Transcriptional Regulation of INSR, the Insulin Receptor Gene


Abstract
1. Insulin Receptor and Insulin Signaling
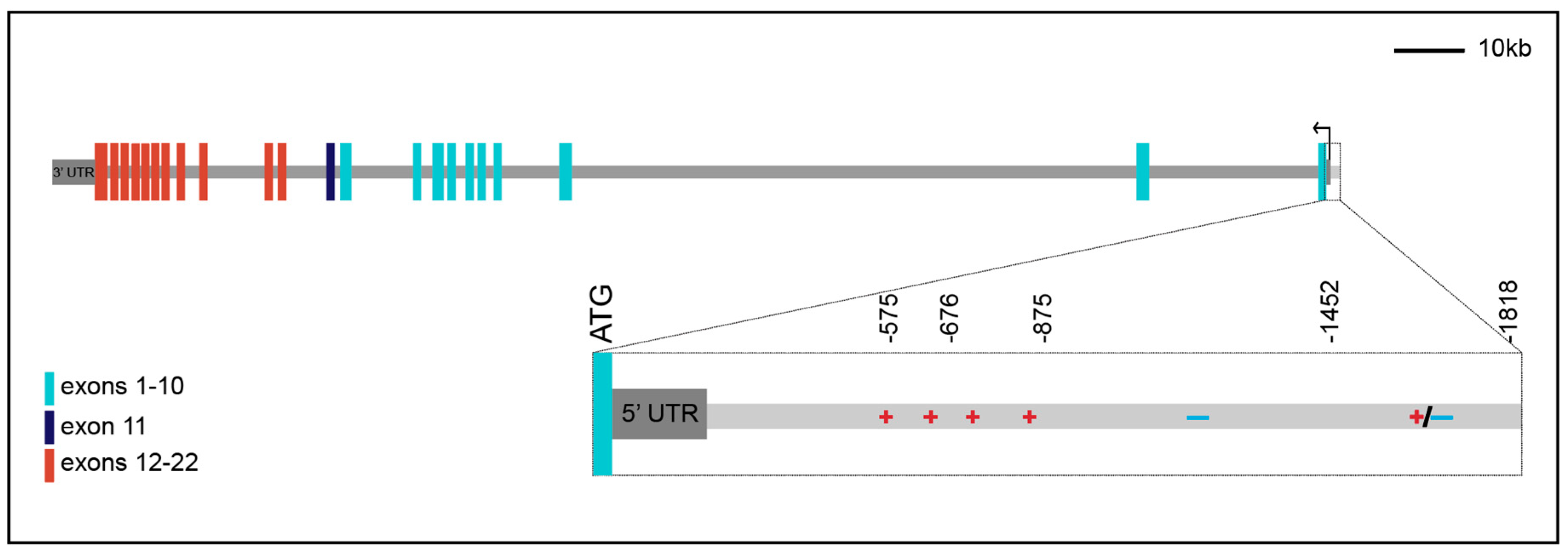
2. The Human Insulin Receptor Gene
3. Promoter Analysis
4. FOXO Feedback Regulation and Control of the Drosophila Insulin Receptor Gene
5. Tissue-Specific Expression of the Insulin Receptor
