentiation of follicular helper T cells. Nat. Immunol. 2014, 15, 657–666. [Google Scholar] [CrossRef]
- Huang, H.; Jeon, M.S.; Liao, L.; Yang, C.; Elly, C.; Yates, J.R., 3rd; Liu, Y.C. K33-linked polyubiquitination of T cell receptor-zeta regulates proteolysis-independent T cell signaling. Immunity 2010, 33, 60–70. [Google Scholar] [CrossRef]
- Giamboi-Miraglia, A.; Cianfarani, F.; Cattani, C.; Lena, A.M.; Serra, V.; Campione, E.; Terrinoni, A.; Zambruno, G.; Odorisio, T.; Di Daniele, N.; et al. The E3 ligase Itch knockout mice show hyperproliferation and wound healing alteration. FEBS J 2015, 282, 4435–4449. [Google Scholar] [CrossRef]
- Stermer, A.R.; Myers, J.L.; Murphy, C.J.; Di Bona, K.R.; Matesic, L.; Richburg, J.H. Female mice with loss-of-function ITCH display an altered reproductive phenotype. Exp. Biol. Med. (Maywood) 2016, 241, 367–374. [Google Scholar] [CrossRef]
- Marino, A.; Menghini, R.; Fabrizi, M.; Casagrande, V.; Mavilio, M.; Stoehr, R.; Candi, E.; Mauriello, A.; Moreno-Navarrete, J.M.; Gomez-Serrano, M.; et al. ITCH deficiency protects from diet-induced obesity. Diabetes 2014, 63, 550–561. [Google Scholar] [CrossRef]
- Chang, L.; Kamata, H.; Solinas, G.; Luo, J.L.; Maeda, S.; Venuprasad, K.; Liu, Y.C.; Karin, M. The E3 ubiquitin ligase itch couples JNK activation to TNFalpha-induced cell death by inducing c-FLIP(L) turnover. Cell 2006, 124, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, C.; Matesic, L.E.; Flavell, R.A. The E3 ligase Itch is a negative regulator of the homeostasis and function of hematopoietic stem cells. Nat. Immunol. 2011, 12, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Thien, C.B.; Langdon, W.Y. c-Cbl and Cbl-b ubiquitin ligases: Substrate diversity and the negative regulation of signalling responses. Biochem. J. 2005, 391, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, B.; Ahmad, G.; Nadeau, S.; Zutshi, N.; An, W.; Scheffe, S.; Dong, L.; Feng, D.; Goetz, B.; Arya, P.; et al. Protein tyrosine kinase regulation by ubiquitination: Critical roles of Cbl-family ubiquitin ligases. Biochim. Biophys. Acta 2013, 1833, 122–139. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Neff, L.; Baron, R.; Levy, J.B. Tyrosine phosphorylation and translocation of the c-cbl protein after activation of tyrosine kinase signaling pathways. J. Biol. Chem. 1995, 270, 14347–14351. [Google Scholar] [CrossRef] [PubMed]
- Naramura, M.; Kole, H.K.; Hu, R.J.; Gu, H. Altered thymic positive selection and intracellular signals in Cbl-deficient mice. Proc. Natl. Acad. Sci. USA 1998, 95, 15547–15552. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.A.; Schnall, R.G.; Venter, D.J.; Barnett, L.; Bertoncello, I.; Thien, C.B.; Langdon, W.Y.; Bowtell, D.D. Tissue hyperplasia and enhanced T-cell signalling via ZAP-70 in c-Cbl-deficient mice. Mol. Cell. Biol. 1998, 18, 4872–4882. [Google Scholar] [CrossRef]
- Naramura, M.; Jang, I.K.; Kole, H.; Huang, F.; Haines, D.; Gu, H. c-Cbl and Cbl-b regulate T cell responsiveness by promoting ligand-induced TCR down-modulation. Nat. Immunol. 2002, 3, 1192–1199. [Google Scholar] [CrossRef]
- Bachmaier, K.; Krawczyk, C.; Kozieradzki, I.; Kong, Y.Y.; Sasaki, T.; Oliveira-dos-Santos, A.; Mariathasan, S.; Bouchard, D.; Wakeham, A.; Itie, A.; et al. Negative regulation of lymphocyte activation and autoimmunity by the molecular adaptor Cbl-b. Nature 2000, 403, 211–216. [Google Scholar] [CrossRef]
- Wang, H.Y.; Altman, Y.; Fang, D.; Elly, C.; Dai, Y.; Shao, Y.; Liu, Y.C. Cbl promotes ubiquitination of the T cell receptor zeta through an adaptor function of Zap-70. J. Biol. Chem. 2001, 276, 26004–26011. [Google Scholar] [CrossRef]
- D’Oro, U.; Munitic, I.; Chacko, G.; Karpova, T.; McNally, J.; Ashwell, J.D. Regulation of constitutive TCR internalization by the zeta-chain. J. Immunol. 2002, 169, 6269–6278. [Google Scholar] [CrossRef] [PubMed]
- Lutz-Nicoladoni, C.; Wolf, D.; Sopper, S. Modulation of Immune Cell Functions by the E3 Ligase Cbl-b. Front. Oncol. 2015, 5, 58. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.O.; Housley, W.J.; Bhowmick, S.; Cone, R.E.; Rajan, T.V.; Forouhar, F.; Clark, R.B. Cbl-b(-/-) T cells demonstrate in vivo resistance to regulatory T cells but a context-dependent resistance to TGF-beta. J. Immunol. 2010, 185, 2051–2058. [Google Scholar] [CrossRef] [PubMed]
- Chiusaroli, R.; Sanjay, A.; Henriksen, K.; Engsig, M.T.; Horne, W.C.; Gu, H.; Baron, R. Deletion of the gene encoding c-Cbl alters the ability of osteoclasts to migrate, delaying resorption and ossification of cartilage during the development of long bones. Dev. Biol. 2003, 261, 537–547. [Google Scholar] [CrossRef]
- Molero, J.C.; Jensen, T.E.; Withers, P.C.; Couzens, M.; Herzog, H.; Thien, C.B.; Langdon, W.Y.; Walder, K.; Murphy, M.A.; Bowtell, D.D.; et al. c-Cbl-deficient mice have reduced adiposity, higher energy expenditure, and improved peripheral insulin action. J. Clin. Investig. 2004, 114, 1326–1333. [Google Scholar] [CrossRef]
- Li, X.; Gadzinsky, A.; Gong, L.; Tong, H.; Calderon, V.; Li, Y.; Kitamura, D.; Klein, U.; Langdon, W.Y.; Hou, F.; et al. Cbl Ubiquitin Ligases Control B Cell Exit from the Germinal-Center Reaction. Immunity 2018, 48, 530–541.e536. [Google Scholar] [CrossRef]
- Gustin, S.E.; Thien, C.B.; Langdon, W.Y. Cbl-b is a negative regulator of inflammatory cytokines produced by IgE-activated mast cells. J. Immunol. 2006, 177, 5980–5989. [Google Scholar] [CrossRef]
- Naramura, M.; Nandwani, N.; Gu, H.; Band, V.; Band, H. Rapidly fatal myeloproliferative disorders in mice with deletion of Casitas B-cell lymphoma (Cbl) and Cbl-b in hematopoietic stem cells. Proc. Natl. Acad. Sci. USA 2010, 107, 16274–16279. [Google Scholar] [CrossRef]
