Non-Coding RNA and Tumor Development in Neurofibromatosis Type 1: ANRIL Rs2151280 Is Associated with Optic Glioma Development and a Mild Phenotype in Neurofibromatosis Type 1 Patients

 , , , , ,
, , , , ,  and
and
Abstract
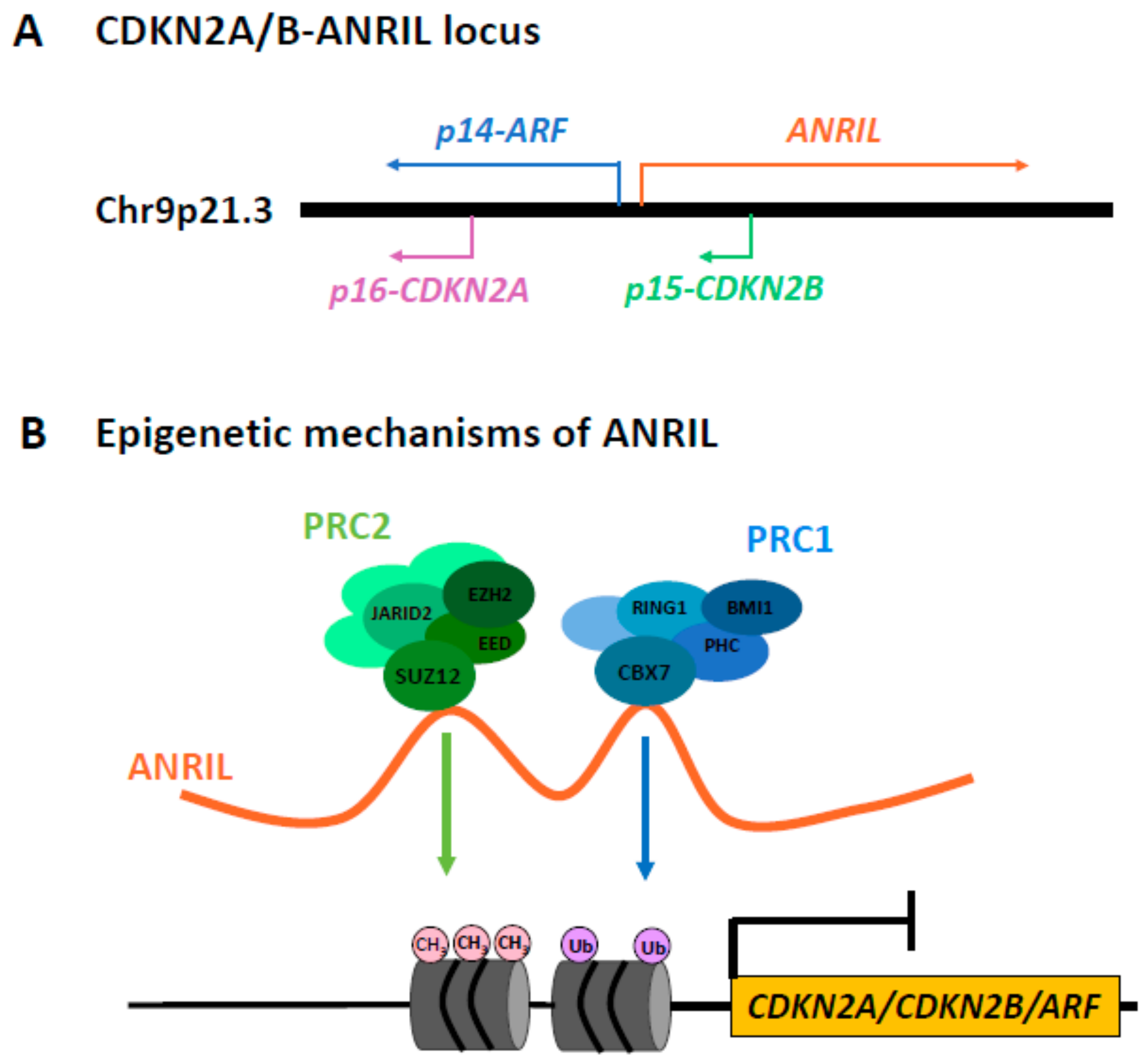
:1. Introduction
2. Materials and Methods
2.1. Review of Literature
2.2. Human Subjects
2.3. Patients and Phenotypic Data
2.4. DNA and RNA Extraction
2.5. Genotyping of Single Nucleotide Polymorphism Rs2151280
2.6. Sequencing Analysis
2.7. Reverse Transcription (RT) and Quantitative Real-Time PCR (qPCR)
2.8. LOH Analysis
2.9. Statistical Analysis
3. Results
3.1. Non-Coding RNA and Tumor Susceptibility in Neurofibromatosis Type 1
3.2. Non-Coding RNA and Neurofibroma Development
3.3. The ANRIL Rs2151280 is a Susceptibility Marker for Optic Glioma Development and Mild Phenotype in NF1 Patients
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Anastasiadou, E.; Jacob, L.S.; Slack, F.J. Non-coding RNA networks in cancer. Nat. Rev. Cancer 2017, 18, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Bejerano, G.; Pheasant, M.; Makunin, I.; Stephen, S.; Kent, W.J.; Mattick, J.S.; Haussler, D. Ultraconserved elements in the human genome. Science 2004, 304, 1321–1325. [Google Scholar] [CrossRef] [PubMed]
- Johnsson, P.; Lipovich, L.; Grandér, D.; Morris, K.V. Evolutionary conservation of long non-coding RNAs; Sequence, structure, function. Biochim. Biophys. Acta-Gen. Subj. 2014, 1840, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Feingold, E.A.; Good, P.J.; Guyer, M.S.; Kamholz, S.; Liefer, L.; Wetterstrand, K.; Collins, F.S.; Gingeras, T.R.; Kampa, D.; Sekinger, E.A.; et al. The ENCODE (ENCyclopedia of DNA Elements) Project. Science 2004, 306, 636–640. [Google Scholar]
- Ebert, M.S.; Sharp, P.A. Roles for microRNAs in conferring robustness to biological processes. Cell 2012, 149, 515–524. [Google Scholar] [CrossRef]
- Yamamura, S.; Imai-Sumida, M.; Tanaka, Y.; Dahiya, R. Interaction and cross-talk between non-coding RNAs. Cell. Mol. Life Sci. 2018, 75, 467–484. [Google Scholar] [CrossRef]
- Moncini, S.; Bevilacqua, A.; Venturin, M.; Fallini, C.; Ratti, A.; Nicolin, A.; Riva, P. The 3′ untranslated region of human Cyclin-Dependent Kinase 5 Regulatory subunit 1 contains regulatory elements affecting transcript stability. BMC Mol. Biol. 2007, 8, 111. [Google Scholar] [CrossRef]
- Zuccotti, P.; Colombrita, C.; Moncini, S.; Barbieri, A.; Lunghi, M.; Gelfi, C.; De Palma, S.; Nicolin, A.; Ratti, A.; Venturin, M.; et al. HnRNPA2/B1 and nELAV proteins bind to a specific U-rich element in CDK5R1 3’-UTR and oppositely regulate its expression. Biochim. Biophys. Acta-Gene Regul. Mech. 2014, 1839, 506–516. [Google Scholar] [CrossRef]
- Upadhyaya, M. Genetic basis of tumorigenesis in NF1 malignant peripheral nerve sheath tumors. Front. Biosci. 2011, 16, 937–951. [Google Scholar] [CrossRef]
- Sedani, A.; Cooper, D.N.; Upadhyaya, M. An emerging role for microRNAs in NF1 tumorigenesis. Hum. Genom. 2012, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Pasmant, E.; Sabbagh, A.; Vidaud, M.; Bièche, I. ANRIL, a long, noncoding RNA, is an unexpected major hotspot in GWAS. FASEB J. 2011, 25, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Pasmant, E.; Sabbagh, A.; Masliah-Planchon, J.; Ortonne, N.; Laurendeau, I.; Melin, L.; Ferkal, S.; Hernandez, L.; Leroy, K.; Valeyrie-Allanore, L.; et al. Role of noncoding RNA ANRIL in genesis of plexiform neurofibromas in neurofibromatosis type 1. J. Natl. Cancer Inst. 2011, 103, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Jin, Z.; Chen, Y.; Xu, H.; Ma, C.; Hong, X.; Li, Y.; Zhao, G. Knockdown of long non-coding RNA ANRIL inhibits proliferation, migration, and invasion but promotes apoptosis of human glioma cells by upregulation of miR-34a. J. Cell. Biochem. 2018, 119, 2708–2718. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, S.; Thompson, P.; Frayling, I.; Upadhyaya, M. Molecular diagnosis of neurofibromatosis type 1, 2 Years experience. Fam. Cancer 2007, 6, 21–34. [Google Scholar] [CrossRef]
- Ablon, J. Gender response to neurofibromatosis. Soc. Sci. Med. 1996, 42, 99–110. [Google Scholar] [CrossRef]
- Riccardi, V.M.; Riccardi, S.L. Von Recklinghausen neurofibromatosis: New perspectives. Tex. Med. 1982, 78, 43–44. [Google Scholar]
- Ferrari, L.; Scuvera, G.; Tucci, A.; Bianchessi, D.; Rusconi, F.; Menni, F.; Battaglioli, E.; Milani, D.; Riva, P. Identification of an atypical microdeletion generating the RNF135-SUZ12 chimeric gene and causing a position effect in an NF1 patient with overgrowth. Hum. Genet. 2017, 136, 1329–1339. [Google Scholar] [CrossRef]
- Lee, W.; Teckie, S.; Wiesner, T.; Ran, L.; Prieto Granada, C.N.; Lin, M.; Zhu, S.; Cao, Z.; Liang, Y.; Sboner, A.; et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat. Genet. 2014, 46, 1227–1232. [Google Scholar] [CrossRef]
- Staedtke, V.; Bai, R.Y.; Blakeley, J.O. Cancer of the peripheral nerve in neurofibromatosis type 1. Neurotherapeutics 2017, 14, 298–306. [Google Scholar] [CrossRef]
- Masliah-Planchon, J.; Pasmant, E.; Luscan, A.; Laurendeau, I.; Ortonne, N.; Hivelin, M.; Varin, J.; Valeyrie-Allanore, L.; Dumaine, V.; Lantieri, L.; et al. MicroRNAome profiling in benign and malignant neurofibromatosis type 1-associated nerve sheath tumors: Evidences of PTEN pathway alterations in early NF1 tumorigenesis. BMC Genom. 2013, 14, 474. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.M.; Grosshans, H.; Shingara, J.; Byrom, M.; Jarvis, R.; Cheng, A.; Labourier, E.; Reinert, K.L.; Brown, D.; Slack, F.J. RAS is regulated by the let-7 microRNA family. Cell 2005, 120, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Chai, G.; Liu, N.; Ma, J.; Li, H.; Oblinger, J.L.; Prahalad, A.K.; Gong, M.; Chang, L.S.; Wallace, M.; Muir, D.; et al. MicroRNA-10b regulates tumorigenesis in neurofibromatosis type 1. Cancer Sci. 2010, 101, 1997–2004. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhao, Y.; Liu, C.; Chen, X.; Qi, Y.; Jiang, Y.; Zou, C.; Zhang, X.; Liu, S.; Wang, X.; et al. Analysis of MiR-195 and MiR-497 expression, regulation and role in breast cancer. Clin. Cancer Res. 2011, 17, 1722–1730. [Google Scholar] [CrossRef]
- Lewis, B.P.; Shih, I.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of mammalian microRNA targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef]
- Poliseno, L.; Salmena, L.; Riccardi, L.; Fornari, A.; Song, M.S.; Hobbs, R.M.; Sportoletti, P.; Varmeh, S.; Egia, A.; Fedele, G.; et al. Identification of the miR-106b∼25 microRNA cluster as a proto-oncogenic PTEN-targeting intron that cooperates with its host gene MCM7 in transformation. Sci. Signal. 2010, 3, ra29. [Google Scholar] [CrossRef]
- Shi, W.; Gerster, K.; Alajez, N.M.; Tsang, J.; Waldron, L.; Pintilie, M.; Hui, A.B.; Sykes, J.; P’ng, C.; Miller, N.; et al. MicroRNA-301 mediates proliferation and invasion in human breast cancer. Cancer Res. 2011, 71, 2926–2937. [Google Scholar] [CrossRef]
- Yong, S.L.; Dutta, A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev. 2007, 21, 1025–1030. [Google Scholar]
- Ma, L.; Young, J.; Prabhala, H.; Pan, E.; Mestdagh, P.; Muth, D.; Teruya-Feldstein, J.; Reinhardt, F.; Onder, T.T.; Valastyan, S.; et al. MiR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat. Cell Biol. 2010, 12, 247–256. [Google Scholar] [CrossRef]
- Nagel, R.; Le Sage, C.; Diosdado, B.; Van Der Waal, M.; Oude Vrielink, J.A.F.; Bolijn, A.; Meijer, G.A.; Agami, R. Regulation of the adenomatous polyposis coli gene by the miR-135 family in colorectal cancer. Cancer Res. 2008, 68, 5795–5802. [Google Scholar] [CrossRef]
- Peter, M.E. Let-7 and miR-200 microRNAs: Guardians against pluripotency and cancer progression. Cell Cycle 2009, 8, 843–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotkrua, P.; Akiyama, Y.; Hashimoto, Y.; Otsubo, T.; Yuasa, Y. MiR-9 downregulates CDX2 expression in gastric cancer cells. Int. J. Cancer 2011, 129, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Witmer, P.D.W.; Casey, E.; Valle, D.; Sukumar, S. DNA methylation regulates microRNA expression. Cancer Biol. Ther. 2007, 6, 1290–1294. [Google Scholar] [CrossRef] [PubMed]
- Yekta, S.; Shih, I.H.; Bartel, D.P. MicroRNA-directed cleavage of HOXB8 mRNA. Science 2004, 304, 594–596. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Ding, L.; Bennewith, K.L.; Tong, R.T.; Welford, S.M.; Ang, K.K.; Story, M.; Le, Q.T.; Giaccia, A.J. Hypoxia-inducible mir-210 regulates normoxic gene expression involved in tumor initiation. Mol. Cell 2009, 35, 856–867. [Google Scholar] [CrossRef] [PubMed]
- Schultz, J.; Lorenz, P.; Gross, G.; Ibrahim, S.; Kunz, M. MicroRNA let-7b targets important cell cycle molecules in malignant melanoma cells and interferes with anchorage-independent growth. Cell Res. 2008, 18, 549–557. [Google Scholar] [CrossRef] [Green Version]
- Trompeter, H.I.; Abbad, H.; Iwaniuk, K.M.; Hafner, M.; Renwick, N.; Tuschl, T.; Schira, J.; Müller, H.W.; Wernet, P. MicroRNAs MiR-17, MiR-20a, and MiR-106b Act in concert to modulate E2F activity on cell cycle arrest during neuronal lineage differentiation of USSC. PLoS ONE 2011, 6, e16138. [Google Scholar] [CrossRef]
- Wu, J.; Qian, J.; Li, C.; Kwok, L.; Cheng, F.; Liu, P.; Perdomo, C.; Kotton, D.; Vaziri, C.; Anderlind, C.; et al. miR-129 regulates cell proliferation by downregulating Cdk6 expression. Cell Cycle 2010, 9, 1809–1818. [Google Scholar] [CrossRef] [Green Version]
- Xu, T.; Zhu, Y.; Xiong, Y.; Ge, Y.Y.; Yun, J.P.; Zhuang, S.M. MicroRNA-195 suppresses tumorigenicity and regulates G 1 /S transition of human hepatocellular carcinoma cells. Hepatology 2009, 50, 113–121. [Google Scholar] [CrossRef]
- Subramanian, S.; Thayanithy, V.; West, R.B.; Lee, C.H.; Beck, A.H.; Zhu, S.; Downs-Kelly, E.; Montgomery, K.; Goldblum, J.R.; Hogendoorn, P.C.W.; et al. Genome-wide transcriptome analyses reveal p53 inactivation mediated loss of miR-34a expression in malignant peripheral nerve sheath tumours. J. Pathol. 2010, 220, 58–70. [Google Scholar] [CrossRef]
- Lee, Y.B.; Bantounas, I.; Lee, D.Y.; Phylactou, L.; Caldwell, M.A.; Uney, J.B. Twist-1 regulates the miR-199a/214 cluster during development. Nucleic Acids Res. 2009, 37, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Gong, M.; Ma, J.; Li, M.; Zhou, M.; Hock, J.M.; Yu, X. MicroRNA-204 critically regulates carcinogenesis in malignant peripheral nerve sheath tumors. Neuro Oncol. 2012, 14, 1007–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itani, S.; Kunisada, T.; Morimoto, Y.; Yoshida, A.; Sasaki, T.; Ito, S.; Ouchida, M.; Sugihara, S.; Shimizu, K.; Ozaki, T. MicroRNA-21 correlates with tumorigenesis in malignant peripheral nerve sheath tumor (MPNST) via programmed cell death protein 4 (PDCD4). J. Cancer Res. Clin. Oncol. 2012, 138, 1501–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Presneau, N.; Eskandarpour, M.; Shemais, T.; Henderson, S.; Halai, D.; Tirabosco, R.; Flanagan, A.M. MicroRNA profiling of peripheral nerve sheath tumours identifies miR-29c as a tumour suppressor gene involved in tumour progression. Br. J. Cancer 2013, 108, 964–972. [Google Scholar] [CrossRef] [PubMed]
- Listernick, R.; Louis, D.N.; Packer, R.J.; Gutmann, D.H. Optic pathway gliomas in children with neurofibromatosis 1, Consensus statement from the NF1 optic pathway glioma task force. Ann. Neurol. 1997, 41, 143–149. [Google Scholar] [CrossRef]
- Ho, C.Y.; Bar, E.; Giannini, C.; Marchionni, L.; Karajannis, M.A.; Zagzag, D.; Gutmann, D.H.; Eberhart, C.G.; Rodriguez, F.J. MicroRNA profiling in pediatric pilocytic astrocytoma reveals biologically relevant targets, including PBX3, NFIB, and METAP2. Neuro Oncol. 2013, 15, 69–82. [Google Scholar] [CrossRef]
- Darrigo Júnior, L.G.; Lira, R.C.P.; Fedatto, P.F.; Marco Antonio, D.S.; Valera, E.T.; Aguiar, S.; Yunes, J.A.; Brandalise, S.R.; Neder, L.; Saggioro, F.P.; et al. MicroRNA profile of pediatric pilocytic astrocytomas identifies two tumor-specific signatures when compared to non-neoplastic white matter. J. Neurooncol. 2019, 141, 373–382. [Google Scholar] [CrossRef]
- Tchernev, G.; Chokoeva, A.A.; Patterson, J.W.; Bakardzhiev, I.; Wollina, U.; Tana, C. Plexiform Neurofibroma: A Case Report. Medicine (Baltimore) 2016, 95, e2663. [Google Scholar] [CrossRef]
- Drak Alsibai, K.; Vacher, S.; Meseure, D.; Nicolas, A.; Lae, M.; Schnitzler, A.; Chemlali, W.; Cros, J.; Longchampt, E.; Cacheux, W.; et al. High positive correlations between ANRIL and p16-CDKN2A/p15-CDKN2B/p14-ARF gene cluster overexpression in multi-tumor types suggest deregulated activation of an ANRIL–ARF bidirectional promoter. Non-Coding RNA 2019, 5, 44. [Google Scholar] [CrossRef]
- Yap, K.L.; Li, S.; Muñoz-Cabello, A.M.; Raguz, S.; Zeng, L.; Mujtaba, S.; Gil, J.; Walsh, M.J.; Zhou, M.M. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol. Cell 2010, 38, 662–674. [Google Scholar] [CrossRef]
- Margueron, R.; Li, G.; Sarma, K.; Blais, A.; Zavadil, J.; Woodcock, C.L.; Dynlacht, B.D.; Reinberg, D. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol. Cell 2008, 32, 503–518. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Li, X.; Song, Y.; Zhang, P.; Xiao, Y.; Xing, Y. Long non-coding RNA ANRIL is up-regulated in bladder cancer and regulates bladder cancer cell proliferation and apoptosis through the intrinsic pathway. Biochem. Biophys. Res. Commun. 2015, 467, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.J.; Lin, Y.Y.; Ding, J.X.; Feng, W.W.; Jin, H.Y.; Hua, K.Q. Long non-coding RNA ANRIL predicts poor prognosis and promotes invasion/metastasis in serous ovarian cancer. Int. J. Oncol. 2015, 46, 2497–2505. [Google Scholar] [CrossRef] [PubMed]
- Naemura, M.; Murasaki, C.; Inoue, Y.; Okamoto, H.; Kotake, Y. Long noncoding RNA ANRIL regulates proliferation of non-small cell lung cancer and cervical cancer cells. Anticancer Res. 2015, 35, 5377–5382. [Google Scholar]
- Meseure, D.; Vacher, S.; Alsibai, K.D.; Nicolas, A.; Chemlali, W.; Caly, M.; Lidereau, R.; Pasmant, E.; Callens, C.; Bieche, I. Expression of ANRIL-polycomb complexes-CDKN2A/B/ARF genes in breast tumors: Identification of a two-gene (EZH2/CBX7) signature with independent prognostic value. Mol. Cancer Res. 2016, 14, 623–633. [Google Scholar] [CrossRef]
- Zhang, E.B.; Kong, R.; Yin, D.D.; You, L.H.; Sun, M.; Han, L.; Xu, T.P.; Xia, R.; Yang, J.S.; De, W.; et al. Long noncoding RNA ANRIL indicates a poor prognosis of gastric cancer and promotes tumor growth by epigenetically silencing of miR-99a/miR-449a. Oncotarget 2014, 5, 2276–2292. [Google Scholar] [CrossRef]
- Chen, D.; Zhang, Z.; Mao, C.; Zhou, Y.; Yu, L.; Yin, Y.; Wu, S.; Mou, X.; Zhu, Y. ANRIL inhibits p15INK4b through the TGFβ1 signaling pathway in human esophageal squamous cell carcinoma. Cell. Immunol. 2014, 289, 91–96. [Google Scholar] [CrossRef]
- Nie, F.Q.; Sun, M.; Yang, J.S.; Xie, M.; Xu, T.P.; Xia, R.; Liu, Y.W.; Liu, X.H.; Zhang, E.B.; Lu, K.H.; et al. Long noncoding RNA ANRIL promotes non-small cell lung cancer cell proliferation and inhibits apoptosis by silencing KLF2 and P21 expression. Mol. Cancer Ther. 2015, 14, 268–277. [Google Scholar] [CrossRef]
- Huang, M.D.; Chen, W.M.; Qi, F.Z.; Xia, R.; Sun, M.; Xu, T.P.; Yin, L.; Zhang, E.B.; De, W.; Shu, Y.Q. Long non-coding RNA ANRIL is upregulated in hepatocellular carcinoma and regulates cell apoptosis by epigenetic silencing of KLF2. J. Hematol. Oncol. 2015, 8, 57. [Google Scholar] [CrossRef]
- Cunnington, M.S.; Koref, M.S.; Mayosi, B.M.; Burn, J.; Keavney, B. Chromosome 9p21 SNPs associated with multiple disease phenotypes correlate with ANRIL expression. PLoS Genet. 2010, 6, e1000899. [Google Scholar] [CrossRef]
- Pasmant, E.; Laurendeau, I.; Héron, D.; Vidaud, M.; Vidaud, D.; Bièche, I. Characterization of a germ-line deletion, including the entire INK4/ARF locus, in a melanoma-neural system tumor family: Identification of ANRIL, an antisense noncoding RNA whose expression coclusters with ARF. Cancer Res. 2007, 67, 3963–3969. [Google Scholar] [CrossRef] [PubMed]
- Mußotter, T.; Kluwe, L.; Högel, J.; Nguyen, R.; Cooper, D.N.; Mautner, V.F.; Kehrer-Sawatzki, H. Non-coding RNA ANRIL and the number of plexiform neurofibromas in patients with NF1 microdeletions. BMC Med. Genet. 2012, 13, 98. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Hsieh, C.-H.; Alonso, L.C. ANRIL: A lncRNA at the CDKN2A/B locus with roles in cancer and metabolic disease. Front. Endocrinol. (Lausanne) 2018, 9, 405. [Google Scholar] [CrossRef] [PubMed]
- Beert, E.; Brems, H.; Daniëls, B.; de Wever, I.; van Calenbergh, F.; Schoenaers, J.; Debiec-Rychter, M.; Gevaert, O.; de Raedt, T.; van den Bruel, A.; et al. Atypical neurofibromas in neurofibromatosis type 1 are premalignant tumors. Genes Chromosom. Cancer 2011, 50, 1021–1032. [Google Scholar] [CrossRef]
- Tano, K.; Akimitsu, N. Long non-coding RNAs in cancer progression. Front. Genet. 2012, 3, 219. [Google Scholar] [CrossRef] [Green Version]
- Kotake, Y.; Nakagawa, T.; Kitagawa, K.; Suzuki, S.; Liu, N.; Kitagawa, M.; Xiong, Y. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15 INK4B tumor suppressor gene. Oncogene 2011, 30, 1956–1962. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Clinical Characterization | N° (%) |
|---|---|
| N° pts | 104 |
| Gender (M/F) | 42/62 |
| Median Age (Range) | 35 (2–71) |
| DNF | 91/104 (86) |
| PNF | 39/104 (38) |
| OPG | 24/104 (23) |
| Other CNS Tumors | 17/104 (16) |
| Extra CNSTumors | 15/104 (14) |
| Clinical scores | N° (%) |
| Riccardi Score 1 | 9/104 (9) |
| Riccardi Score 2 | 32/104(31) |
| Riccardi Score 3 | 24/104 (23) |
| Riccardi Score 4 | 39/104 (38) |
| Ablon Score 1 | 17/104 (16) |
| Ablon Score 2 | 39/104 (38) |
| Ablon Score 3 | 33/104 (32) |
| Ablon Score 4 | 15/104 (14) |
| Molecular Characterization | N° (%) |
| Mutation detected | 100/104 (96) |
| Point mutation | 58/104 (56) |
| Intragenic deletion/duplication | 42/104 (40) |
| SAMPLES | f(C) | f(T) | f(CC) | f(CT) | f(TT) |
|---|---|---|---|---|---|
| Complete casuistry (n = 104) | 0.52 | 0.48 | (n = 24) 0.23 | (n = 61) 0.59 | (n = 19) 0.18 |
| DNF (n = 91) | 0.51 | 0.49 | (n = 19) 0.21 | (n = 56) 0.62 | (n = 16) 0.18 |
| PNF (n = 39) | 0.54 | 0.46 | (n = 9) 0.23 | (n = 24) 0.62 | (n = 6) 0.15 |
| OPG (n = 24) | 0.62 | 0.38 | (n = 10) 0.42 | (n = 10) 0.42 | (n = 4) 0.17 |
| Other tumors (n = 17) | 0.56 | 0.44 | (n = 4) 0.24 | (n = 10) 0.59 | (n = 3) 0.18 |
| Unaffected Controls | 0.5 | 0.5 | 0.2 | 0.6 | 0.2 |
| General population | 0.5 | 0.5 | 0.27 | 0.46 | 0.27 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tritto, V.; Ferrari, L.; Esposito, S.; Zuccotti, P.; Bianchessi, D.; Natacci, F.; Saletti, V.; Eoli, M.; Riva, P. Non-Coding RNA and Tumor Development in Neurofibromatosis Type 1: ANRIL Rs2151280 Is Associated with Optic Glioma Development and a Mild Phenotype in Neurofibromatosis Type 1 Patients. Genes 2019, 10, 892. https://doi.org/10.3390/genes10110892
Tritto V, Ferrari L, Esposito S, Zuccotti P, Bianchessi D, Natacci F, Saletti V, Eoli M, Riva P. Non-Coding RNA and Tumor Development in Neurofibromatosis Type 1: ANRIL Rs2151280 Is Associated with Optic Glioma Development and a Mild Phenotype in Neurofibromatosis Type 1 Patients. Genes. 2019; 10(11):892. https://doi.org/10.3390/genes10110892
Chicago/Turabian StyleTritto, Viviana, Luca Ferrari, Silvia Esposito, Paola Zuccotti, Donatella Bianchessi, Federica Natacci, Veronica Saletti, Marica Eoli, and Paola Riva. 2019. "Non-Coding RNA and Tumor Development in Neurofibromatosis Type 1: ANRIL Rs2151280 Is Associated with Optic Glioma Development and a Mild Phenotype in Neurofibromatosis Type 1 Patients" Genes 10, no. 11: 892. https://doi.org/10.3390/genes10110892
APA StyleTritto, V., Ferrari, L., Esposito, S., Zuccotti, P., Bianchessi, D., Natacci, F., Saletti, V., Eoli, M., & Riva, P. (2019). Non-Coding RNA and Tumor Development in Neurofibromatosis Type 1: ANRIL Rs2151280 Is Associated with Optic Glioma Development and a Mild Phenotype in Neurofibromatosis Type 1 Patients. Genes, 10(11), 892. https://doi.org/10.3390/genes10110892