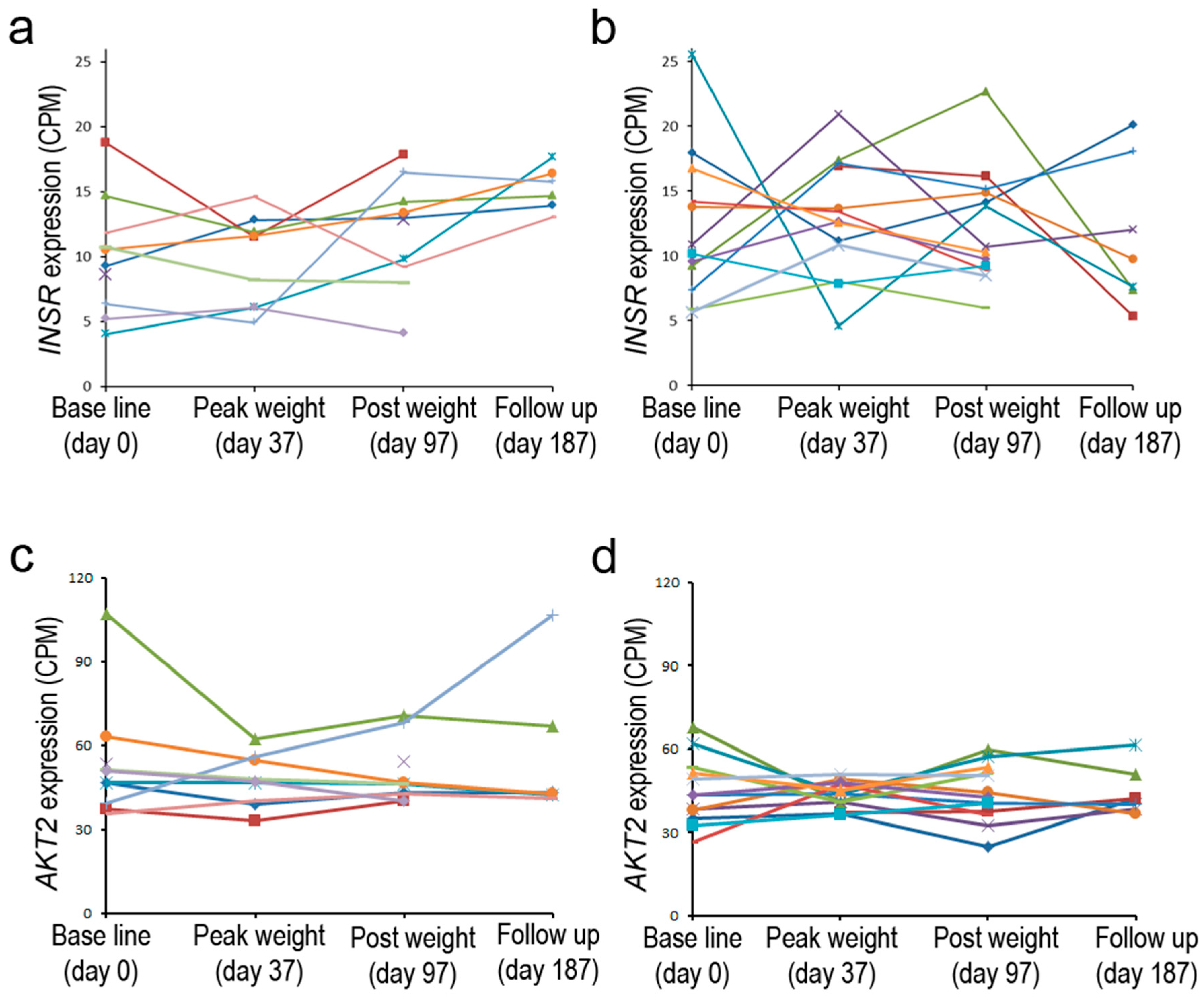
6. Insulin Receptor Expression in Obesity, Insulin Resistance, and Diabetes Mellitus
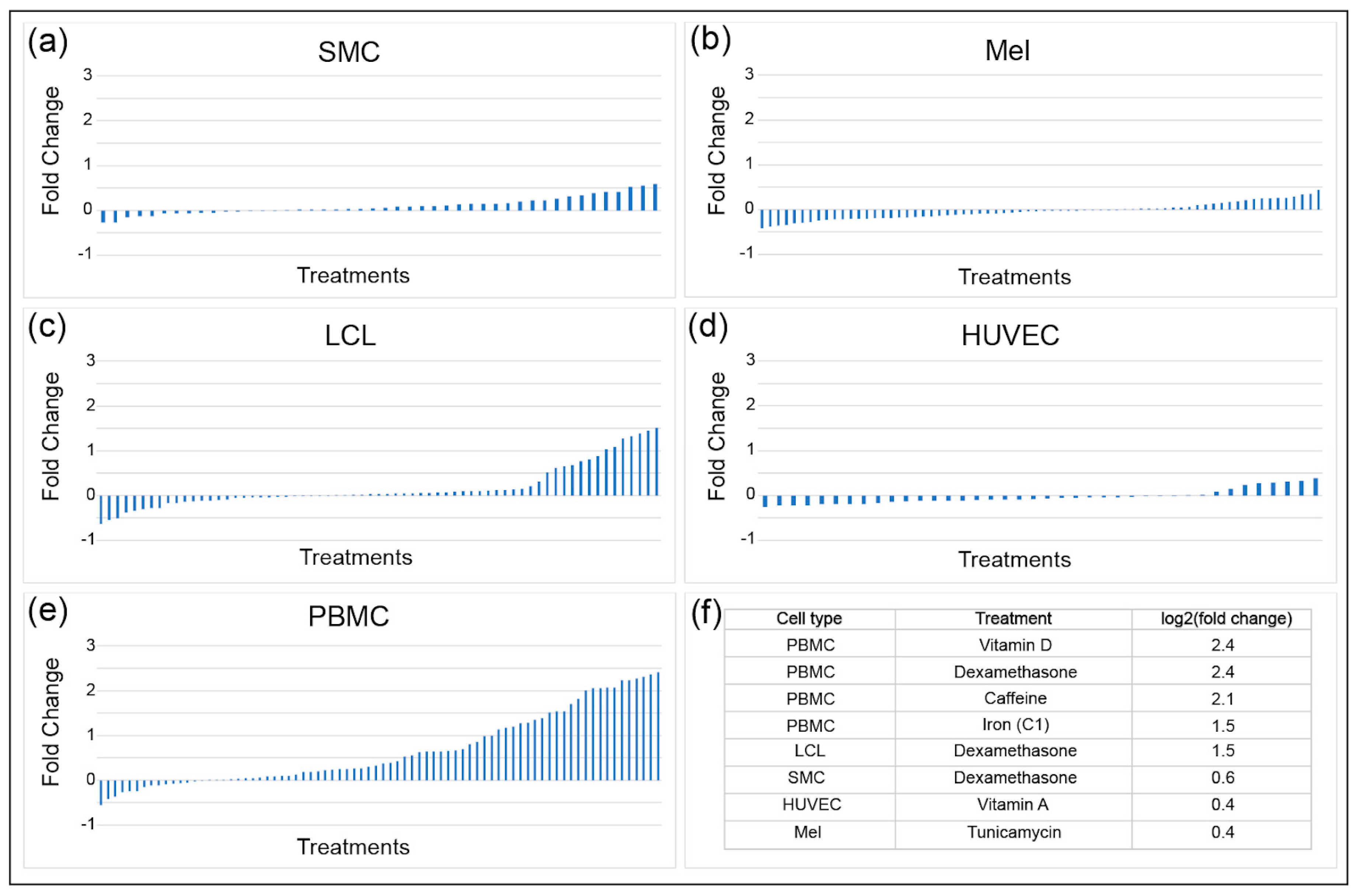
7. Evidence for Specific Regulation of INSR Transcription by Endogenous and Exogenous Stimuli
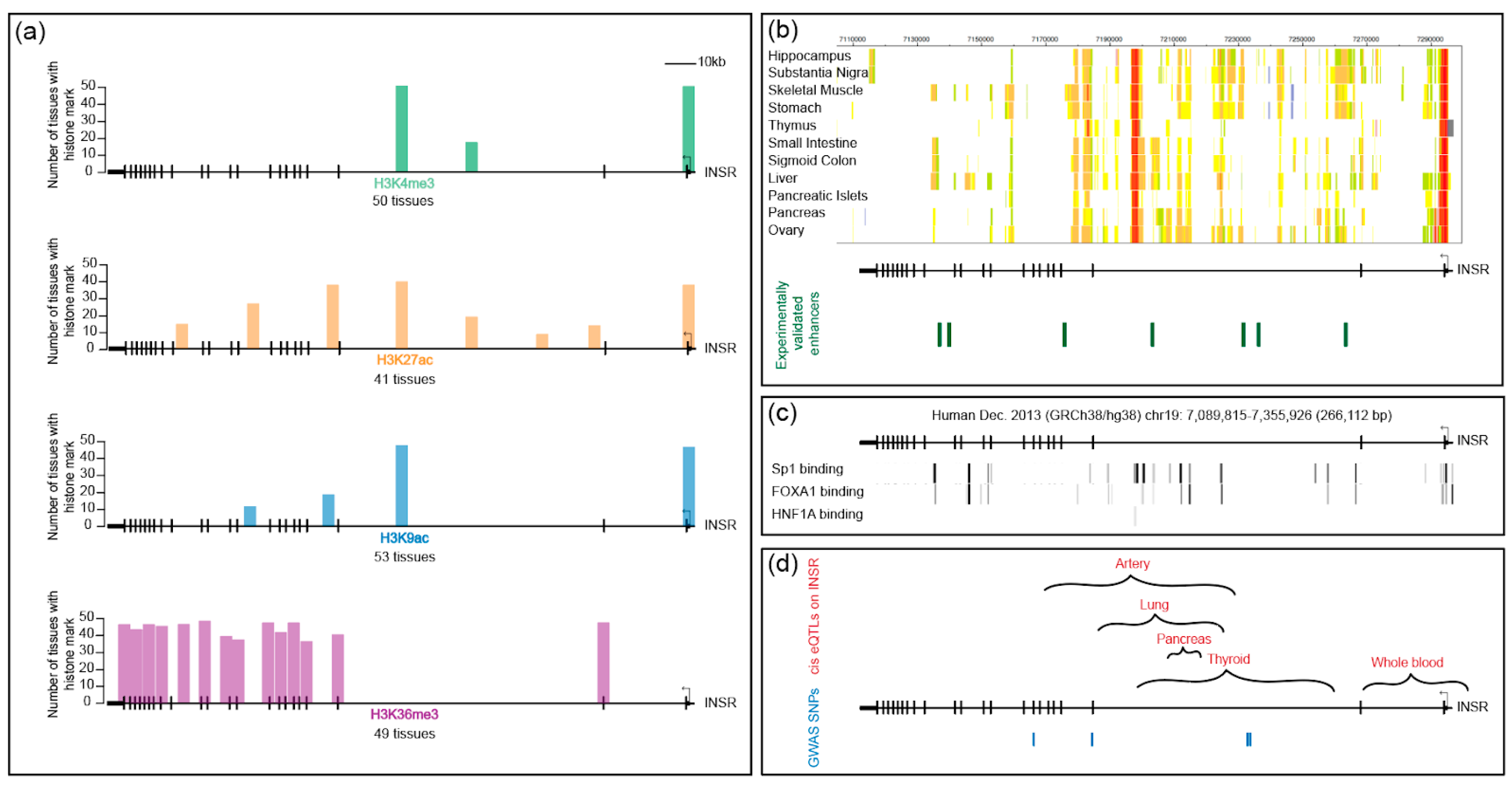
8. Omics Approaches to Studying the INSR Chromatin Landscape
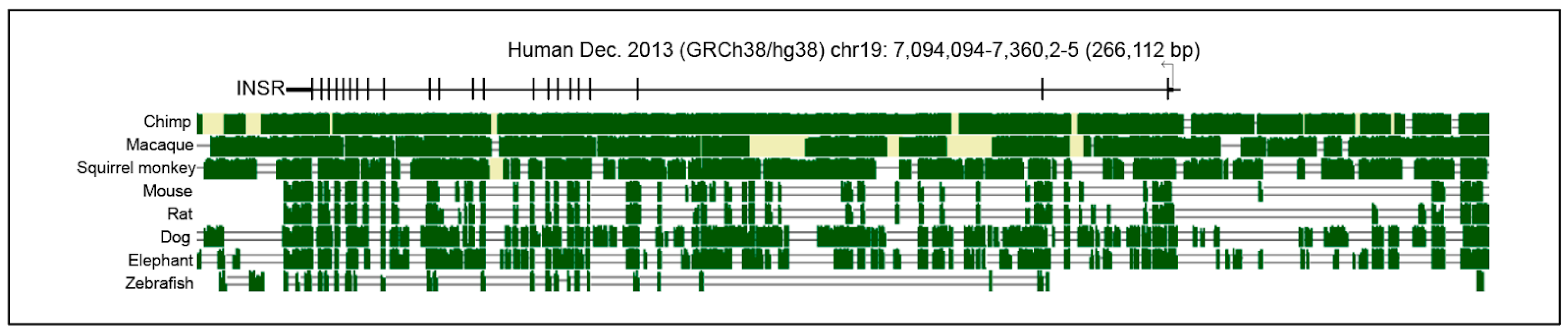
9. Evolutionary Adaptation of Insulin Receptor Signaling
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Olson, T.S.; Bamberger, M.J.; Lane, M.D. Post-translational changes in tertiary and quaternary structure of the insulin proreceptor. J. Biol. Chem. 1988, 263, 7342–7351. [Google Scholar] [PubMed]
- Belfiore, A.; Malaguarnera, R.; Vella, V.; Lawrence, M.C.; Sciacca, L.; Frasca, F.; Morrione, A.; Vigneri, R. Insulin Receptor isoforms in physiology and disease: An updated view. Endocr. Rev. 2017, 38, 379–431. [Google Scholar] [CrossRef] [PubMed]
- Haeusler, R.A.; McGraw, T.E.; Accili, D. Biochemical and cellular properties of Insulin Receptor signalling. Nat. Rev. Mol. