Alternative Lengthening of Telomeres (ALT) in Tumors and Pluripotent Stem Cells

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Telomere Function in Maintaining Genomic Stability
3. Telomere Maintenance by Telomerase or ALT
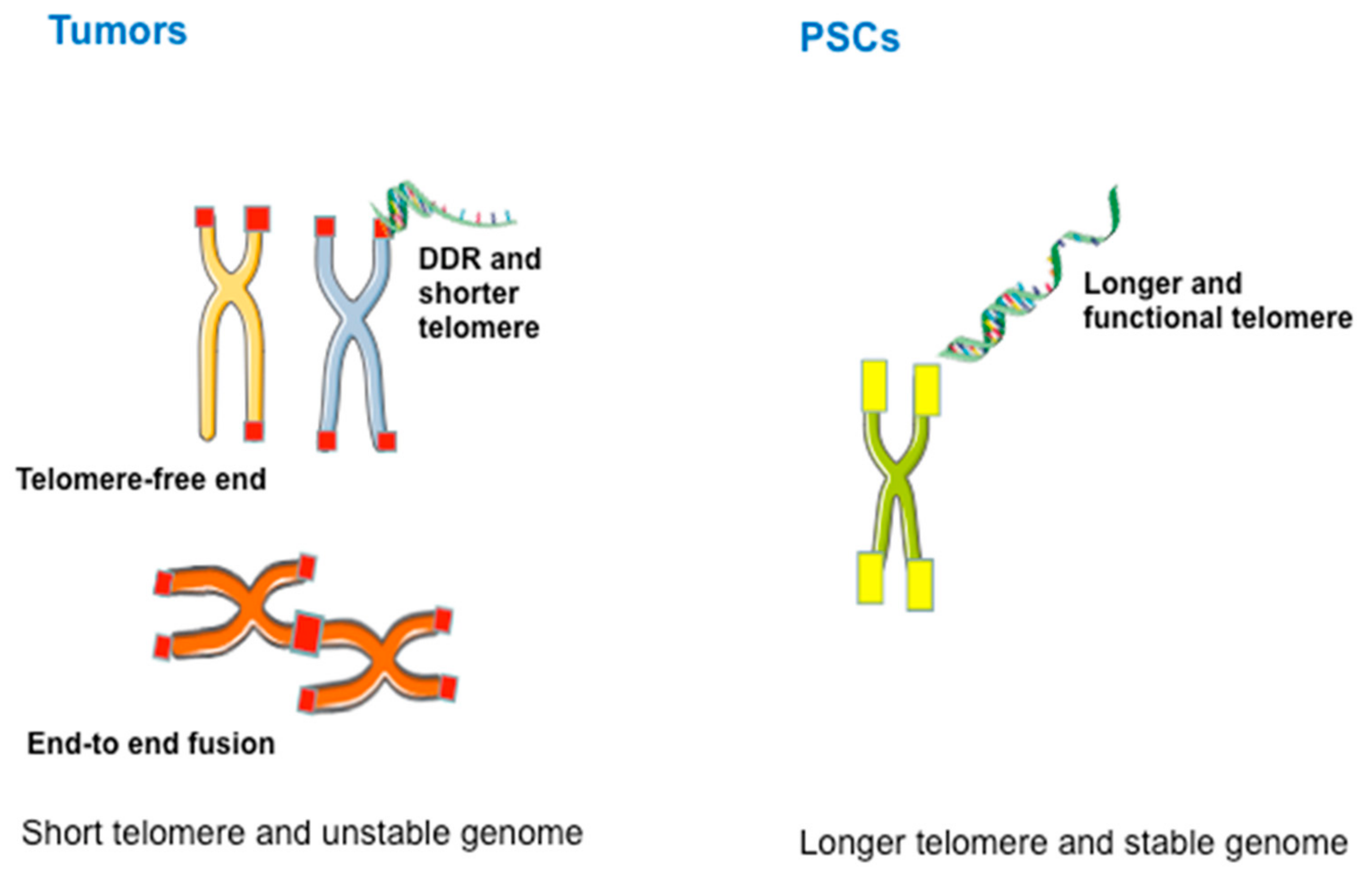
4. Characteristics and Diagnosis of ALT
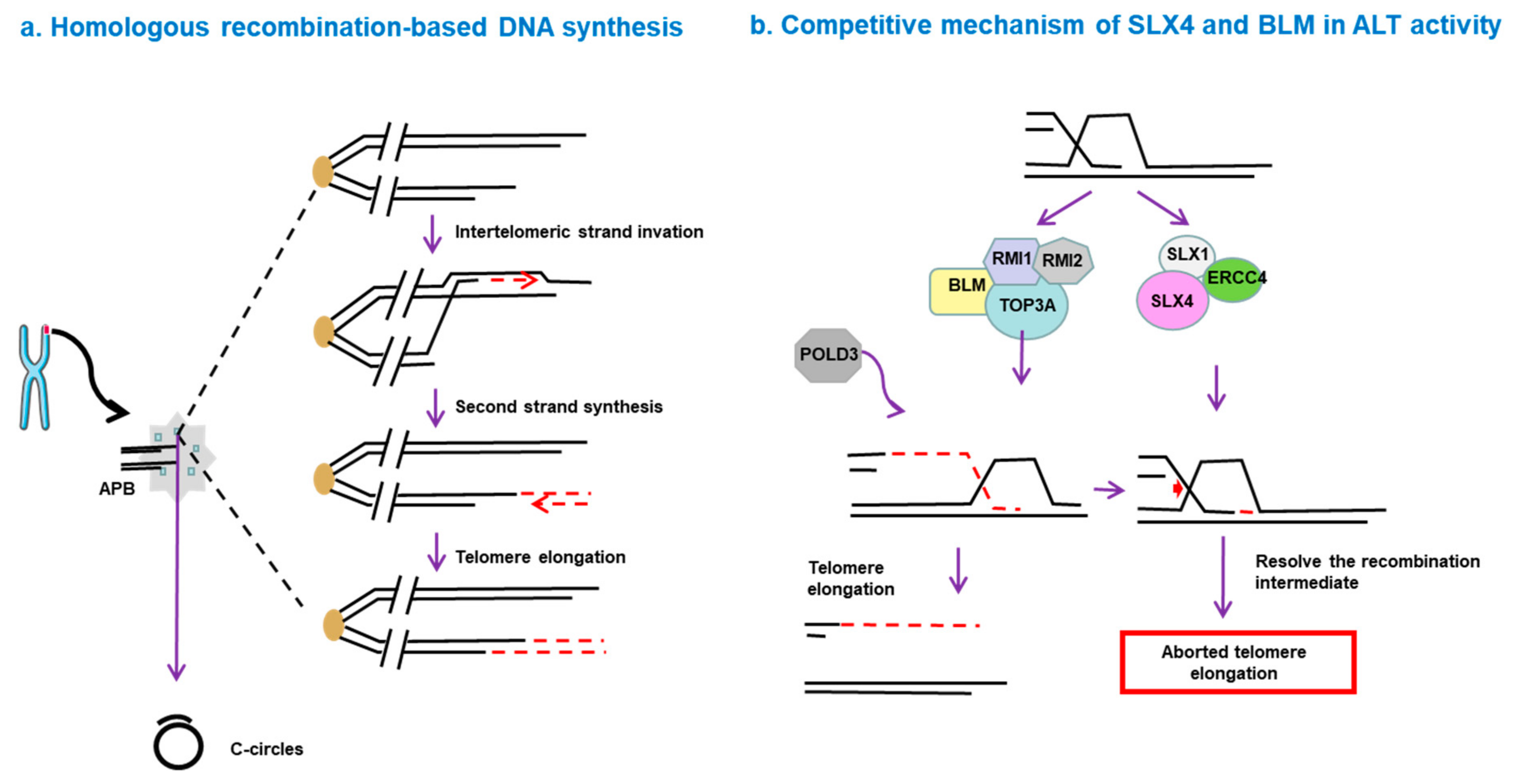
5. How Is ALT Activated?
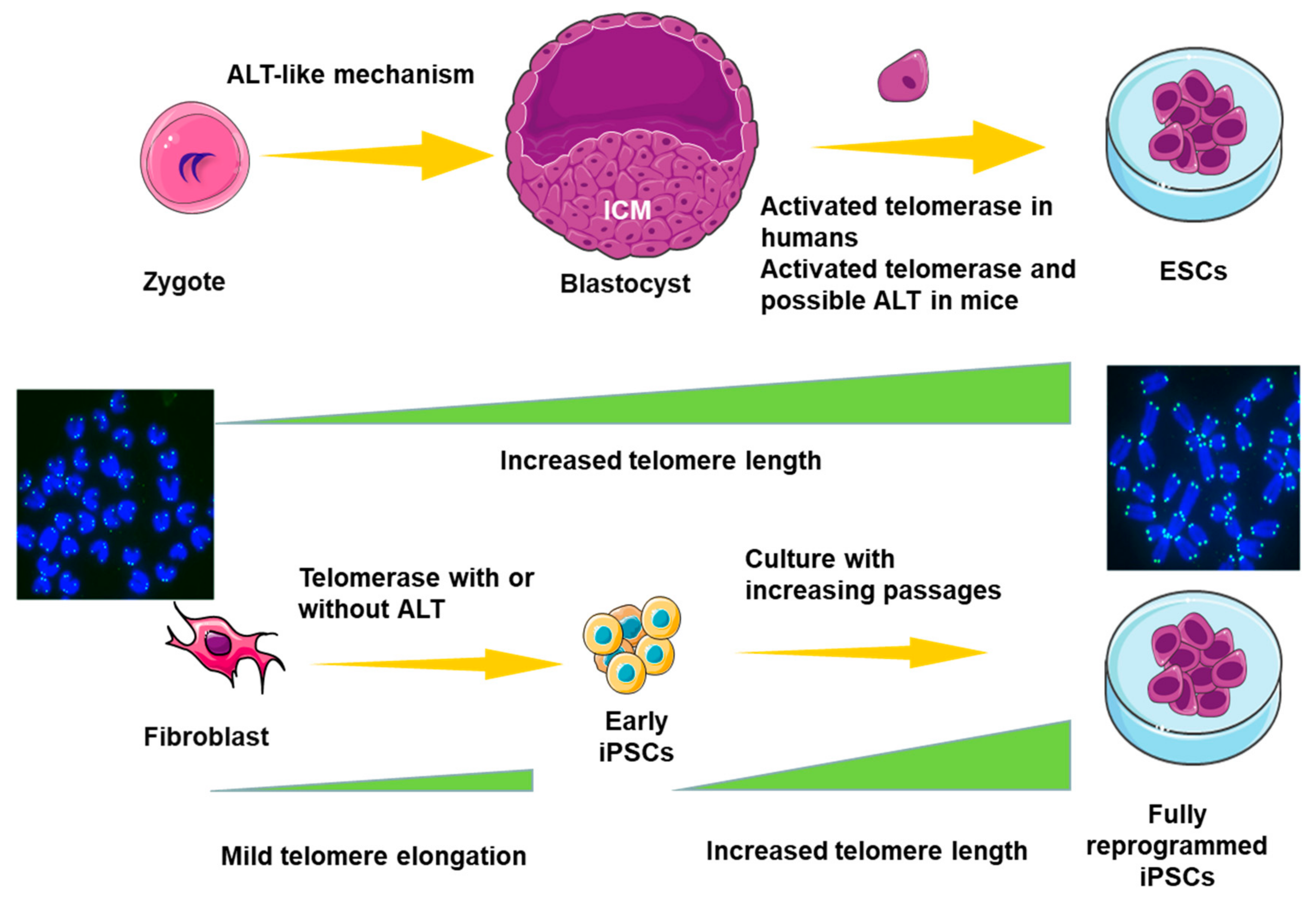
6. ALT in Pluripotent Stem Cells (PSCs)
7. Potential Therapeutic Strategy Targeting ALT
8. Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Makarov, V.L.; Hirose, Y.; Langmore, J.P. Long G tails at both ends of human chromosomes suggest a C strand degradation mechanism for telomere shortening. Cell 1997, 88, 657–666. [Google Scholar] [CrossRef]
- Wright, W.E.; Tesmer, V.M.; Huffman, K.E.; Levene, S.D.; Shay, J.W. Normal human chromosomes have long G-rich telomeric overhangs at one end. Genes Dev. 1997, 11, 2801–2809. [Google Scholar] [CrossRef] [PubMed]
- Sfeir, A.; de Lange, T. Removal of shelterin reveals the telomere end-protection problem. Science 2012, 336, 593–597. [Google Scholar] [CrossRef] [PubMed]
- Griffith, J.D.; Comeau, L.; Rosenfield, S.; Stansel, R.M.; Bianchi, A.; Moss, H.; de Lange, T. Mammalian telomeres end in a large duplex loop. Cell 1999, 97, 503–514. [Google Scholar] [CrossRef]
- De Lange, T. Shelterin-Mediated Telomere Protection. Annu. Rev. Genet. 2018, 52, 223–247. [Google Scholar] [CrossRef]
- Hayashi, M.T. Telomere biology in aging and cancer: Early history and perspectives. Genes Genet. Syst. 2018, 92, 107–118. [Google Scholar] [CrossRef]
- Jafri, M.A.; Ansari, S.A.; Alqahtani, M.H.; Shay, J.W. Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome Med. 2016, 8, 69. [Google Scholar] [CrossRef]
- Olovnikov, A.M. A theory of marginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J. Theor. Biol. 1973, 41, 181–190. [Google Scholar] [CrossRef]
- Levy, M.Z.; Allsopp, R.C.; Futcher, A.B.; Greider, C.W.; Harley, C.B. Telomere end-replication problem and cell aging. J. Mol. Biol. 1992, 225, 951–960. [Google Scholar] [CrossRef]
- Hewitt, G.; Jurk, D.; Marques, F.D.; Correia-Melo, C.; Hardy, T.; Gackowska, A.; Anderson, R.; Taschuk, M.; Mann, J.; Passos, J.F. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 2012, 3, 708. [Google Scholar] [CrossRef]
- Anderson, B.H.; Kasher, P.R.; Mayer, J.; Szynkiewicz, M.; Jenkinson, E.M.; Bhaskar, S.S.; Urquhart, J.E.; Daly, S.B.; Dickerson, J.E.; O’Sullivan, J.; et al. Mutations in CTC1, encoding conserved telomere maintenance component 1, cause Coats plus. Nat. Genet. 2012, 44, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Simon, A.J.; Lev, A.; Zhang, Y.; Weiss, B.; Rylova, A.; Eyal, E.; Kol, N.; Barel, O.; Cesarkas, K.; Soudack, M.; et al. Mutations in STN1 cause Coats plus syndrome and are associated with genomic and telomere defects. J. Exp. Med. 2016, 213, 1429–1440. [Google Scholar] [CrossRef] [PubMed]
- Larizza, L.; Magnani, I.; Roversi, G. Rothmund-Thomson syndrome and RECQL4 defect: Splitting and lumping. Cancer Lett. 2006, 232, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Benarroch-Popivker, D.; Pisano, S.; Mendez-Bermudez, A.; Lototska, L.; Kaur, P.; Bauwens, S.; Djerbi, N.; Latrick, C.M.; Fraisier, V.; Pei, B.; et al. TRF2-Mediated Control of Telomere DNA Topology as a Mechanism for Chromosome-End Protection. Mol. Cell 2016, 61, 274–286. [Google Scholar] [CrossRef]
- Gong, Y.; de Lange, T. A Shld1-controlled POT1a provides support for repression of ATR signaling at telomeres through RPA exclusion. Mol. Cell 2010, 40, 377–387. [Google Scholar] [CrossRef]
- Munoz-Lorente, M.A.; Cano-Martin, A.C.; Blasco, M.A. Mice with hyper-long telomeres show less metabolic aging and longer lifespans. Nat. Commun. 2019, 10, 4723. [Google Scholar] [CrossRef]
- Perez-Romero, C.A.; Lalonde, M.; Chartrand, P.; Cusanelli, E. Induction and relocalization of telomeric repeat-containing RNAs during diauxic shift in budding yeast. Curr. Genet. 2018, 64, 1117–1127. [Google Scholar] [CrossRef]
- Deng, Z.; Norseen, J.; Wiedmer, A.; Riethman, H.; Lieberman, P.M. TERRA RNA binding to TRF2 facilitates heterochromatin formation and ORC recruitment at telomeres. Mol. Cell 2009, 35, 403–415. [Google Scholar] [CrossRef]
- Sarek, G.; Vannier, J.B.; Panier, S.; Petrini, J.H.J.; Boulton, S.J. TRF2 recruits RTEL1 to telomeres in S phase to promote t-loop unwinding. Mol. Cell 2015, 57, 622–635. [Google Scholar] [CrossRef]
- O’Sullivan, R.J.; Karlseder, J. Telomeres: Protecting chromosomes against genome instability. Nat. Rev. Mol. Cell Biol. 2010, 11, 171–181. [Google Scholar] [CrossRef]
- Schaffitzel, C.; Berger, I.; Postberg, J.; Hanes, J.; Lipps, H.J.; Pluckthun, A. In vitro generated antibodies specific for telomeric guanine-quadruplex DNA react with Stylonychia lemnae macronuclei. Proc. Natl. Acad. Sci. USA 2001, 98, 8572–8577. [Google Scholar] [CrossRef] [PubMed]
- Paeschke, K.; Simonsson, T.; Postberg, J.; Rhodes, D.; Lipps, H.J. Telomere end-binding proteins control the formation of G-quadruplex DNA structures in vivo. Nat. Struct. Mol. Biol. 2005, 12, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Schertzer, M.; Wu, X.; Gertsenstein, M.; Selig, S.; Kammori, M.; Pourvali, R.; Poon, S.; Vulto, I.; Chavez, E.; et al. Regulation of murine telomere length by Rtel: An essential gene encoding a helicase-like protein. Cell 2004, 117, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Sampathi, S.; Dai, H.; Liu, C.; Zhou, M.; Hu, J.; Huang, Q.; Campbell, J.; Shin-Ya, K.; Zheng, L.; et al. Mammalian DNA2 helicase/nuclease cleaves G-quadruplex DNA and is required for telomere integrity. EMBO J. 2013, 32, 1425–1439. [Google Scholar] [CrossRef] [PubMed]
- Paeschke, K.; Bochman, M.L.; Garcia, P.D.; Cejka, P.; Friedman, K.L.; Kowalczykowski, S.C.; Zakian, V.A. Pif1 family helicases suppress genome instability at G-quadruplex motifs. Nature 2013, 497, 458–462. [Google Scholar] [CrossRef]
- Bodnar, A.G.; Ouellette, M.; Frolkis, M.; Holt, S.E.; Chiu, C.P.; Morin, G.B.; Harley, C.B.; Shay, J.W.; Lichtsteiner, S.; Wright, W.E. Extension of life-span by introduction of telomerase into normal human cells. Science 1998, 279, 349–352. [Google Scholar] [CrossRef]
- Heaphy, C.M.; de Wilde, R.F.; Jiao, Y.; Klein, A.P.; Edil, B.H.; Shi, C.; Bettegowda, C.; Rodriguez, F.J.; Eberhart, C.G.; Hebbar, S.; et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science 2011, 333, 425. [Google Scholar] [CrossRef]
- Cerone, M.A.; Londono-Vallejo, J.A.; Bacchetti, S. Telomere maintenance by telomerase and by recombination can coexist in human cells. Hum. Mol. Genet. 2001, 10, 1945–1952. [Google Scholar] [CrossRef]
- Xin, H.; Liu, D.; Wan, M.; Safari, A.; Kim, H.; Sun, W.; O’Connor, M.S.; Songyang, Z. TPP1 is a homologue of ciliate TEBP-beta and interacts with POT1 to recruit telomerase. Nature 2007, 445, 559–562. [Google Scholar] [CrossRef]
- Gavory, G.; Farrow, M.; Balasubramanian, S. Minimum length requirement of the alignment domain of human telomerase RNA to sustain catalytic activity in vitro. Nucleic Acids Res. 2002, 30, 4470–4480. [Google Scholar] [CrossRef]