- Mohapatra, B.; Zutshi, N.; An, W.; Goetz, B.; Arya, P.; Bielecki, T.A.; Mushtaq, I.; Storck, M.D.; Meza, J.L.; Band, V.; et al. An essential role of CBL and CBL-B ubiquitin ligases in mammary stem cell maintenance. Development 2017, 144, 1072–1086. [Google Scholar] [CrossRef]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-beta: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Taipale, J.; Miyazono, K.; Heldin, C.H.; Keski-Oja, J. Latent transforming growth factor-beta 1 associates to fibroblast extracellular matrix via latent TGF-beta binding protein. J. Cell Biol. 1994, 124, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Itoh, F.; Divecha, N.; Brocks, L.; Oomen, L.; Janssen, H.; Calafat, J.; Itoh, S.; Dijke Pt, P. The FYVE domain in Smad anchor for receptor activation (SARA) is sufficient for localization of SARA in early endosomes and regulates TGF-beta/Smad signalling. Genes Cells Devoted Mol. Cell. Mech. 2002, 7, 321–331. [Google Scholar] [CrossRef]
- Miura, S.; Takeshita, T.; Asao, H.; Kimura, Y.; Murata, K.; Sasaki, Y.; Hanai, J.I.; Beppu, H.; Tsukazaki, T.; Wrana, J.L.; et al. Hgs (Hrs), a FYVE domain protein, is involved in Smad signaling through cooperation with SARA. Mol. Cell. Biol. 2000, 20, 9346–9355. [Google Scholar] [CrossRef]
- Massague, J.; Seoane, J.; Wotton, D. Smad transcription factors. Genes Dev. 2005, 19, 2783–2810. [Google Scholar] [CrossRef]
- Janknecht, R.; Wells, N.J.; Hunter, T. TGF-beta-stimulated cooperation of smad proteins with the coactivators CBP/p300. Genes Dev. 1998, 12, 2114–2119. [Google Scholar] [CrossRef]
- Pei, D.; Shu, X.; Gassama-Diagne, A.; Thiery, J.P. Mesenchymal-epithelial transition in development and reprogramming. Nat. Cell Biol. 2019, 21, 44–53. [Google Scholar] [CrossRef]
- Moustakas, A.; Kardassis, D. Regulation of the human p21/WAF1/Cip1 promoter in hepatic cells by functional interactions between Sp1 and Smad family members. Proc. Natl. Acad. Sci. USA 1998, 95, 6733–6738. [Google Scholar] [CrossRef]
- Nakao, A.; Afrakhte, M.; Moren, A.; Nakayama, T.; Christian, J.L.; Heuchel, R.; Itoh, S.; Kawabata, M.; Heldin, N.E.; Heldin, C.H.; et al. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature 1997, 389, 631–635. [Google Scholar] [CrossRef]
- Chong, P.A.; Lin, H.; Wrana, J.L.; Forman-Kay, J.D. An expanded WW domain recognition motif revealed by the interaction between Smad7 and the E3 ubiquitin ligase Smurf2. J. Biol. Chem. 2006, 281, 17069–17075. [Google Scholar] [CrossRef]
- Kavsak, P.; Rasmussen, R.K.; Causing, C.G.; Bonni, S.; Zhu, H.; Thomsen, G.H.; Wrana, J.L. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol. Cell 2000, 6, 1365–1375. [Google Scholar] [CrossRef]
- Zhou, F.; Li, F.; Xie, F.; Zhang, Z.; Huang, H.; Zhang, L. TRAF4 mediates activation of TGF-beta signaling and is a biomarker for oncogenesis in breast cancer. Sci. China Life Sci. 2014, 57, 1172–1176. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Jin, C.; Tang, Y.; Tang, L.Y.; Zhang, Y.E. Ubiquitination of tumor necrosis factor receptor-associated factor 4 (TRAF4) by Smad ubiquitination regulatory factor 1 (Smurf1) regulates motility of breast epithelial and cancer cells. J. Biol. Chem. 2013, 288, 21784–21792. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhou, F.; Garcia de Vinuesa, A.; de Kruijf, E.M.; Mesker, W.E.; Hui, L.; Drabsch, Y.; Li, Y.; Bauer, A.; Rousseau, A.; et al. TRAF4 promotes TGF-beta receptor signaling and drives breast cancer metastasis. Mol. Cell 2013, 51, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chang, C.; Gehling, D.J.; Hemmati-Brivanlou, A.; Derynck, R. Regulation of Smad degradation and activity by Smurf2, an E3 ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2001, 98, 974–979. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.Y.; Yamashita, M.; Coussens, N.P.; Tang, Y.; Wang, X.; Li, C.; Deng, C.X.; Cheng, S.Y.; Zhang, Y.E. Ablation of Smurf2 reveals an inhibition in TGF-beta signalling through multiple mono-ubiquitination of Smad3. EMBO J. 2011, 30, 4777–4789. [Google Scholar] [CrossRef]
- Bonni, S.; Wang, H.R.; Causing, C.G.; Kavsak, P.; Stroschein, S.L.; Luo, K.; Wrana, J.L. TGF-beta induces assembly of a Smad2-Smurf2 ubiquitin ligase complex that targets SnoN for degradation. Nat. Cell Biol. 2001, 3, 587–595. [Google Scholar] [CrossRef]
- Alarcon, C.; Zaromytidou, A.I.; Xi, Q.; Gao, S.; Yu, J.; Fujisawa, S.; Barlas, A.; Miller, A.N.; Manova-Todorova, K.; Macias, M.J.; et al. Nuclear CDKs drive Smad transcriptional activation and turnover in BMP and TGF-beta pathways. Cell 2009, 139, 757–769. [Google Scholar] [CrossRef]
- Aragon, E.; Goerner, N.; Zaromytidou, A.I.; Xi, Q.; Escobedo, A.; Massague, J.; Macias, M.J. A Smad action turnover switch operated by WW domain readers of a phosphoserine code. Genes Dev. 2011, 25, 1275–1288. [Google Scholar] [CrossRef]
- Bai, Y.; Yang, C.; Hu, K.; Elly, C.; Liu, Y.C. Itch E3 ligase-mediated regulation of TGF-beta signaling by modulating smad2 phosphorylation. Mol. Cell 2004, 15, 825–831. [Google Scholar] [CrossRef]
- Park, S.H.; Jung, E.H.; Kim, G.Y.; Kim, B.C.; Lim, J.H.; Woo, C.H. Itch E3 ubiquitin ligase positively regulates TGF-beta signaling to EMT via Smad7 ubiquitination. Mol. Cells 2015, 38, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Pefani, D.E.; Pankova, D.; Abraham, A.G.; Grawenda, A.M.; Vlahov, N.; Scrace, S.; O’Neill, E. TGF-beta Targets the Hippo Pathway Scaffold RASSF1A to Facilitate YAP/SMAD2 Nuclear Translocation. Mol. Cell 2016, 63, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Koinuma, D.; Shinozaki, M.; Komuro, A.; Goto, K.; Saitoh, M.; Hanyu, A.; Ebina, M.; Nukiwa, T.; Miyazawa, K.; Imamura, T.; et al. Arkadia amplifies TGF-beta superfamily signalling through degradation of Smad7. EMBO J. 2003, 22, 6458–6470. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.Y.; Li, X.Z.; Peng, Y.M.; Liu, H.; Liu, Y.H. Arkadia-Smad7-mediated positive regulation of TGF-beta signaling in a rat model of tubulointerstitial fibrosis. Am. J. Nephrol. 2007, 27, 176–183. [Google Scholar] [CrossRef]
- Liu, W.; Rui, H.; Wang, J.; Lin, S.; He, Y.; Chen, M.; Li, Q.; Ye, Z.; Zhang, S.; Chan, S.C.; et al. Axin is a scaffold protein in TGF-beta signaling that promotes degradation of Smad7 by Arkadia. EMBO J. 2006, 25, 1646–1658. [Google Scholar] [CrossRef]
- Zhang, L.; Huang, H.; Zhou, F.; Schimmel, J.; Pardo, C.G.; Zhang, T.; Barakat, T.S.; Sheppard, K.A.; Mickanin, C.; Porter, J.A.; et al. RNF12 controls embryonic stem cell fate and morphogenesis in zebrafish embryos by targeting Smad7 for degradation. Mol. Cell 2012, 46, 650–661. [Google Scholar] [CrossRef]
- Nagano, Y.; Mavrakis, K.J.; Lee, K.L.; Fujii, T.; Koinuma, D.; Sase, H.; Yuki, K.; Isogaya, K.; Saitoh, M.; Imamura, T.; et al. Arkadia induces degradation of SnoN and c-Ski to enhance transforming growth factor-beta signaling. J. Biol. Chem. 2007, 282, 20492–20501. [Google Scholar] [CrossRef]
- Inoue, Y.; Imamura, T. Regulation of TGF-beta family signaling by E3 ubiquitin ligases. Cancer Sci. 2008, 99, 2107–2112. [Google Scholar] [CrossRef]
- Le Scolan, E.; Zhu, Q.; Wang, L.; Bandyopadhyay, A.; Javelaud, D.; Mauviel, A.; Sun, L.; Luo, K. Transforming growth factor-beta suppresses the ability of Ski to inhibit tumor metastasis by inducing its degradation. Cancer Res. 2008, 68, 3277–3285. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, Y.; Gao, R.; Yang, X.; Yan, X.; Wang, C.; Jiang, S.; Yu, L. RLIM interacts with Smurf2 and promotes TGF-beta induced U2OS cell migration. Biochem. Biophys. Res. Commun. 2011, 414, 181–185. [Google Scholar] [CrossRef]
- Gruber, T.; Hinterleitner, R.; Hermann-Kleiter, N.; Meisel, M.; Kleiter, I.; Wang, C.M.; Viola, A.; Pfeifhofer-Obermair, C.; Baier, G. Cbl-b mediates TGFbeta sensitivity by downregulating inhibitory SMAD7 in primary T cells. J. Mol. Cell Biol. 2013, 5, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Jin, K.; Shao, L.; Fan, Y.; Tu, Y.; Li, Y.; Yang, B.; van Dam, H.; Ten Dijke, P.; Weng, H.; et al. FAF1 phosphorylation by AKT accumulates TGF-beta type II receptor and drives breast cancer metastasis. Nat. Commun. 2017, 8, 15021. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Dorn, D.C.; Erdjument-Bromage, H.; Tempst, P.; Moore, M.A.; Massague, J. Hematopoiesis controlled by distinct TIF1gamma and Smad4 branches of the TGFbeta pathway. Cell 2006, 125, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Morsut, L.; Yan, K.P.; Enzo, E.; Aragona, M.; Soligo, S.M.; Wendling, O.; Mark, M.; Khetchoumian, K.; Bressan, G.; Chambon, P.; et al. Negative control of Smad activity by ectodermin/Tif1gamma patterns the mammalian embryo. Development 2010, 137, 2571–2578. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xi, Q.; Wang, Z.; Zaromytidou, A.I.; Zhang, X.H.; Chow-Tsang, L.F.; Liu, J.X.; Kim, H.; Barlas, A.; Manova-Todorova, K.; Kaartinen, V.; et al. A poised chromatin platform for TGF-beta access to master regulators. Cell 2011, 147, 1511–1524. [Google Scholar] [CrossRef] [PubMed]
- Agricola, E.; Randall, R.A.; Gaarenstroom, T.; Dupont, S.; Hill, C.S. Recruitment of TIF1gamma to chromatin via its PHD finger-bromodomain activates its ubiquitin ligase and transcriptional repressor activities. Mol. Cell 2011, 43, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Tracy Cai, X.; Li, H.; Safyan, A.; Gawlik, J.; Pyrowolakis, G.; Jasper, H. AWD regulates timed activation of BMP signaling in intestinal stem cells to maintain tissue homeostasis. Nat. Commun. 2019, 10, 2988. [Google Scholar] [CrossRef]
- McCabe, B.D.; Hom, S.; Aberle, H.; Fetter, R.D.; Marques, G.; Haerry, T.E.; Wan, H.; O’Connor, M.B.; Goodman, C.S.; Haghighi, A.P. Highwire regulates presynaptic BMP signaling essential for synaptic growth. Neuron 2004, 41, 891–905. [Google Scholar] [CrossRef]
- Liang, M.; Liang, Y.Y.; Wrighton, K.; Ungermannova, D.; Wang, X.P.; Brunicardi, F.C.; Liu, X.; Feng, X.H.; Lin, X. Ubiquitination and proteolysis of cancer-derived Smad4 mutants by SCFSkp2. Mol. Cell. Biol. 2004, 24, 7524–7537. [Google Scholar] [CrossRef]
- Yang, X.; Li, C.; Xu, X.; Deng, C. The tumor suppressor SMAD4/DPC4 is essential for epiblast proliferation and mesoderm induction in mice. Proc. Natl. Acad. Sci. USA 1998, 95, 3667–3672. [Google Scholar] [CrossRef]
- Shull, M.M.; Ormsby, I.; Kier, A.B.; Pawlowski, S.; Diebold, R.J.; Yin, M.; Allen, R.; Sidman, C.; Proetzel, G.; Calvin, D.; et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature 1992, 359, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Mavrakis, K.J.; Andrew, R.L.; Lee, K.L.; Petropoulou, C.; Dixon, J.E.; Navaratnam, N.; Norris, D.P.; Episkopou, V. Arkadia enhances Nodal/TGF-beta signaling by coupling phospho-Smad2/3 activity and turnover. PLoS Biol. 2007, 5, e67. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Ying, S.X.; Zhang, G.M.; Li, C.; Cheng, S.Y.; Deng, C.X.; Zhang, Y.E. Ubiquitin ligase Smurf1 controls osteoblast activity and bone homeostasis by targeting MEKK2 for degradation. Cell 2005, 121, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Ramkumar, C.; Kong, Y.; Trabucco, S.E.; Gerstein, R.M.; Zhang, H. Smurf2 regulates hematopoietic stem cell self-renewal and aging. Aging Cell 2014, 13, 478–486. [Google Scholar] [CrossRef]
- Zhao, L.; Huang, J.; Guo, R.; Wang, Y.; Chen, D.; Xing, L. Smurf1 inhibits mesenchymal stem cell proliferation and differentiation into osteoblasts through JunB degradation. J. Bone Min. Res. 2010, 25, 1246–1256. [Google Scholar] [CrossRef]
- Guo, R.; Yamashita, M.; Zhang, Q.; Zhou, Q.; Chen, D.; Reynolds, D.G.; Awad, H.A.; Yanoso, L.; Zhao, L.; Schwarz, E.M.; et al. Ubiquitin ligase Smurf1 mediates tumor necrosis factor-induced systemic bone loss by promoting proteasomal degradation of bone morphogenetic signaling proteins. J. Biol. Chem. 2008, 283, 23084–23092. [Google Scholar] [CrossRef]
- Zhu, K.; Tang, Y.; Xu, X.; Dang, H.; Tang, L.Y.; Wang, X.; Wang, X.W.; Zhang, Y.E. Non-proteolytic ubiquitin modification of PPARgamma by Smurf1 protects the liver from steatosis. PLoS Biol. 2018, 16, e3000091. [Google Scholar] [CrossRef]
- Ramkumar, C.; Kong, Y.; Cui, H.; Hao, S.; Jones, S.N.; Gerstein, R.M.; Zhang, H. Smurf2 regulates the senescence response and suppresses tumorigenesis in mice. Cancer Res. 2012, 72, 2714–2719. [Google Scholar] [CrossRef]
- Ramkumar, C.; Cui, H.; Kong, Y.; Jones, S.N.; Gerstein, R.M.; Zhang, H. Smurf2 suppresses B-cell proliferation and lymphomagenesis by mediating ubiquitination and degradation of YY1. Nat. Commun. 2013, 4, 2598. [Google Scholar] [CrossRef]
- Sriramachandran, A.M.; Meyer-Teschendorf, K.; Pabst, S.; Ulrich, H.D.; Gehring, N.H.; Hofmann, K.; Praefcke, G.J.K.; Dohmen, R.J. Arkadia/RNF111 is a SUMO-targeted ubiquitin ligase with preference for substrates marked with SUMO1-capped SUMO2/3 chain. Nat. Commun. 2019, 10, 3678. [Google Scholar] [CrossRef]
- Sharma, V.; Antonacopoulou, A.G.; Tanaka, S.; Panoutsopoulos, A.A.; Bravou, V.; Kalofonos, H.P.; Episkopou, V. Enhancement of TGF-beta signaling responses by the E3 ubiquitin ligase Arkadia provides tumor suppression in colorectal cancer. Cancer Res. 2011, 71, 6438–6449. [Google Scholar] [CrossRef] [PubMed]
- Episkopou, V.; Arkell, R.; Timmons, P.M.; Walsh, J.J.; Andrew, R.L.; Swan, D. Induction of the mammalian node requires Arkadia function in the extraembryonic lineages. Nature 2001, 410, 825–830. [Google Scholar] [CrossRef] [PubMed]
- Carrano, A.C.; Eytan, E.; Hershko, A.; Pagano, M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat. Cell. Biol. 1999, 1, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, L.M.; Yeh, K.H.; Lee, S.J.; Sun, H.; Zhang, H. p27(Kip1) ubiquitination and degradation is regulated by the SCF(Skp2) complex through phosphorylated Thr187 in p27. Curr. Biol. 1999, 9, 661–664. [Google Scholar] [CrossRef]
- Ganoth, D.; Bornstein, G.; Ko, T.K.; Larsen, B.; Tyers, M.; Pagano, M.; Hershko, A. The cell-cycle regulatory protein Cks1 is required for SCF(Skp2)-mediated ubiquitinylation of p27. Nat. Cell. Biol. 2001, 3, 321–324. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.; Nagahama, H.; Minamishima, Y.A.; Matsumoto, M.; Nakamichi, I.; Kitagawa, K.; Shirane, M.; Tsunematsu, R.; Tsukiyama, T.; Ishida, N.; et al. Targeted disruption of Skp2 results in accumulation of cyclin E and p27(Kip1), polyploidy and centrosome overduplication. EMBO J. 2000, 19, 2069–2081. [Google Scholar] [CrossRef] [PubMed]
- Kossatz, U.; Dietrich, N.; Zender, L.; Buer, J.; Manns, M.P.; Malek, N.P. Skp2-dependent degradation of p27kip1 is essential for cell cycle progression. Genes Dev. 2004, 18, 2602–2607. [Google Scholar] [CrossRef]
- Minamishima, Y.A.; Nakayama, K.; Nakayama, K. Recovery of liver mass without proliferation of hepatocytes after partial hepatectomy in Skp2-deficient mice. Cancer Res. 2002, 62, 995–999. [Google Scholar]
- Yoshida, K.; Nakayama, K.; Nagahama, H.; Harada, T.; Harada, C.; Imaki, J.; Matsuda, A.; Yamamoto, K.; Ito, M.; Ohno, S.; et al. Involvement of p27(KIP1) degradation by Skp2 in the regulation of proliferation in response to wounding of corneal epithelium. Investig. Ophthalmol. Vis. Sci. 2002, 43, 364–370. [Google Scholar]
- Ge, Z.; Guo, X.; Li, J.; Hartman, M.; Kawasawa, Y.I.; Dovat, S.; Song, C. Clinical significance of high c-MYC and low MYCBP2 expression and their association with Ikaros dysfunction in adult acute lymphoblastic leukemia. Oncotarget 2015, 6, 42300–42311. [Google Scholar] [CrossRef]
- Lewcock, J.W.; Genoud, N.; Lettieri, K.; Pfaff, S.L. The ubiquitin ligase Phr1 regulates axon outgrowth through modulation of microtubule dynamics. Neuron 2007, 56, 604–620. [Google Scholar] [CrossRef] [PubMed]
- Bloom, A.J.; Miller, B.R.; Sanes, J.R.; DiAntonio, A. The requirement for Phr1 in CNS axon tract formation reveals the corticostriatal boundary as a choice point for cortical axons. Genes Dev. 2007, 21, 2593–2606. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Kim, S.; Bahl, S.; Li, L.; Burande, C.F.; Smith, N.; James, M.; Beauchamp, R.L.; Bhide, P.; DiAntonio, A.; et al. The E3 ubiquitin ligase protein associated with Myc (Pam) regulates mammalian/mechanistic target of rapamycin complex 1 (mTORC1) signaling in vivo through N- and C-terminal domains. J. Biol. Chem. 2012, 287, 30063–30072. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Witt, R.M.; Santos, T.M.; Polizzano, C.; Sabatini, B.L.; Ramesh, V. Pam (Protein associated with Myc) functions as an E3 ubiquitin ligase and regulates TSC/mTOR signaling. Cell Signal 2008, 20, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Babetto, E.; Beirowski, B.; Russler, E.V.; Milbrandt, J.; DiAntonio, A. The Phr1 ubiquitin ligase promotes injury-induced axon self-destruction. Cell Rep. 2013, 3, 1422–1429. [Google Scholar] [CrossRef] [PubMed]
- Chretien, M.L.; Legouge, C.; Martin, R.Z.; Hammann, A.; Trad, M.; Aucagne, R.; Largeot, A.; Bastie, J.N.; Delva, L.; Quere, R. Trim33/Tif1gamma is involved in late stages of granulomonopoiesis in mice. Exp. Hematol. 2016, 44, 727–739.e726. [Google Scholar] [CrossRef]
- Kim, J.; Kaartinen, V. Generation of mice with a conditional allele for Trim33. Genesis 2008, 46, 329–333. [Google Scholar] [CrossRef]
- Ferri, F.; Parcelier, A.; Petit, V.; Gallouet, A.S.; Lewandowski, D.; Dalloz, M.; van den Heuvel, A.; Kolovos, P.; Soler, E.; Squadrito, M.L.; et al. TRIM33 switches off Ifnb1 gene transcription during the late phase of macrophage activation. Nat. Commun. 2015, 6, 8900. [Google Scholar] [CrossRef]
- Kusy, S.; Gault, N.; Ferri, F.; Lewandowski, D.; Barroca, V.; Jaracz-Ros, A.; Losson, R.; Romeo, P.H. Adult hematopoiesis is regulated by TIF1gamma, a repressor of TAL1 and PU.1 transcriptional activity. Cell Stem Cell 2011, 8, 412–425. [Google Scholar] [CrossRef]
- Bai, X.; Trowbridge, J.J.; Riley, E.; Lee, J.A.; DiBiase, A.; Kaartinen, V.M.; Orkin, S.H.; Zon, L.I. TiF1-gamma plays an essential role in murine hematopoiesis and regulates transcriptional elongation of erythroid genes. Dev. Biol. 2013, 373, 422–430. [Google Scholar] [CrossRef]
- Quere, R.; Saint-Paul, L.; Carmignac, V.; Martin, R.Z.; Chretien, M.L.; Largeot, A.; Hammann, A.; Pais de Barros, J.P.; Bastie, J.N.; Delva, L. Tif1gamma regulates the TGF-beta1 receptor and promotes physiological aging of hematopoietic stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, 10592–10597. [Google Scholar] [CrossRef] [PubMed]
- Gontan, C.; Achame, E.M.; Demmers, J.; Barakat, T.S.; Rentmeester, E.; van, I.W.; Grootegoed, J.A.; Gribnau, J. RNF12 initiates X-chromosome inactivation by targeting REX1 for degradation. Nature 2012, 485, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, L.; Panning, B.; Leung, K.N. Activators and repressors: A balancing act for X-inactivation. Semin. Cell Dev. Biol. 2016, 56, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Bossenz, M.; Chung, Y.; Ma, H.; Byron, M.; Taniguchi-Ishigaki, N.; Zhu, X.; Jiao, B.; Hall, L.L.; Green, M.R.; et al. Maternal Rnf12/RLIM is required for imprinted X-chromosome inactivation in mice. Nature 2010, 467, 977–981. [Google Scholar] [CrossRef] [PubMed]
- Kammoun, M.; Maas, E.; Criem, N.; Gribnau, J.; Zwijsen, A.; Vermeesch, J.R. RLIM enhances BMP signalling mediated fetal lung development in mice. bioRxiv 2018. [Google Scholar] [CrossRef]
- Shin, J.; Wallingford, M.C.; Gallant, J.; Marcho, C.; Jiao, B.; Byron, M.; Bossenz, M.; Lawrence, J.B.; Jones, S.N.; Mager, J.; et al. RLIM is dispensable for X-chromosome inactivation in the mouse embryonic epiblast. Nature 2014, 511, 86–89. [Google Scholar] [CrossRef]
- Wang, F.; Shin, J.; Shea, J.M.; Yu, J.; Boskovic, A.; Byron, M.; Zhu, X.; Shalek, A.K.; Regev, A.; Lawrence, J.B.; et al. Regulation of X-linked gene expression during early mouse development by Rlim. Elife 2016, 5. [Google Scholar] [CrossRef]
- Jiao, B.; Ma, H.; Shokhirev, M.N.; Drung, A.; Yang, Q.; Shin, J.; Lu, S.; Byron, M.; Kalantry, S.; Mercurio, A.M.; et al. Paternal RLIM/Rnf12 is a survival factor for milk-producing alveolar cells. Cell 2012, 149, 630–641. [Google Scholar] [CrossRef]
- Chen, W.; Jiang, X.; Luo, Z. WWP2: A multifunctional ubiquitin ligase gene. Pathol. Oncol. Res. 2014, 20, 799–803. [Google Scholar] [CrossRef]
- Zou, W.; Chen, X.; Shim, J.H.; Huang, Z.; Brady, N.; Hu, D.; Drapp, R.; Sigrist, K.; Glimcher, L.H.; Jones, D. The E3 ubiquitin ligase Wwp2 regulates craniofacial development through mono-ubiquitylation of Goosecoid. Nat. Cell Biol. 2011, 13, 59–65. [Google Scholar] [CrossRef]
- Yang, Y.; Liao, B.; Wang, S.; Yan, B.; Jin, Y.; Shu, H.B.; Wang, Y.Y. E3 ligase WWP2 negatively regulates TLR3-mediated innate immune response by targeting TRIF for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2013, 110, 5115–5120. [Google Scholar] [CrossRef]
- Li, H.; Zhang, P.; Zhang, Q.; Li, C.; Zou, W.; Chang, Z.; Cui, C.P.; Zhang, L. WWP2 is a physiological ubiquitin ligase for phosphatase and tensin homolog (PTEN) in mice. J. Biol. Chem. 2018, 293, 8886–8899. [Google Scholar] [CrossRef]
- Ambrozkiewicz, M.C.; Schwark, M.; Kishimoto-Suga, M.; Borisova, E.; Hori, K.; Salazar-Lazaro, A.; Rusanova, A.; Altas, B.; Piepkorn, L.; Bessa, P.; et al. Polarity Acquisition in Cortical Neurons Is Driven by Synergistic Action of Sox9-Regulated Wwp1 and Wwp2 E3 Ubiquitin Ligases and Intronic miR-140. Neuron 2018, 100, 1097–1115.e1015. [Google Scholar] [CrossRef]
- Guruharsha, K.G.; Kankel, M.W.; Artavanis-Tsakonas, S. The Notch signalling system: Recent insights into the complexity of a conserved pathway. Nat. Rev. Genet. 2012, 13, 654–666. [Google Scholar] [CrossRef]
- Nowell, C.S.; Radtke, F. Notch as a tumour suppressor. Nat. Rev. Cancer 2017, 17, 145–159. [Google Scholar] [CrossRef]
- Zavadil, J.; Cermak, L.; Soto-Nieves, N.; Bottinger, E.P. Integration of TGF-beta/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. EMBO J. 2004, 23, 1155–1165. [Google Scholar] [CrossRef]
- Niimi, H.; Pardali, K.; Vanlandewijck, M.; Heldin, C.H.; Moustakas, A. Notch signaling is necessary for epithelial growth arrest by TGF-beta. J. Cell Biol. 2007, 176, 695–707. [Google Scholar] [CrossRef]
- Bray, S.J. Notch signalling in context. Nat. Rev. Mol. Cell Biol. 2016, 17, 722–735. [Google Scholar] [CrossRef]
- Haines, N.; Irvine, K.D. Glycosylation regulates Notch signalling. Nat. Rev. Mol. Cell Biol. 2003, 4, 786–797. [Google Scholar] [CrossRef]
- Logeat, F.; Bessia, C.; Brou, C.; LeBail, O.; Jarriault, S.; Seidah, N.G.; Israel, A. The Notch1 receptor is cleaved constitutively by a furin-like convertase. Proc. Natl. Acad. Sci. USA 1998, 95, 8108–8112. [Google Scholar] [CrossRef]
- Blobel, C.P. Metalloprotease-disintegrins: Links to cell adhesion and cleavage of TNF alpha and Notch. Cell 1997, 90, 589–592. [Google Scholar] [CrossRef]
- Pan, D.; Rubin, G.M. Kuzbanian controls proteolytic processing of Notch and mediates lateral inhibition during Drosophila and vertebrate neurogenesis. Cell 1997, 90, 271–280. [Google Scholar] [CrossRef]
- Itoh, M.; Kim, C.H.; Palardy, G.; Oda, T.; Jiang, Y.J.; Maust, D.; Yeo, S.Y.; Lorick, K.; Wright, G.J.; Ariza-McNaughton, L.; et al. Mind bomb is a ubiquitin ligase that is essential for efficient activation of Notch signaling by Delta. Dev. Cell 2003, 4, 67–82. [Google Scholar] [CrossRef]
- De Strooper, B.; Annaert, W.; Cupers, P.; Saftig, P.; Craessaerts, K.; Mumm, J.S.; Schroeter, E.H.; Schrijvers, V.; Wolfe, M.S.; Ray, W.J.; et al. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature 1999, 398, 518–522. [Google Scholar] [CrossRef]
- Wu, L.; Aster, J.C.; Blacklow, S.C.; Lake, R.; Artavanis-Tsakonas, S.; Griffin, J.D. MAML1, a human homologue of Drosophila mastermind, is a transcriptional co-activator for NOTCH receptors. Nat. Genet. 2000, 26, 484–489. [Google Scholar] [CrossRef]
- Tamura, K.; Taniguchi, Y.; Minoguchi, S.; Sakai, T.; Tun, T.; Furukawa, T.; Honjo, T. Physical interaction between a novel domain of the receptor Notch and the transcription factor RBP-J kappa/Su(H). Curr. Biol. 1995, 5, 1416–1423. [Google Scholar] [CrossRef]
- Nagel, A.C.; Krejci, A.; Tenin, G.; Bravo-Patino, A.; Bray, S.; Maier, D.; Preiss, A. Hairless-mediated repression of notch target genes requires the combined activity of Groucho and CtBP corepressors. Mol. Cell. Biol. 2005, 25, 10433–10441. [Google Scholar] [CrossRef]
- Oswald, F.; Winkler, M.; Cao, Y.; Astrahantseff, K.; Bourteele, S.; Knochel, W.; Borggrefe, T. RBP-Jkappa/SHARP recruits CtIP/CtBP corepressors to silence Notch target genes. Mol. Cell. Biol. 2005, 25, 10379–10390. [Google Scholar] [CrossRef]
- Qiu, L.; Joazeiro, C.; Fang, N.; Wang, H.Y.; Elly, C.; Altman, Y.; Fang, D.; Hunter, T.; Liu, Y.C. Recognition and ubiquitination of Notch by Itch, a hect-type E3 ubiquitin ligase. J. Biol. Chem. 2000, 275, 35734–35737. [Google Scholar] [CrossRef]
- Puca, L.; Chastagner, P.; Meas-Yedid, V.; Israel, A.; Brou, C. Alpha-arrestin 1 (ARRDC1) and beta-arrestins cooperate to mediate Notch degradation in mammals. J. Cell Sci. 2013, 126, 4457–4468. [Google Scholar] [CrossRef]
- McGill, M.A.; McGlade, C.J. Mammalian numb proteins promote Notch1 receptor ubiquitination and degradation of the Notch1 intracellular domain. J. Biol. Chem. 2003, 278, 23196–23203. [Google Scholar] [CrossRef]
- Nie, J.; McGill, M.A.; Dermer, M.; Dho, S.E.; Wolting, C.D.; McGlade, C.J. LNX functions as a RING type E3 ubiquitin ligase that targets the cell fate determinant Numb for ubiquitin-dependent degradation. EMBO J. 2002, 21, 93–102. [Google Scholar] [CrossRef]
- Mund, T.; Graeb, M.; Mieszczanek, J.; Gammons, M.; Pelham, H.R.; Bienz, M. Disinhibition of the HECT E3 ubiquitin ligase WWP2 by polymerized Dishevelled. Open Biol. 2015, 5, 150185. [Google Scholar] [CrossRef]
- Jung, J.G.; Stoeck, A.; Guan, B.; Wu, R.C.; Zhu, H.; Blackshaw, S.; Shih Ie, M.; Wang, T.L. Notch3 interactome analysis identified WWP2 as a negative regulator of Notch3 signaling in ovarian cancer. PLoS Genet. 2014, 10, e1004751. [Google Scholar] [CrossRef]
- Sorensen, E.B.; Conner, S.D. gamma-secretase-dependent cleavage initiates notch signaling from the plasma membrane. Traffic 2010, 11, 1234–1245. [Google Scholar] [CrossRef]
- Zheng, L.; Conner, S.D. PI5P4Kgamma functions in DTX1-mediated Notch signaling. Proc. Natl. Acad. Sci. USA 2018, 115, E1983–E1990. [Google Scholar] [CrossRef]
- Fryer, C.J.; White, J.B.; Jones, K.A. Mastermind recruits CycC:CDK8 to phosphorylate the Notch ICD and coordinate activation with turnover. Mol. Cell 2004, 16, 509–520. [Google Scholar] [CrossRef]
- Rallis, C.; Pinchin, S.M.; Ish-Horowicz, D. Cell-autonomous integrin control of Wnt and Notch signalling during somitogenesis. Development 2010, 137, 3591–3601. [Google Scholar] [CrossRef]
- Ruel, L.; Bourouis, M.; Heitzler, P.; Pantesco, V.; Simpson, P. Drosophila shaggy kinase and rat glycogen synthase kinase-3 have conserved activities and act downstream of Notch. Nature 1993, 362, 557–560. [Google Scholar] [CrossRef]
- Espinosa, L.; Ingles-Esteve, J.; Aguilera, C.; Bigas, A. Phosphorylation by glycogen synthase kinase-3 beta down-regulates Notch activity, a link for Notch and Wnt pathways. J. Biol. Chem. 2003, 278, 32227–32235. [Google Scholar] [CrossRef]
- Zhou, T.; Yi, F.; Wang, Z.; Guo, Q.; Liu, J.; Bai, N.; Li, X.; Dong, X.; Ren, L.; Cao, L.; et al. The Functions of DNA Damage Factor RNF8 in the Pathogenesis and Progression of Cancer. Int. J. Biol. Sci. 2019, 15, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, S.; Sczaniecka, M.; McLaren, L.; Russell, F.; Gladstone, K.; Hupp, T.; Wallace, M. Non-degradative ubiquitination of the Notch1 receptor by the E3 ligase MDM2 activates the Notch signalling pathway. Biochem. J. 2013, 450, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Ronai, Z.A. Rewired Notch/p53 by Numb’ing Mdm2. J. Cell Biol. 2018, 217, 445–446. [Google Scholar] [CrossRef] [PubMed]
- Deblandre, G.A.; Lai, E.C.; Kintner, C. Xenopus neuralized is a ubiquitin ligase that interacts with XDelta1 and regulates Notch signaling. Dev. Cell 2001, 1, 795–806. [Google Scholar] [CrossRef]
- Kramer, H. Neuralized: Regulating notch by putting away delta. Dev. Cell 2001, 1, 725–726. [Google Scholar] [CrossRef]
- Fischer, A.; Schumacher, N.; Maier, M.; Sendtner, M.; Gessler, M. The Notch target genes Hey1 and Hey2 are required for embryonic vascular development. Genes Dev. 2004, 18, 901–911. [Google Scholar] [CrossRef]
- Zhang, N.; Gridley, T. Defects in somite formation in lunatic fringe-deficient mice. Nature 1998, 394, 374–377. [Google Scholar] [CrossRef]
- Swiatek, P.J.; Lindsell, C.E.; del Amo, F.F.; Weinmaster, G.; Gridley, T. Notch1 is essential for postimplantation development in mice. Genes Dev. 1994, 8, 707–719. [Google Scholar] [CrossRef]
- Koo, B.K.; Yoon, M.J.; Yoon, K.J.; Im, S.K.; Kim, Y.Y.; Kim, C.H.; Suh, P.G.; Jan, Y.N.; Kong, Y.Y. An obligatory role of mind bomb-1 in notch signaling of mammalian development. PLoS ONE 2007, 2, e1221. [Google Scholar] [CrossRef]
- Koo, B.K.; Lim, H.S.; Song, R.; Yoon, M.J.; Yoon, K.J.; Moon, J.S.; Kim, Y.W.; Kwon, M.C.; Yoo, K.W.; Kong, M.P.; et al. Mind bomb 1 is essential for generating functional Notch ligands to activate Notch. Development 2005, 132, 3459–3470. [Google Scholar] [CrossRef]
- Barsi, J.C.; Rajendra, R.; Wu, J.I.; Artzt, K. Mind bomb1 is a ubiquitin ligase essential for mouse embryonic development and Notch signaling. Mech. Dev. 2005, 122, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.H.; Bellon, M.; Nicot, C. FBXW7: A critical tumor suppressor of human cancers. Mol. Cancer 2018, 17, 115. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.H.; Perez-Losada, J.; Wu, D.; Delrosario, R.; Tsunematsu, R.; Nakayama, K.I.; Brown, K.; Bryson, S.; Balmain, A. Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature 2004, 432, 775–779. [Google Scholar] [CrossRef] [PubMed]
- Busino, L.; Millman, S.E.; Scotto, L.; Kyratsous, C.A.; Basrur, V.; O’Connor, O.; Hoffmann, A.; Elenitoba-Johnson, K.S.; Pagano, M. Fbxw7alpha- and GSK3-mediated degradation of p100 is a pro-survival mechanism in multiple myeloma. Nat. Cell Biol. 2012, 14, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Rodriguez-Barrueco, R.; Yu, J.; Do, C.; Silva, J.M.; Gautier, J. MYC is a critical target of FBXW7. Oncotarget 2015, 6, 3292–3305. [Google Scholar] [CrossRef] [PubMed]
- Tetzlaff, M.T.; Yu, W.; Li, M.; Zhang, P.; Finegold, M.; Mahon, K.; Harper, J.W.; Schwartz, R.J.; Elledge, S.J. Defective cardiovascular development and elevated cyclin E and Notch proteins in mice lacking the Fbw7 F-box protein. Proc. Natl. Acad. Sci. USA 2004, 101, 3338–3345. [Google Scholar] [CrossRef]
- Tsunematsu, R.; Nakayama, K.; Oike, Y.; Nishiyama, M.; Ishida, N.; Hatakeyama, S.; Bessho, Y.; Kageyama, R.; Suda, T.; Nakayama, K.I. Mouse Fbw7/Sel-10/Cdc4 is required for notch degradation during vascular development. J. Biol. Chem. 2004, 279, 9417–9423. [Google Scholar] [CrossRef]
- Onoyama, I.; Tsunematsu, R.; Matsumoto, A.; Kimura, T.; de Alboran, I.M.; Nakayama, K.; Nakayama, K.I. Conditional inactivation of Fbxw7 impairs cell-cycle exit during T cell differentiation and results in lymphomatogenesis. J. Exp. Med. 2007, 204, 2875–2888. [Google Scholar] [CrossRef]
- Matsuoka, S.; Oike, Y.; Onoyama, I.; Iwama, A.; Arai, F.; Takubo, K.; Mashimo, Y.; Oguro, H.; Nitta, E.; Ito, K.; et al. Fbxw7 acts as a critical fail-safe against premature loss of hematopoietic stem cells and development of T-ALL. Genes Dev. 2008, 22, 986–991. [Google Scholar] [CrossRef]
- Sancho, R.; Jandke, A.; Davis, H.; Diefenbacher, M.E.; Tomlinson, I.; Behrens, A. F-box and WD repeat domain-containing 7 regulates intestinal cell lineage commitment and is a haploinsufficient tumor suppressor. Gastroenterology 2010, 139, 929–941. [Google Scholar] [CrossRef]
- Matsumoto, A.; Onoyama, I.; Sunabori, T.; Kageyama, R.; Okano, H.; Nakayama, K.I. Fbxw7-dependent degradation of Notch is required for control of “stemness” and neuronal-glial differentiation in neural stem cells. J. Biol. Chem. 2011, 286, 13754–13764. [Google Scholar] [CrossRef] [PubMed]
- Onoyama, I.; Suzuki, A.; Matsumoto, A.; Tomita, K.; Katagiri, H.; Oike, Y.; Nakayama, K.; Nakayama, K.I. Fbxw7 regulates lipid metabolism and cell fate decisions in the mouse liver. J. Clin. Investig. 2011, 121, 342–354. [Google Scholar] [CrossRef] [PubMed]
- Krebs, L.T.; Iwai, N.; Nonaka, S.; Welsh, I.C.; Lan, Y.; Jiang, R.; Saijoh, Y.; O’Brien, T.P.; Hamada, H.; Gridley, T. Notch signaling regulates left-right asymmetry determination by inducing Nodal expression. Genes Dev. 2003, 17, 1207–1212. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.J.; Koo, B.K.; Song, R.; Jeong, H.W.; Shin, J.; Kim, Y.W.; Kong, Y.Y.; Suh, P.G. Mind bomb-1 is essential for intraembryonic hematopoiesis in the aortic endothelium and the subaortic patches. Mol. Cell. Biol. 2008, 28, 4794–4804. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.; Lee, D.; Hong, S.; Park, S.G.; Song, M.R. The E3 ligase Mind bomb-1 (Mib1) modulates Delta-Notch signaling to control neurogenesis and gliogenesis in the developing spinal cord. J. Biol. Chem. 2013, 288, 2580–2592. [Google Scholar] [CrossRef] [PubMed]
- Horn, S.; Kobberup, S.; Jorgensen, M.C.; Kalisz, M.; Klein, T.; Kageyama, R.; Gegg, M.; Lickert, H.; Lindner, J.; Magnuson, M.A.; et al. Mind bomb 1 is required for pancreatic beta-cell formation. Proc. Natl. Acad. Sci. USA 2012, 109, 7356–7361. [Google Scholar] [CrossRef] [PubMed]
- Luxan, G.; Casanova, J.C.; Martinez-Poveda, B.; Prados, B.; D’Amato, G.; MacGrogan, D.; Gonzalez-Rajal, A.; Dobarro, D.; Torroja, C.; Martinez, F.; et al. Mutations in the NOTCH pathway regulator MIB1 cause left ventricular noncompaction cardiomyopathy. Nat. Med. 2013, 19, 193–201. [Google Scholar] [CrossRef]
- Kim, Y.W.; Koo, B.K.; Jeong, H.W.; Yoon, M.J.; Song, R.; Shin, J.; Jeong, D.C.; Kim, S.H.; Kong, Y.Y. Defective Notch activation in microenvironment leads to myeloproliferative disease. Blood 2008, 112, 4628–4638. [Google Scholar] [CrossRef]
- Song, R.; Kim, Y.W.; Koo, B.K.; Jeong, H.W.; Yoon, M.J.; Yoon, K.J.; Jun, D.J.; Im, S.K.; Shin, J.; Kong, M.P.; et al. Mind bomb 1 in the lymphopoietic niches is essential for T and marginal zone B cell development. J. Exp. Med. 2008, 205, 2525–2536. [Google Scholar] [CrossRef]
- Hsu, T.S.; Mo, S.T.; Hsu, P.N.; Lai, M.Z. c-FLIP is a target of the E3 ligase deltex1 in gastric cancer. Cell Death Dis. 2018, 9, 135. [Google Scholar] [CrossRef]
- Hsiao, H.W.; Liu, W.H.; Wang, C.J.; Lo, Y.H.; Wu, Y.H.; Jiang, S.T.; Lai, M.Z. Deltex1 is a target of the transcription factor NFAT that promotes T cell anergy. Immunity 2009, 31, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Storck, S.; Delbos, F.; Stadler, N.; Thirion-Delalande, C.; Bernex, F.; Verthuy, C.; Ferrier, P.; Weill, J.C.; Reynaud, C.A. Normal immune system development in mice lacking the Deltex-1 RING finger domain. Mol. Cell. Biol. 2005, 25, 1437–1445. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hsiao, H.W.; Hsu, T.S.; Liu, W.H.; Hsieh, W.C.; Chou, T.F.; Wu, Y.J.; Jiang, S.T.; Lai, M.Z. Deltex1 antagonizes HIF-1alpha and sustains the stability of regulatory T cells in vivo. Nat. Commun. 2015, 6, 6353. [Google Scholar] [CrossRef] [PubMed]
- Ou-Yang, H.F.; Zhang, H.W.; Wu, C.G.; Zhang, P.; Zhang, J.; Li, J.C.; Hou, L.H.; He, F.; Ti, X.Y.; Song, L.Q.; et al. Notch signaling regulates the FOXP3 promoter through RBP-J- and Hes1-dependent mechanisms. Mol. Cell Biochem. 2009, 320, 109–114. [Google Scholar] [CrossRef]
- Burghardt, S.; Claass, B.; Erhardt, A.; Karimi, K.; Tiegs, G. Hepatocytes induce Foxp3(+) regulatory T cells by Notch signaling. J. Leukoc. Biol. 2014, 96, 571–577. [Google Scholar] [CrossRef]
- Panier, S.; Durocher, D. Regulatory ubiquitylation in response to DNA double-strand breaks. DNA Repair. 2009, 8, 436–443. [Google Scholar] [CrossRef]
- Kolas, N.K.; Chapman, J.R.; Nakada, S.; Ylanko, J.; Chahwan, R.; Sweeney, F.D.; Panier, S.; Mendez, M.; Wildenhain, J.; Thomson, T.M.; et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science 2007, 318, 1637–1640. [Google Scholar] [CrossRef]
- Li, L.; Halaby, M.J.; Hakem, A.; Cardoso, R.; El Ghamrasni, S.; Harding, S.; Chan, N.; Bristow, R.; Sanchez, O.; Durocher, D.; et al. Rnf8 deficiency impairs class switch recombination, spermatogenesis, and genomic integrity and predisposes for cancer. J. Exp. Med. 2010, 207, 983–997. [Google Scholar] [CrossRef]
- Lu, L.Y.; Wu, J.; Ye, L.; Gavrilina, G.B.; Saunders, T.L.; Yu, X. RNF8-dependent histone modifications regulate nucleosome removal during spermatogenesis. Dev. Cell 2010, 18, 371–384. [Google Scholar] [CrossRef]
- Ouyang, S.; Song, Y.; Tian, Y.; Chen, Y.; Yu, X.; Wang, D. RNF8 deficiency results in neurodegeneration in mice. Neurobiol. Aging 2015, 36, 2850–2860. [Google Scholar] [CrossRef]
- Valnegri, P.; Huang, J.; Yamada, T.; Yang, Y.; Mejia, L.A.; Cho, H.Y.; Oldenborg, A.; Bonni, A. RNF8/UBC13 ubiquitin signaling suppresses synapse formation in the mammalian brain. Nat. Commun. 2017, 8, 1271. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Senturk, E.; Manfredi, J.J. Mdm2 and tumorigenesis: Evolving theories and unsolved mysteries. Genes Cancer 2012, 3, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Mendrysa, S.M.; McElwee, M.K.; Michalowski, J.; O’Leary, K.A.; Young, K.M.; Perry, M.E. mdm2 Is critical for inhibition of p53 during lymphopoiesis and the response to ionizing irradiation. Mol. Cell. Biol. 2003, 23, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.N.; Roe, A.E.; Donehower, L.A.; Bradley, A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 1995, 378, 206–208. [Google Scholar] [CrossRef]
- Mendrysa, S.M.; O’Leary, K.A.; McElwee, M.K.; Michalowski, J.; Eisenman, R.N.; Powell, D.A.; Perry, M.E. Tumor suppression and normal aging in mice with constitutively high p53 activity. Genes Dev. 2006, 20, 16–21. [Google Scholar] [CrossRef]
- Bult, C.J.; Blake, J.A.; Smith, C.L.; Kadin, J.A.; Richardson, J.E.; Mouse Genome Database, G. Mouse Genome Database (MGD) 2019. Nucleic Acids Res. 2019, 47, D801–D806. [Google Scholar] [CrossRef]
- Amberger, J.S.; Bocchini, C.A.; Schiettecatte, F.; Scott, A.F.; Hamosh, A. OMIM.org: Online Mendelian Inheritance in Man (OMIM(R)), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015, 43, D789–D798. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baloghova, N.; Lidak, T.; Cermak, L. Ubiquitin Ligases Involved in the Regulation of Wnt, TGF-β, and Notch Signaling Pathways and Their Roles in Mouse Development and Homeostasis. Genes 2019, 10, 815. https://doi.org/10.3390/genes10100815
Baloghova N, Lidak T, Cermak L. Ubiquitin Ligases Involved in the Regulation of Wnt, TGF-β, and Notch Signaling Pathways and Their Roles in Mouse Development and Homeostasis. Genes. 2019; 10(10):815. https://doi.org/10.3390/genes10100815
Chicago/Turabian StyleBaloghova, Nikol, Tomas Lidak, and Lukas Cermak. 2019. "Ubiquitin Ligases Involved in the Regulation of Wnt, TGF-β, and Notch Signaling Pathways and Their Roles in Mouse Development and Homeostasis" Genes 10, no. 10: 815. https://doi.org/10.3390/genes10100815
APA StyleBaloghova, N., Lidak, T., & Cermak, L. (2019). Ubiquitin Ligases Involved in the Regulation of Wnt, TGF-β, and Notch Signaling Pathways and Their Roles in Mouse Development and Homeostasis. Genes, 10(10), 815. https://doi.org/10.3390/genes10100815