The Critical Role of Hypoxic Microenvironment and Epigenetic Deregulation in Esophageal Cancer Radioresistance

 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Biological Basis of Cancer Radiation Therapy
Tumor-Associated Microenvironment: Hypoxia, HIF-1α, and Radioresistance
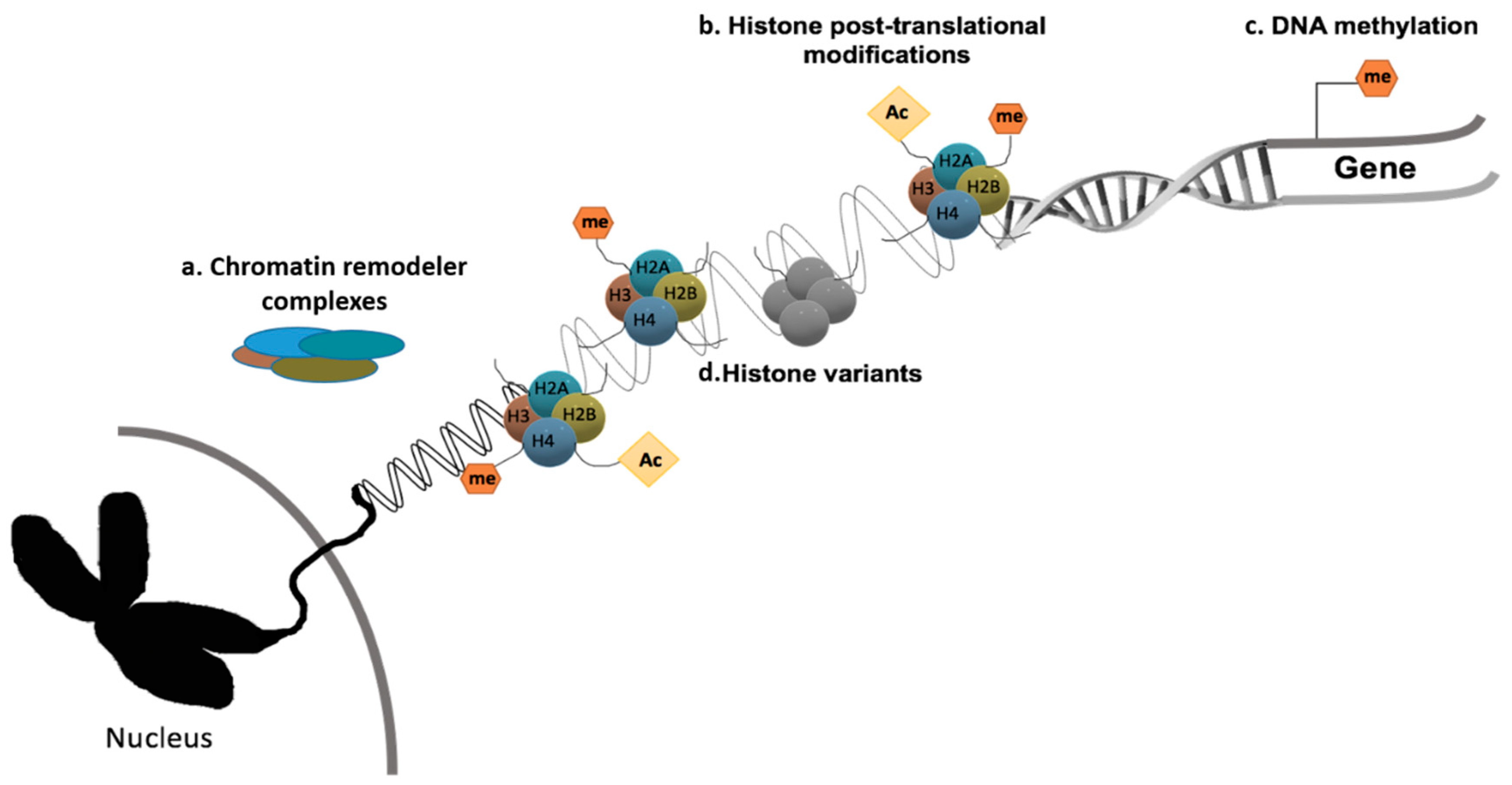
3. Cancer Epigenetics: A Brief Overview
3.1. Epigenetic Remodeling and Hypoxic Microenvironment
3.2. Epigenetic Modifications and Radiosensitivity
4. Emerging Novel Therapeutic Targets through Epigenetic Chromatin Modulation under Hypoxia
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Miller, K.D.; Siegel, R.L.; Lin, C.C.; Mariotto, A.B.; Kramer, J.L.; Rowland, J.H.; Stein, K.D.; Alteri, R.; Jemal, A. Cancer treatment and survivorship statistics, 2016. CA Cancer J. Clin. 2016, 66, 271–289. [Google Scholar] [CrossRef]
- Napier, K.J.; Scheerer, M.; Misra, S. Esophageal cancer: A Review of epidemiology, pathogenesis, staging workup and treatment modalities. World. J. Gastrointest. Oncol. 2014, 6, 112–120. [Google Scholar] [CrossRef]
- Arnold, M.; Soerjomataram, I.; Ferlay, J.; Forman, D. Global incidence of oesophageal cancer by histological subtype in 2012. Gut 2015, 64, 381–387. [Google Scholar] [CrossRef]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Pickens, A.; Orringer, M.B. Geographical distribution and racial disparity in esophageal cancer. Ann. Thorac. Surg. 2003, 76, S1367–S1369. [Google Scholar] [CrossRef]
- Engel, L.S.; Chow, W.H.; Vaughan, T.L.; Gammon, M.D.; Risch, H.A.; Stanford, J.L.; Schoenberg, J.B.; Mayne, S.T.; Dubrow, R.; Rotterdam, H.; et al. Population attributable risks of esophageal and gastric cancers. J. Natl. Cancer Inst. 2003, 95, 1404–1413. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.D.; Cassano, A.D.; Neifeld, J.P. Neoadjuvant therapy for esophageal cancer. World J. Gastrointest. Oncol. 2014, 6, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Campbell, N.P.; Villaflor, V.M. Neoadjuvant treatment of esophageal cancer. World J. Gastroenterol. 2010, 16, 3793–3803. [Google Scholar] [CrossRef] [PubMed]
- Koshy, M.; Greenwald, B.D.; Hausner, P.; Krasna, M.J.; Horiba, N.; Battafarano, R.J.; Burrows, W.; Suntharalingam, M. Outcomes after trimodality therapy for esophageal cancer: The impact of histology on failure patterns. Am. J. Clin. Oncol. 2011, 34, 259–264. [Google Scholar] [CrossRef]
- Wald, O.; Smaglo, B.; Mok, H.; Groth, S.S. Future directions in esophageal cancer therapy. Ann. Cardiothorac. Surg. 2017, 6, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Lovly, C.M.; Salama, A.K.; Salgia, R. Tumor Heterogeneity and Therapeutic Resistance. Am. Soc. Clin. Oncol. Educ. Book 2016, 35, e585–e593. [Google Scholar] [CrossRef] [PubMed]