- Greider, C.W.; Blackburn, E.H. A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature 1989, 337, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Lue, N.F.; Chan, J.; Wright, W.E.; Hurwitz, J. The CDC13-STN1-TEN1 complex stimulates Pol alpha activity by promoting RNA priming and primase-to-polymerase switch. Nat. Commun. 2014, 5, 5762. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Stewart, J.A.; Kasbek, C.; Zhao, Y.; Wright, W.E.; Price, C.M. Human CST has independent functions during telomere duplex replication and C-strand fill-in. Cell Rep. 2012, 2, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, R.T.; Veronese, N.; Pea, A.; Nottegar, A.; Smith, L.; Pilati, C.; Demurtas, J.; Fassan, M.; Cheng, L.; Luchini, C. Alternative lengthening of telomeres (ALT) influences survival in soft tissue sarcomas: A systematic review with meta-analysis. BMC Cancer 2019, 19, 232–238. [Google Scholar] [CrossRef]
- Henson, J.D.; Hannay, J.A.; McCarthy, S.W.; Royds, J.A.; Yeager, T.R.; Robinson, R.A.; Wharton, S.B.; Jellinek, D.A.; Arbuckle, S.M.; Yoo, J.; et al. A robust assay for alternative lengthening of telomeres in tumors shows the significance of alternative lengthening of telomeres in sarcomas and astrocytomas. Clin. Cancer Res. 2005, 11, 217–225. [Google Scholar]
- Zhou, J.; Reddy, M.V. MR Imaging Characteristics Associate with Tumor-Associated Macrophages in Glioblastoma and Provide an Improved Signature for Survival Prognostication. AJNR. Am. J. Neuroradiol. 2018, 39, 252–259. [Google Scholar] [CrossRef]
- Pompili, L.; Leonetti, C.; Biroccio, A.; Salvati, E. Diagnosis and treatment of ALT tumors: Is Trabectedin a new therapeutic option? J. Exp. Clin. Cancer Res. 2017, 36, 189–198. [Google Scholar] [CrossRef]
- Henson, J.D.; Reddel, R.R. Assaying and investigating alternative lengthening of telomeres activity in human cells and cancers. FEBS Lett. 2010, 584, 3800–3811. [Google Scholar] [CrossRef]
- Heaphy, C.M.; Subhawong, A.P.; Hong, S.M.; Goggins, M.G.; Montgomery, E.A.; Gabrielson, E.; Netto, G.J.; Epstein, J.I.; Lotan, T.L.; Westra, W.H.; et al. Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am. J. Pathol. 2011, 179, 1608–1615. [Google Scholar] [CrossRef]
- Xue, Y.; Li, L.; Zhang, D.; Wu, K.; Chen, Y.; Zeng, J.; Wang, X.; He, D. Twisted epithelial-to-mesenchymal transition promotes progression of surviving bladder cancer T24 cells with hTERT-dysfunction. PLoS ONE 2011, 6, e27748. [Google Scholar] [CrossRef]
- Perrem, K.; Bryan, T.M.; Englezou, A.; Hackl, T.; Moy, E.L.; Reddel, R.R. Repression of an alternative mechanism for lengthening of telomeres in somatic cell hybrids. Oncogene 1999, 18, 3383–3390. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Hwang, S.S.; Liesa, M.; Gan, B.; Sahin, E.; Jaskelioff, M.; Ding, Z.; Ying, H.; Boutin, A.T.; Zhang, H.; et al. Antitelomerase therapy provokes ALT and mitochondrial adaptive mechanisms in cancer. Cell 2012, 148, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Khoo, C.M.; Naylor, M.L.; Maser, R.S.; DePinho, R.A. Telomere-based crisis: Functional differences between telomerase activation and ALT in tumor progression. Genes Dev. 2003, 17, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Pickett, H.A.; Reddel, R.R. Molecular mechanisms of activity and derepression of alternative lengthening of telomeres. Nat. Struct. Mol. Biol. 2015, 22, 875–880. [Google Scholar] [CrossRef]
- Episkopou, H.; Draskovic, I.; Van Beneden, A.; Tilman, G.; Mattiussi, M.; Gobin, M.; Arnoult, N.; Londono-Vallejo, A.; Decottignies, A. Alternative Lengthening of Telomeres is characterized by reduced compaction of telomeric chromatin. Nucleic Acids Res. 2014, 42, 4391–4405. [Google Scholar] [CrossRef]
- Gibbons, R.J.; McDowell, T.L.; Raman, S.; O’Rourke, D.M.; Garrick, D.; Ayyub, H.; Higgs, D.R. Mutations in ATRX, encoding a SWI/SNF-like protein, cause diverse changes in the pattern of DNA methylation. Nat. Genet. 2000, 24, 368–371. [Google Scholar] [CrossRef]
- Nan, X.; Hou, J.; Maclean, A.; Nasir, J.; Lafuente, M.J.; Shu, X.; Kriaucionis, S.; Bird, A. Interaction between chromatin proteins MECP2 and ATRX is disrupted by mutations that cause inherited mental retardation. Proc. Natl. Acad. Sci. USA 2007, 104, 2709–2714. [Google Scholar] [CrossRef]
- Watson, L.A.; Solomon, L.A.; Li, J.R.; Jiang, Y.; Edwards, M.; Shin-ya, K.; Beier, F.; Berube, N.G. Atrx deficiency induces telomere dysfunction, endocrine defects, and reduced life span. J. Clin. Investig. 2013, 123, 2049–2063. [Google Scholar] [CrossRef]
- Li, F.; Deng, Z.; Zhang, L.; Wu, C.; Jin, Y.; Hwang, I.; Vladimirova, O.; Xu, L.; Yang, L.; Lu, B.; et al. ATRX loss induces telomere dysfunction and necessitates induction of alternative lengthening of telomeres during human cell immortalization. EMBO J. 2019, 38, e96659. [Google Scholar] [CrossRef]
- Cesare, A.J.; Reddel, R.R. Alternative lengthening of telomeres: Models, mechanisms and implications. Nat. Rev. Genet. 2010, 11, 319–330. [Google Scholar] [CrossRef]
- Bryan, T.M.; Englezou, A.; Gupta, J.; Bacchetti, S.; Reddel, R.R. Telomere elongation in immortal human cells without detectable telomerase activity. EMBO J. 1995, 14, 4240–4248. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Lan, J.; Wang, C.; Wu, Q.; Zhu, Y.; Lai, X.; Sun, J.; Jin, C.; Huang, H. PML3 interacts with TRF1 and is essential for ALT-associated PML bodies assembly in U2OS cells. Cancer Lett. 2010, 291, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Amorim, J.P.; Santos, G.; Vinagre, J.; Soares, P. The Role of ATRX in the Alternative Lengthening of Telomeres (ALT) Phenotype. Genes 2016, 7, 66. [Google Scholar] [CrossRef] [PubMed]
- Yeager, T.R.; Neumann, A.A.; Englezou, A.; Huschtscha, L.I.; Noble, J.R.; Reddel, R.R. Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res. 1999, 59, 4175–4179. [Google Scholar] [PubMed]
- Osterwald, S.; Deeg, K.I.; Chung, I.; Parisotto, D.; Worz, S.; Rohr, K.; Erfle, H.; Rippe, K. PML induces compaction, TRF2 depletion and DNA damage signaling at telomeres and promotes their alternative lengthening. J. Cell Sci. 2015, 128, 1887–1900. [Google Scholar] [CrossRef]
- Henson, J.D.; Cao, Y.; Huschtscha, L.I.; Chang, A.C.; Au, A.Y.; Pickett, H.A.; Reddel, R.R. DNA C-circles are specific and quantifiable markers of alternative-lengthening-of-telomeres activity. Nat. Biotechnol. 2009, 27, 1181–1185. [Google Scholar] [CrossRef]
- Cesare, A.J.; Griffith, J.D. Telomeric DNA in ALT cells is characterized by free telomeric circles and heterogeneous t-loops. Mol. Cell Biol. 2004, 24, 9948–9957. [Google Scholar] [CrossRef]
- Cesare, A.J.; Kaul, Z.; Cohen, S.B.; Napier, C.E.; Pickett, H.A.; Neumann, A.A.; Reddel, R.R. Spontaneous occurrence of telomeric DNA damage response in the absence of chromosome fusions. Nat. Struct. Mol. Biol. 2009, 16, 1244–1251. [Google Scholar] [CrossRef]
- Mender, I.; Shay, J.W. Telomere Dysfunction Induced Foci (TIF) Analysis. Bio-Protocol 2015, 5, e1656. [Google Scholar] [CrossRef]
- Dilley, R.L.; Verma, P.; Cho, N.W.; Winters, H.D.; Wondisford, A.R.; Greenberg, R.A. Break-induced telomere synthesis underlies alternative telomere maintenance. Nature 2016, 539, 54–58. [Google Scholar] [CrossRef]
- Henson, J.D.; Neumann, A.A.; Yeager, T.R.; Reddel, R.R. Alternative lengthening of telomeres in mammalian cells. Oncogene 2002, 21, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Cesare, A.J.; Heaphy, C.M.; O’Sullivan, R.J. Visualization of Telomere Integrity and Function In Vitro and In Vivo Using Immunofluorescence Techniques. Curr. Protoc. Cytom. 2015, 73, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.M.; Dagg, R.A.; Henson, J.D.; Au, A.Y.; Royds, J.A.; Reddel, R.R. Detection of alternative lengthening of telomeres by telomere quantitative PCR. Nucleic Acids Res. 2013, 41, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Roumelioti, F.M.; Sotiriou, S.K.; Katsini, V.; Chiourea, M.; Halazonetis, T.D. Alternative lengthening of human telomeres is a conservative DNA replication process with features of break-induced replication. EMBO Rep. 2016, 17, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Cox, K.E.; Marechal, A.; Flynn, R.L. SMARCAL1 Resolves Replication Stress at ALT Telomeres. Cell Rep. 2016, 14, 1032–1040. [Google Scholar] [CrossRef]
- Agathangelou, K.; Apostolou, Z.; Garinis, G.A. Nuclear DNA Damage and Ageing. Subcell Biochem. 2018, 90, 309–322. [Google Scholar] [CrossRef]
- Fallet, E.; Jolivet, P.; Soudet, J.; Lisby, M.; Gilson, E.; Teixeira, M.T. Length-dependent processing of telomeres in the absence of telomerase. Nucleic Acids Res. 2014, 42, 3648–3665. [Google Scholar] [CrossRef]
- Tacconi, E.M.; Tarsounas, M. How homologous recombination maintains telomere integrity. Chromosoma 2015, 124, 119–130. [Google Scholar] [CrossRef]
- Liu, H.; Xie, Y.; Zhang, Z.; Mao, P.; Liu, J.; Ma, W.; Zhao, Y. Telomeric Recombination Induced by DNA Damage Results in Telomere Extension and Length Heterogeneity. Neoplasia 2018, 20, 905–916. [Google Scholar] [CrossRef]
- Zhang, J.M.; Yadav, T.; Ouyang, J.; Lan, L.; Zou, L. Alternative Lengthening of Telomeres through Two Distinct Break-Induced Replication Pathways. Cell Rep. 2019, 26, 955–968. [Google Scholar] [CrossRef]
- Verma, P.; Dilley, R.L.; Zhang, T.; Gyparaki, M.T.; Li, Y.; Greenberg, R.A. RAD52 and SLX4 act nonepistatically to ensure telomere stability during alternative telomere lengthening. Genes Dev. 2019, 33, 221–235. [Google Scholar] [CrossRef] [PubMed]
- Benson, F.E.; Baumann, P.; West, S.C. Synergistic actions of Rad51 and Rad52 in recombination and DNA repair. Nature 1998, 391, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, R.; Minocherhomji, S.; Hickson, I.D. RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Mol. Cell 2016, 64, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Makarova, A.V.; Stodola, J.L.; Burgers, P.M. A four-subunit DNA polymerase ζ complex containing Pol δ accessory subunits is essential for PCNA-mediated mutagenesis. Nucleic Acids Res. 2012, 40, 11618–11626. [Google Scholar] [CrossRef]
- Sobinoff, A.P.; Allen, J.A.; Neumann, A.A.; Yang, S.F.; Walsh, M.E.; Henson, J.D.; Reddel, R.R. BLM and SLX4 play opposing roles in recombination-dependent replication at human telomeres. EMBO J. 2017, 36, 2907–2919. [Google Scholar] [CrossRef]
- Teichroeb, J.H.; Kim, J.; Betts, D.H. The role of telomeres and telomerase reverse transcriptase isoforms in pluripotency induction and maintenance. RNA Biol. 2016, 13, 707–719. [Google Scholar] [CrossRef]
- Huang, Y.; Liang, P.; Liu, D.; Huang, J.; Songyang, Z. Telomere regulation in pluripotent stem cells. Protein Cell 2014, 5, 194–202. [Google Scholar] [CrossRef]
- Flores, I.; Canela, A.; Vera, E.; Tejera, A.; Cotsarelis, G.; Blasco, M.A. The longest telomeres: A general signature of adult stem cell compartments. Genes Dev. 2008, 22, 654–667. [Google Scholar] [CrossRef]
- Liu, L. Linking Telomere Regulation to Stem Cell Pluripotency. Trends Genet. 2017, 33, 16–33. [Google Scholar] [CrossRef]
- Pickett, H.A.; Cesare, A.J.; Johnston, R.L.; Neumann, A.A.; Reddel, R.R. Control of telomere length by a trimming mechanism that involves generation of t-circles. EMBO J. 2009, 28, 799–809. [Google Scholar] [CrossRef]
- Guo, R.; Ye, X.; Yang, J.; Zhou, Z.; Tian, C.; Wang, H.; Wang, H.; Fu, H.; Liu, C.; Zeng, M.; et al. Feeders facilitate telomere maintenance and chromosomal stability of embryonic stem cells. Nat. Commun. 2018, 9, 2620–2635. [Google Scholar] [CrossRef] [PubMed]
- Zalzman, M.; Falco, G.; Sharova, L.V.; Nishiyama, A.; Thomas, M.; Lee, S.L.; Stagg, C.A.; Hoang, H.G.; Yang, H.T.; Indig, F.E.; et al. Zscan4 regulates telomere elongation and genomic stability in ES cells. Nature 2010, 464, 858–863. [Google Scholar] [CrossRef] [PubMed]
- Nakai-Futatsugi, Y.; Niwa, H. Zscan4 Is Activated after Telomere Shortening in Mouse Embryonic Stem Cells. Stem Cell Rep. 2016, 6, 483–495. [Google Scholar] [CrossRef]
- Dan, J.; Liu, Y.; Liu, N.; Chiourea, M.; Okuka, M.; Wu, T.; Ye, X.; Mou, C.; Wang, L.; Wang, L.; et al. Rif1 maintains telomere length homeostasis of ESCs by mediating heterochromatin silencing. Dev. Cell 2014, 29, 7–19. [Google Scholar] [CrossRef]
- Shi, T.; Bunker, R.D.; Mattarocci, S.; Ribeyre, C.; Faty, M.; Gut, H.; Scrima, A.; Rass, U.; Rubin, S.M.; Shore, D.; et al. Rif1 and Rif2 shape telomere function and architecture through multivalent Rap1 interactions. Cell 2013, 153, 1340–1353. [Google Scholar] [CrossRef] [PubMed]
- Buonomo, S.B.; Wu, Y.; Ferguson, D.; de Lange, T. Mammalian Rif1 contributes to replication stress survival and homology-directed repair. J. Cell Biol. 2009, 187, 385–398. [Google Scholar] [CrossRef]
- Chapman, J.R.; Barral, P.; Vannier, J.B.; Borel, V.; Steger, M.; Tomas-Loba, A.; Sartori, A.A.; Adams, I.R.; Batista, F.D.; Boulton, S.J. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol. Cell 2013, 49, 858–871. [Google Scholar] [CrossRef]
- Zeng, S.; Liu, L.; Sun, Y.; Xie, P.; Hu, L.; Yuan, D.; Chen, D.; Ouyang, Q.; Lin, G.; Lu, G. Telomerase-mediated telomere elongation from human blastocysts to embryonic stem cells. J. Cell Sci. 2014, 127, 752–762. [Google Scholar] [CrossRef]
- Huang, J.; Wang, F.; Okuka, M.; Liu, N.; Ji, G.; Ye, X.; Zuo, B.; Li, M.; Liang, P.; Ge, W.W.; et al. Association of telomere length with authentic pluripotency of ES/iPS cells. Cell Res. 2011, 21, 779–792. [Google Scholar] [CrossRef]
- Liu, L.; Bailey, S.M.; Okuka, M.; Munoz, P.; Li, C.; Zhou, L.; Wu, C.; Czerwiec, E.; Sandler, L.; Seyfang, A.; et al. Telomere lengthening early in development. Nat. Cell Biol. 2007, 9, 1436–1441. [Google Scholar] [CrossRef]
- Varela, E.; Schneider, R.P.; Ortega, S.; Blasco, M.A. Different telomere-length dynamics at the inner cell mass versus established embryonic stem (ES) cells. Proc. Natl. Acad. Sci. USA 2011, 108, 15207–15212. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Yin, Y.; Ye, X.; Liu, K.; Zhu, H.; Wang, L.; Chiourea, M.; Okuka, M.; Ji, G.; Dan, J.; et al. Molecular insights into the heterogeneity of telomere reprogramming in induced pluripotent stem cells. Cell Res. 2012, 22, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Lv, W.; Ye, X.; Wang, L.; Zhang, M.; Yang, H.; Okuka, M.; Zhou, C.; Zhang, X.; Liu, L.; et al. Zscan4 promotes genomic stability during reprogramming and dramatically improves the quality of iPS cells as demonstrated by tetraploid complementation. Cell Res. 2013, 23, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Hwang, I.; Choi, K.A.; Jeong, H.; Lee, J.Y.; Hong, S. Generation of induced pluripotent stem cells without genetic defects by small molecules. Biomaterials 2015, 39, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Tian, C.-L.; Ye, X.; Sheng, X.; Wang, H.; Liu, Y.; Liu, L. Dynamics of Telomere Rejuvenation during Chemical Induction to Pluripotent Stem Cells. Stem Cell Rep. 2018, 11, 70–87. [Google Scholar] [CrossRef]
- Marion, R.M.; Strati, K.; Li, H.; Tejera, A.; Schoeftner, S.; Ortega, S.; Serrano, M.; Blasco, M.A. Telomeres acquire embryonic stem cell characteristics in induced pluripotent stem cells. Cell Stem Cell 2009, 4, 141–154. [Google Scholar] [CrossRef]
- Wong, L.H. Epigenetic regulation of telomere chromatin integrity in pluripotent embryonic stem cells. Epigenomics 2010, 2, 639–655. [Google Scholar] [CrossRef]
- Meshorer, E.; Yellajoshula, D.; George, E.; Scambler, P.J.; Brown, D.T.; Misteli, T. Hyperdynamic plasticity of chromatin proteins in pluripotent embryonic stem cells. Dev. Cell 2006, 10, 105–116. [Google Scholar] [CrossRef]
- Garcia-Cao, M.; O’Sullivan, R.; Peters, A.H.; Jenuwein, T.; Blasco, M.A. Epigenetic regulation of telomere length in mammalian cells by the Suv39h1 and Suv39h2 histone methyltransferases. Nat. Genet. 2004, 36, 94–99. [Google Scholar] [CrossRef]
- Benetti, R.; Gonzalo, S.; Jaco, I.; Schotta, G.; Klatt, P.; Jenuwein, T.; Blasco, M.A. Suv4-20h deficiency results in telomere elongation and derepression of telomere recombination. J. Cell Biol. 2007, 178, 925–960. [Google Scholar] [CrossRef]
- Lu, F.; Liu, Y.; Jiang, L.; Yamaguchi, S.; Zhang, Y. Role of Tet proteins in enhancer activity and telomere elongation. Genes Dev. 2014, 28, 2103–2119. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Guo, R.; Wang, H.; Ye, X.; Zhou, Z.; Dan, J.; Wang, H.; Gong, P.; Deng, W.; Yin, Y.; et al. Tet Enzymes Regulate Telomere Maintenance and Chromosomal Stability of Mouse ESCs. Cell Rep. 2016, 15, 1809–1821. [Google Scholar] [CrossRef]
- Le, R.; Kou, Z.; Jiang, Y.; Li, M.; Huang, B.; Liu, W.; Li, H.; Kou, X.; He, W.; Rudolph, K.L.; et al. Enhanced telomere rejuvenation in pluripotent cells reprogrammed via nuclear transfer relative to induced pluripotent stem cells. Cell Stem Cell 2014, 14, 27–39. [Google Scholar] [CrossRef] [PubMed]
- De Vitis, M.; Berardinelli, F.; Sgura, A. Telomere Length Maintenance in Cancer: At the Crossroad between Telomerase and Alternative Lengthening of Telomeres (ALT). Int. J. Mol. Sci. 2018, 19, 606. [Google Scholar] [CrossRef] [PubMed]
- Dyer, M.A.; Qadeer, Z.A.; Valle-Garcia, D.; Bernstein, E. Atrx and daxx: Mechanisms and mutations. Cold Spring Harbor Perspect. Med. 2017, 7, a026567. [Google Scholar] [CrossRef] [PubMed]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tonjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Shi, C.; Edil, B.H.; de Wilde, R.F.; Klimstra, D.S.; Maitra, A.; Schulick, R.D.; Tang, L.H.; Wolfgang, C.L.; Choti, M.A.; et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 2011, 331, 1199–1203. [Google Scholar] [CrossRef]
- Lovejoy, C.A.; Li, W.; Reisenweber, S.; Thongthip, S.; Bruno, J.; de Lange, T.; De, S.; Petrini, J.H.; Sung, P.A.; Jasin, M.; et al. Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLoS Genet. 2012, 8, 1002772. [Google Scholar] [CrossRef]
- Han, M.; Napier, C.E. Synthetic lethality of cytolytic HSV-1 in cancer cells with ATRX and PML deficiency. J. Cell Sci. 2019, 132, 222349–222358. [Google Scholar] [CrossRef]
- Chen, Y.J.; Hakin-Smith, V.; Teo, M.; Xinarianos, G.E.; Jellinek, D.A.; Carroll, T.; McDowell, D.; MacFarlane, M.R.; Boet, R.; Baguley, B.C.; et al. Association of mutant TP53 with alternative lengthening of telomeres and favorable prognosis in glioma. Cancer Res. 2006, 66, 6473–6476. [Google Scholar] [CrossRef]
- Celeghin, A.; Giunco, S.; Freguja, R.; Zangrossi, M.; Nalio, S.; Dolcetti, R.; De Rossi, A. Short-term inhibition of TERT induces telomere length-independent cell cycle arrest and apoptotic response in EBV-immortalized and transformed B cells. Cell Death Dis. 2016, 7, 2562–2566. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Sheng, J.F.; Cai, L.; Xu, Y.; Liao, H.; Tao, Z.Z. The Telomerase and Alternative Lengthening of Telomeres Mechanisms Regulate Laryngeal Cancer Cell Apoptosis via the PI3K/Akt Pathway. ORL 2018, 80, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Salvati, E.; Leonetti, C.; Rizzo, A.; Scarsella, M.; Mottolese, M.; Galati, R.; Sperduti, I.; Stevens, M.F.; D’Incalci, M.; Blasco, M.; et al. Telomere damage induced by the G-quadruplex ligand RHPS4 has an antitumor effect. J. Clin. Investig. 2007, 117, 3236–3247. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.; Salvati, E.; Porru, M.; D’Angelo, C.; Stevens, M.F.; D’Incalci, M.; Leonetti, C.; Gilson, E.; Zupi, G.; Biroccio, A. Stabilization of quadruplex DNA perturbs telomere replication leading to the activation of an ATR-dependent ATM signaling pathway. Nucleic Acids Res. 2009, 37, 5353–5364. [Google Scholar] [CrossRef]
- Porru, M.; Zizza, P.; Franceschin, M.; Leonetti, C.; Biroccio, A. EMICORON: A multi-targeting G4 ligand with a promising preclinical profile. Biochim. Biophys. Acta (BBA) 2017, 1861, 1362–1370. [Google Scholar] [CrossRef]
- Zheng, X.H.; Nie, X.; Fang, Y.; Zhang, Z.; Xiao, Y.; Mao, Z.; Liu, H.; Ren, J.; Wang, F.; Xia, L.; et al. A Cisplatin Derivative Tetra-Pt(bpy) as an Oncotherapeutic Agent for Targeting ALT Cancer. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, J.; Wild, A.T.; Wu, W.H.; Shah, R.; Danussi, C.; Riggins, G.J.; Kannan, K.; Sulman, E.P.; Chan, T.A.; et al. G-quadruplex DNA drives genomic instability and represents a targetable molecular abnormality in ATRX-deficient malignant glioma. Nat. Commun. 2019, 10, 943. [Google Scholar] [CrossRef]
- Neidle, S. Quadruplex Nucleic Acids as Novel Therapeutic Targets. J. Med. Chem. 2016, 59, 5987–6011. [Google Scholar] [CrossRef]
- Bojovic, B.; Booth, R.E.; Jin, Y.; Zhou, X.; Crowe, D.L. Alternative lengthening of telomeres in cancer stem cells in vivo. Oncogene 2015, 34, 611–630. [Google Scholar] [CrossRef]
- Hu, Y.; Shi, G.; Zhang, L.; Li, F.; Jiang, Y.; Jiang, S.; Ma, W.; Zhao, Y.; Songyang, Z.; Huang, J. Switch telomerase to ALT mechanism by inducing telomeric DNA damages and dysfunction of ATRX and DAXX. Sci. Rep. 2016, 6, 32280–32289. [Google Scholar] [CrossRef]
- Gocha, A.R.; Nuovo, G.; Iwenofu, O.H.; Groden, J. Human sarcomas are mosaic for telomerase-dependent and telomerase-independent telomere maintenance mechanisms: Implications for telomere-based therapies. Am. J. Pathol. 2013, 182, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Pezzolo, A.; Pistorio, A.; Gambini, C.; Haupt, R.; Ferraro, M.; Erminio, G.; De Bernardi, B.; Garaventa, A.; Pistoia, V. Intratumoral diversity of telomere length in individual neuroblastoma tumors. Oncotarget 2014, 6, 7493–7503. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Peng, M.; Song, Q. The co-expression of telomerase and ALT pathway in human breast cancer tissues. Tumor Biol. 2014, 35, 4087–4093. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Matsumine, A.; Sudo, A. The value of trabectedin in the treatment of soft tissue sarcoma. Ther. Clin. Risk Manag. 2016, 12, 73–79. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, S.; Wang, F.; Liu, L. Alternative Lengthening of Telomeres (ALT) in Tumors and Pluripotent Stem Cells. Genes 2019, 10, 1030. https://doi.org/10.3390/genes10121030
Zhao S, Wang F, Liu L. Alternative Lengthening of Telomeres (ALT) in Tumors and Pluripotent Stem Cells. Genes. 2019; 10(12):1030. https://doi.org/10.3390/genes10121030
Chicago/Turabian StyleZhao, Shuang, Feng Wang, and Lin Liu. 2019. "Alternative Lengthening of Telomeres (ALT) in Tumors and Pluripotent Stem Cells" Genes 10, no. 12: 1030. https://doi.org/10.3390/genes10121030
APA StyleZhao, S., Wang, F., & Liu, L. (2019). Alternative Lengthening of Telomeres (ALT) in Tumors and Pluripotent Stem Cells. Genes, 10(12), 1030. https://doi.org/10.3390/genes10121030