- Peerlings, J.; Van De Voorde, L.; Mitea, C.; Larue, R.; Yaromina, A.; Sandeleanu, S.; Spiegelberg, L.; Dubois, L.; Lambin, P.; Mottaghy, F.M. Hypoxia and hypoxia response-associated molecular markers in esophageal cancer: A systematic review. Methods 2017, 130, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Danhier, P.; Banski, P.; Payen, V.L.; Grasso, D.; Ippolito, L.; Sonveaux, P.; Porporato, P.E. Cancer metabolism in space and time: Beyond the Warburg effect. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 556–572. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Rao, C.M. Epigenetics in cancer: Fundamentals and Beyond. Pharmacol. Therapeutics 2017, 173, 118–134. [Google Scholar] [CrossRef]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33 (Suppl. S3), 245–254. [Google Scholar] [CrossRef]
- Nguyen, M.P.; Lee, S.; Lee, Y.M. Epigenetic regulation of hypoxia inducible factor in diseases and therapeutics. Arch. Pharmacal Res. 2013, 36, 252–263. [Google Scholar] [CrossRef]
- Chi, H.C.; Tsai, C.Y.; Tsai, M.M.; Lin, K.H. Impact of DNA and RNA Methylation on Radiobiology and Cancer Progression. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef]
- Smits, K.M.; Melotte, V.; Niessen, H.E.; Dubois, L.; Oberije, C.; Troost, E.G.; Starmans, M.H.; Boutros, P.C.; Vooijs, M.; van Engeland, M.; et al. Epigenetics in radiotherapy: Where are we heading? Radiolther. Oncol. 2014, 111, 168–177. [Google Scholar] [CrossRef]
- Luo, Y.; Mao, Q.; Wang, X.; Yu, J.; Li, M. Radiotherapy for esophageal carcinoma: Dose, response and survival. Cancer Manag. Res. 2018, 10, 13–21. [Google Scholar] [CrossRef]
- Baskar, R.; Dai, J.; Wenlong, N.; Yeo, R.; Yeoh, K.W. Biological response of cancer cells to radiation treatment. Front. Mol. Biosci. 2014, 1, 24. [Google Scholar] [CrossRef] [PubMed]
- Harrington, K.; Jankowska, P.; Hingorani, M. Molecular biology for the radiation oncologist: The 5Rs of radiobiology meet the hallmarks of cancer. Clin. Oncol. 2007, 19, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Lomax, M.E.; Folkes, L.K.; O′Neill, P. Biological consequences of radiation-induced DNA damage: Relevance to radiotherapy. Clin. Oncol. 2013, 25, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Pawlik, T.M.; Keyomarsi, K. Role of cell cycle in mediating sensitivity to radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, 928–942. [Google Scholar] [CrossRef]
- Yang, J.; Yue, J.B.; Liu, J.; Yu, J.M. Repopulation of tumor cells during fractionated radiotherapy and detection methods (Review). Oncol. Lett. 2014, 7, 1755–1760. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef]
- Hall, E.J.; Giaccia, A.J. Radiobiology for the Radiologist, 7th ed.; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2012; Volume II, pp. 432–447. [Google Scholar]
- Kuwahara, Y.; Roudkenar, M.H.; Urushihara, Y.; Saito, Y.; Tomita, K.; Roushandeh, A.M.; Sato, T.; Kurimasa, A.; Fukumoto, M. Clinically relevant radioresistant cell line: A simple model to understand cancer radioresistance. Med. Mol. Morphol. 2017, 50, 195–204. [Google Scholar] [CrossRef]
- Lv, X.; Li, J.; Zhang, C.; Hu, T.; Li, S.; He, S.; Yan, H.; Tan, Y.; Lei, M.; Wen, M.; et al. The role of hypoxia-inducible factors in tumor angiogenesis and cell metabolism. Genes. Dis. 2017, 4, 19–24. [Google Scholar] [CrossRef]
- Al Tameemi, W.; Dale, T.P.; Al-Jumaily, R.M.K.; Forsyth, N.R. Hypoxia-Modified Cancer Cell Metabolism. Front. Cell Dev. Biol. 2019, 7, 4. [Google Scholar] [CrossRef]
- Chan, D.A.; Giaccia, A.J. Hypoxia, gene expression, and metastasis. Cancer Metastasis Rev. 2007, 26, 333–339. [Google Scholar] [CrossRef]
- Martin, J.D.; Fukumura, D.; Duda, D.G.; Boucher, Y.; Jain, R.K. Reengineering the Tumor Microenvironment to Alleviate Hypoxia and Overcome Cancer Heterogeneity. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Koukourakis, M.I.; Giatromanolaki, A.; Skarlatos, J.; Corti, L.; Blandamura, S.; Piazza, M.; Gatter, K.C.; Harris, A.L. Hypoxia inducible factor (HIF-1a and HIF-2a) expression in early esophageal cancer and response to photodynamic therapy and radiotherapy. Cancer Res. 2001, 61, 1830–1832. [Google Scholar] [PubMed]
- Jing, S.W.; Wang, J.; Xu, Q. Expression of hypoxia inducible factor 1 alpha and its clinical significance in esophageal carcinoma: A meta-analysis. Tumour Biol. 2017, 39, 1010428317717983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, K.; Chiba, I.; Morioka, T.; Shimoji, H.; Tamaki, W.; Takamatsu, R.; Nishimaki, T.; Yoshimi, N.; Murayama, S. Clinical significance of HIF-1alpha expression in patients with esophageal cancer treated with concurrent chemoradiotherapy. Anticancer Res. 2011, 31, 2351–2359. [Google Scholar]
- Matsuyama, T.; Nakanishi, K.; Hayashi, T.; Yoshizumi, Y.; Aiko, S.; Sugiura, Y.; Tanimoto, T.; Uenoyama, M.; Ozeki, Y.; Maehara, T. Expression of hypoxia-inducible factor-1alpha in esophageal squamous cell carcinoma. Cancer Sci. 2005, 96, 176–182. [Google Scholar] [CrossRef]
- Brown, J.M.; Wilson, W.R. Exploiting tumour hypoxia in cancer treatment. Nat. Rev. Cancer 2004, 4, 437–447. [Google Scholar] [CrossRef]
- Yang, X.; Yang, B.; Cai, J.; Zhang, C.; Zhang, Q.; Xu, L.; Qin, Q.; Zhu, H.; Ma, J.; Tao, G.; et al. Berberine enhances radiosensitivity of esophageal squamous cancer by targeting HIF-1alpha in vitro and in vivo. Cancer Biol. 2013, 14, 1068–1073. [Google Scholar] [CrossRef] [Green Version]
- Kato, Y.; Yashiro, M.; Fuyuhiro, Y.; Kashiwagi, S.; Matsuoka, J.; Hirakawa, T.; Noda, S.; Aomatsu, N.; Hasegawa, T.; Matsuzaki, T.; et al. Effects of acute and chronic hypoxia on the radiosensitivity of gastric and esophageal cancer cells. Anticancer Res. 2011, 31, 3369–3375. [Google Scholar]
- Rockwell, S.; Dobrucki, I.T.; Kim, E.Y.; Marrison, S.T.; Vu, V.T. Hypoxia and radiation therapy: Past history, ongoing research, and future promise. Curr. Mol. Med. 2009, 9, 442–458. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef]
- Chew, Y.C.; Camporeale, G.; Kothapalli, N.; Sarath, G.; Zempleni, J. Lysine residues in N-terminal and C-terminal regions of human histone H2A are targets for biotinylation by biotinidase. J. Nutr. Biochem. 2006, 17, 225–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fullgrabe, J.; Kavanagh, E.; Joseph, B. Histone onco-modifications. Oncogene 2011, 30, 3391–3403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, C.L.; Laniel, M.A. Histones and histone modifications. Curr. Biol. 2004, 14, R546–R551. [Google Scholar] [CrossRef] [Green Version]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [Green Version]
- Thinnes, C.C.; England, K.S.; Kawamura, A.; Chowdhury, R.; Schofield, C.J.; Hopkinson, R.J. Targeting histone lysine demethylases - progress, challenges, and the future. Biochim. Biophys. Acta 2014, 1839, 1416–1432. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y. Histone lysine demethylases: Emerging roles in development, physiology and disease. Nat. Rev. Genet. 2007, 8, 829–833. [Google Scholar] [CrossRef]
- Black, J.C.; Van Rechem, C.; Whetstine, J.R. Histone lysine methylation dynamics: Establishment, regulation, and biological impact. Mol. Cell 2012, 48, 491–507. [Google Scholar] [CrossRef] [Green Version]
- D′Oto, A.; Tian, Q.W.; Davidoff, A.M.; Yang, J. Histone demethylases and their roles in cancer epigenetics. J. Med. Oncol. Ther. 2016, 1, 34–40. [Google Scholar] [CrossRef] [Green Version]
- Tao, H.; Chen, Y.Y.; Sun, Z.W.; Chen, H.L.; Chen, M. Silence of HDAC6 suppressed esophageal squamous cell carcinoma proliferation and migration by disrupting chaperone function of HSP90. J Cell. Biochem. 2018, 119, 6623–6632. [Google Scholar] [CrossRef] [Green Version]
- Toh, Y.; Yamamoto, M.; Endo, K.; Ikeda, Y.; Baba, H.; Kohnoe, S.; Yonemasu, H.; Hachitanda, Y.; Okamura, T.; Sugimachi, K. Histone H4 acetylation and histone deacetylase 1 expression in esophageal squamous cell carcinoma. Oncol. Rep. 2003, 10, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.W.; Zhang, H.; Wang, B.L.; Sun, P.; Wang, Y.G.; Zhang, P. Effect of the downregulation of SMYD3 expression by RNAi on RIZ1 expression and proliferation of esophageal squamous cell carcinoma. Oncol. Rep. 2014, 32, 1064–1070. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.L.; Holowatyj, A.; Xu, X.E.; Wu, J.Y.; Wu, Z.Y.; Shen, J.H.; Wang, S.H.; Li, E.M.; Yang, Z.Q.; Xu, L.Y. Histone demethylase GASC1, a potential prognostic and predictive marker in esophageal squamous cell carcinoma. Am. J. Cancer Res. 2013, 3, 509–517. [Google Scholar] [PubMed]
- Sun, X.; Qiu, J.J.; Zhu, S.; Cao, B.; Sun, L.; Li, S.; Li, P.; Zhang, S.; Dong, S. Oncogenic features of PHF8 histone demethylase in esophageal squamous cell carcinoma. PLoS ONE 2013, 8, e77353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Y.; Fu, X.; Deng, Y. Histone demethylase JMJD1C regulates esophageal cancer proliferation Via YAP1 signaling. Am. J. Cancer Res. 2017, 7, 115–124. [Google Scholar]
- Kosumi, K.; Baba, Y.; Sakamoto, A.; Ishimoto, T.; Harada, K.; Nakamura, K.; Kurashige, J.; Hiyoshi, Y.; Iwatsuki, M.; Iwagami, S.; et al. Lysine-specific demethylase-1 contributes to malignant behavior by regulation of invasive activity and metabolic shift in esophageal cancer. Int. J. Cancer 2016, 138, 428–439. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Wang, B.; Zhang, K.; Lei, Z.; Guo, Y.; Xiao, H.; Wang, J.; Fan, L.; Lan, C.; Wei, Y.; et al. High expression of lysine-specific demethylase 1 correlates with poor prognosis of patients with esophageal squamous cell carcinoma. Biochem. Biophys. Res. Commun. 2013, 437, 192–198. [Google Scholar] [CrossRef] [Green Version]
- Kulis, M.; Esteller, M. DNA methylation and cancer. Adv. Genet. 2010, 70, 27–56. [Google Scholar] [CrossRef]
- Gaur, P.; Hunt, C.R.; Pandita, T.K. Emerging therapeutic targets in esophageal adenocarcinoma. Oncotarget 2016, 7, 48644–48655. [Google Scholar] [CrossRef] [Green Version]
- Kaz, A.M.; Grady, W.M. Epigenetic biomarkers in esophageal cancer. Cancer Lett. 2014, 342, 193–199. [Google Scholar] [CrossRef] [Green Version]
- Perez-Perri, J.I.; Acevedo, J.M.; Wappner, P. Epigenetics: New questions on the response to hypoxia. Int. J. Mol. Sci. 2011, 12, 4705–4721. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.P.; Wu, K.J. Epigenetic regulation of hypoxia-responsive gene expression: Focusing on chromatin and DNA modifications. Int. J. Cancer 2014, 134, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Skowronski, K.; Dubey, S.; Rodenhiser, D.; Coomber, B. Ischemia dysregulates DNA methyltransferases and p16INK4a methylation in human colorectal cancer cells. Epigenetics 2010, 5, 547–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.F.; Wu, K.J. Epigenetics, TET proteins, and hypoxia in epithelial-mesenchymal transition and tumorigenesis. Biomedicine 2016, 6, 1–8. [Google Scholar] [CrossRef]
- Tsai, Y.P.; Chen, H.F.; Chen, S.Y.; Cheng, W.C.; Wang, H.W.; Shen, Z.J.; Song, C.; Teng, S.C.; He, C.; Wu, K.J. TET1 regulates hypoxia-induced epithelial-mesenchymal transition by acting as a co-activator. Genome Biol. 2014, 15, 13. [Google Scholar] [CrossRef]
- Kim, M.S.; Kwon, H.J.; Lee, Y.M.; Baek, J.H.; Jang, J.E.; Lee, S.W.; Moon, E.J.; Kim, H.S.; Lee, S.K.; Chung, H.Y.; et al. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat. Med. 2001, 7, 437–443. [Google Scholar] [CrossRef]
- Vavilala, D.T.; Ponnaluri, V.K.; Vadlapatla, R.K.; Pal, D.; Mitra, A.K.; Mukherji, M. Honokiol inhibits HIF pathway and hypoxia-induced expression of histone lysine demethylases. Biochem. Biophys. Res. Commun. 2012, 422, 369–374. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Hypoxia-Inducible Histone Lysine Demethylases: Impact on the Aging Process and Age-Related Diseases. Aging Dis. 2016, 7, 180–200. [Google Scholar] [CrossRef]
- Hancock, R.L.; Dunne, K.; Walport, L.J.; Flashman, E.; Kawamura, A. Epigenetic regulation by histone demethylases in hypoxia. Epigenomics 2015, 7, 791–811. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, S.; Kitadate, A.; Abe, F.; Takahashi, N.; Tagawa, H. Hypoxia-inducible KDM3A addiction in multiple myeloma. Blood Adv. 2018, 2, 323–334. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, D.; Cui, W.; Rosario, G.X.; Scott, R.L.; Dhakal, P.; Renaud, S.J.; Tachibana, M.; Rumi, M.A.; Mason, C.W.; Krieg, A.J.; et al. HIF-KDM3A-MMP12 regulatory circuit ensures trophoblast plasticity and placental adaptations to hypoxia. Proc. Natl. Acad. Sci. USA 2016, 113, E7212–E7221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osawa, T.; Tsuchida, R.; Muramatsu, M.; Shimamura, T.; Wang, F.; Suehiro, J.; Kanki, Y.; Wada, Y.; Yuasa, Y.; Aburatani, H.; et al. Inhibition of histone demethylase JMJD1A improves anti-angiogenic therapy and reduces tumor-associated macrophages. Cancer Res. 2013, 73, 3019–3028. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Chang, R.; Zhong, J.; Pandey, A.; Semenza, G.L. Histone demethylase JMJD2C is a coactivator for hypoxia-inducible factor 1 that is required for breast cancer progression. Proc. Natl. Acad. Sci. USA 2012, 109, E3367–E3376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, W.L.; Janknecht, R. KDM4/JMJD2 histone demethylases: Epigenetic regulators in cancer cells. Cancer Res. 2013, 73, 2936–2942. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Choi, K.; Oh, H.; Park, Y.K.; Park, H. HIF-1-dependent induction of Jumonji domain-containing protein (JMJD) 3 under hypoxic conditions. Mol. Cells 2014, 37, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Tian, Z.; Wang, X.; Pan, S.; Huang, W.; Shen, Y.; Gui, Y.; Duan, X.; Cai, Z. Regulation of histone demethylase KDM6B by hypoxia-inducible factor-2alpha. Acta Biochim. Biophys. Sin. 2015, 47, 106–113. [Google Scholar] [CrossRef] [Green Version]
- Ahrens, T.D.; Timme, S.; Hoeppner, J.; Ostendorp, J.; Hembach, S.; Follo, M.; Hopt, U.T.; Werner, M.; Busch, H.; Boerries, M.; et al. Selective inhibition of esophageal cancer cells by combination of HDAC inhibitors and Azacytidine. Epigenetics 2015, 10, 431–445. [Google Scholar] [CrossRef] [Green Version]
- Beyer, S.; Kristensen, M.M.; Jensen, K.S.; Johansen, J.V.; Staller, P. The histone demethylases JMJD1A and JMJD2B are transcriptional targets of hypoxia-inducible factor HIF. J. Biol. Chem. 2008, 283, 36542–36552. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Wang, Y.; Tan, L.; Fu, X. The pivotal role of DNA methylation in the radio-sensitivity of tumor radiotherapy. Cancer Med. 2018, 7, 3812–3819. [Google Scholar] [CrossRef]
- Miousse, I.R.; Kutanzi, K.R.; Koturbash, I. Effects of ionizing radiation on DNA methylation: From experimental biology to clinical applications. Int. J. Radiat. Biol. 2017, 93, 457–469. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, J.P.; Sato, F.; Greenwald, B.D.; Suntharalingam, M.; Krasna, M.J.; Edelman, M.J.; Doyle, A.; Berki, A.T.; Abraham, J.M.; Mori, Y.; et al. Promoter methylation and response to chemotherapy and radiation in esophageal cancer. Clin. Gastroenterol. Hepatol. 2006, 4, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Sakakura, C.; Miyagawa, K.; Fukuda, K.I.; Nakashima, S.; Yoshikawa, T.; Kin, S.; Nakase, Y.; Ida, H.; Yazumi, S.; Yamagishi, H.; et al. Frequent silencing of RUNX3 in esophageal squamous cell carcinomas is associated with radioresistance and poor prognosis. Oncogene 2007, 26, 5927–5938. [Google Scholar] [CrossRef] [PubMed]
- Maroschik, B.; Gurtler, A.; Kramer, A.; Rossler, U.; Gomolka, M.; Hornhardt, S.; Mortl, S.; Friedl, A.A. Radiation-induced alterations of histone post-translational modification levels in lymphoblastoid cell lines. Radiat. Oncol. 2014, 9, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Yuan, D.; Guo, F.; Chen, X.; Zhu, L.; Zhang, H.; Wang, C.; Shao, C. Chromatin remodeling modulates radiosensitivity of the daughter cells derived from cell population exposed to low- and high-LET irradiation. Oncotarget 2017, 8, 52823–52836. [Google Scholar] [CrossRef] [Green Version]
- Ahuja, N.; Sharma, A.R.; Baylin, S.B. Epigenetic Therapeutics: A New Weapon in the War Against Cancer. Annu. Rev. Med. 2016, 67, 73–89. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.G.; Park, M.T.; Heo, K.; Yang, K.M.; Yi, J.M. Epigenetics meets radiation biology as a new approach in cancer treatment. Int. J. Mol. Sci. 2013, 14, 15059–15073. [Google Scholar] [CrossRef] [Green Version]
- Subramaniam, D.; Thombre, R.; Dhar, A.; Anant, S. DNA methyltransferases: A novel target for prevention and therapy. Front. Oncol. 2014, 4, 80. [Google Scholar] [CrossRef]
- Oronsky, B.; Scicinski, J.; Kim, M.M.; Cabrales, P.; Salacz, M.E.; Carter, C.A.; Oronsky, N.; Lybeck, H.; Lybeck, M.; Larson, C.; et al. Turning on the Radio: Epigenetic Inhibitors as Potential Radiopriming Agents. Biomolecules 2016, 6. [Google Scholar] [CrossRef]
- Jonsson, M.; Ragnum, H.B.; Julin, C.H.; Yeramian, A.; Clancy, T.; Frikstad, K.M.; Seierstad, T.; Stokke, T.; Matias-Guiu, X.; Ree, A.H.; et al. Hypoxia-independent gene expression signature associated with radiosensitisation of prostate cancer cell lines by histone deacetylase inhibition. Br. J. Cancer 2016, 115, 929–939. [Google Scholar] [CrossRef] [Green Version]
- Makita, N.; Ninomiya, I.; Tsukada, T.; Okamoto, K.; Harada, S.; Nakanuma, S.; Sakai, S.; Makino, I.; Kinoshita, J.; Hayashi, H.; et al. Inhibitory effects of valproic acid in DNA double-strand break repair after irradiation in esophageal squamous carcinoma cells. Oncol. Rep. 2015, 34, 1185–1192. [Google Scholar] [CrossRef] [Green Version]
- Fath, D.M.; Kong, X.; Liang, D.; Lin, Z.; Chou, A.; Jiang, Y.; Fang, J.; Caro, J.; Sang, N. Histone deacetylase inhibitors repress the transactivation potential of hypoxia-inducible factors independently of direct acetylation of HIF-alpha. J. Biol. Chem. 2006, 281, 13612–13619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.Y.; Sang, N.L. Histone Deacetylase Inhibitors: The Epigenetic Therapeutics That Repress Hypoxia-Inducible Factors. J. Biomed. Biotechnol. 2011, 2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, E.D.; Gazdar, A.F. Inhibiting the Jumonji family: A potential new clinical approach to targeting aberrant epigenetic mechanisms. Epigenomics 2016, 8, 313–316. [Google Scholar] [CrossRef] [PubMed]
- Banelli, B.; Daga, A.; Forlani, A.; Allemanni, G.; Marubbi, D.; Pistillo, M.P.; Profumo, A.; Romani, M. Small molecules targeting histone demethylase genes (KDMs) inhibit growth of temozolomide-resistant glioblastoma cells. Oncotarget 2017, 8, 34896–34910. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.K.; Bonaldi, T.; Cuomo, A.; Del Rosario, J.R.; Hosfield, D.J.; Kanouni, T.; Kao, S.C.; Lai, C.; Lobo, N.A.; Matuszkiewicz, J.; et al. Design of KDM4 Inhibitors with Antiproliferative Effects in Cancer Models. ACS Med. Chem. Lett. 2017, 8, 869–874. [Google Scholar] [CrossRef]
- Maes, T.; Carceller, E.; Salas, J.; Ortega, A.; Buesa, C. Advances in the development of histone lysine demethylase inhibitors. Curr. Opin. Pharmacol. 2015, 23, 52–60. [Google Scholar] [CrossRef]
- Bayo, J.; Tran, T.A.; Wang, L.; Pena-Llopis, S.; Das, A.K.; Martinez, E.D. Jumonji Inhibitors Overcome Radioresistance in Cancer through Changes in H3K4 Methylation at Double-Strand Breaks. Cell Rep. 2018, 25, 1040–1050. [Google Scholar] [CrossRef] [Green Version]
- Rath, B.H.; Waung, I.; Camphausen, K.; Tofilon, P.J. Inhibition of the Histone H3K27 Demethylase UTX Enhances Tumor Cell Radiosensitivity. Mol. Cancer Ther. 2018, 17, 1070–1078. [Google Scholar] [CrossRef] [Green Version]
- Pippa, S.; Mannironi, C.; Licursi, V.; Bombardi, L.; Colotti, G.; Cundari, E.; Mollica, A.; Coluccia, A.; Naccarato, V.; La Regina, G. Small Molecule Inhibitors of KDM5 Histone Demethylases Increase Radio-Sensitivity of Breast Cancer Cells Over-Expressing JARID1B. Molecules 2019, 24, 1739. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Epigenetic Drug | Target Function | Role in EC Treatment (Yes/No) | RT Response Improvement (Yes/No) | Study with Hypoxia Linked (Yes/No) | Other Models | References |
|---|---|---|---|---|---|---|
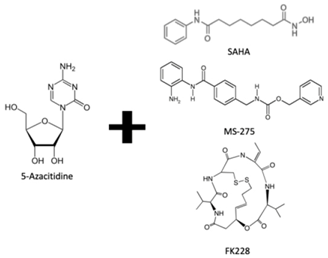 | DNMTi combined with HDACi | Yes | Yes | No | - | [78] |
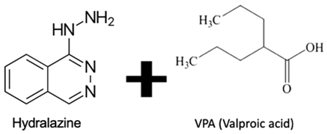 | DNMTi combined with HDACi | No | Yes | No | Cervical cancer | [89] |
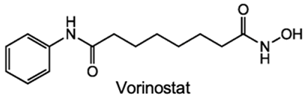 | HDACi | No | Yes | Yes | Prostate cancer | [90] |
 | HDACi | Yes | Yes | No | - | [91] |
 | HDACi | Yes | Yes | No | Prostate cancer and head and neck tumors | [90] |
 | HDACi | No | No | Yes | - | [92] |
 | KDM4 inhibitors | Yes | No | No | - | [96] |
 | LSD inhibitors | No | No | No | Acute myeloid leukemia (AML) and small cell lung carcinoma (SCLC) | [97] |
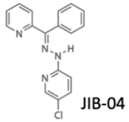 | KDM5B inhibition (JmjC-KDM PAN inhibitor) | No | Yes | No | Lung cancer | [98] |
| EPT103182 | KDM5 subfamily inhibitor | No | No | No | Multiple myeloma | [97] |
 | KDM5 subfamily inhibitors | No | Yes | No | Breast cancer | [100] |
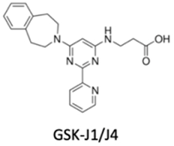 | KDM6 subfamily inhibitor | No | Yes | No | Glioblastoma, breast and lung adenocarcinoma | [99] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Macedo-Silva, C.; Miranda-Gonçalves, V.; Henrique, R.; Jerónimo, C.; Bravo, I. The Critical Role of Hypoxic Microenvironment and Epigenetic Deregulation in Esophageal Cancer Radioresistance. Genes 2019, 10, 927. https://doi.org/10.3390/genes10110927
Macedo-Silva C, Miranda-Gonçalves V, Henrique R, Jerónimo C, Bravo I. The Critical Role of Hypoxic Microenvironment and Epigenetic Deregulation in Esophageal Cancer Radioresistance. Genes. 2019; 10(11):927. https://doi.org/10.3390/genes10110927
Chicago/Turabian StyleMacedo-Silva, Catarina, Vera Miranda-Gonçalves, Rui Henrique, Carmen Jerónimo, and Isabel Bravo. 2019. "The Critical Role of Hypoxic Microenvironment and Epigenetic Deregulation in Esophageal Cancer Radioresistance" Genes 10, no. 11: 927. https://doi.org/10.3390/genes10110927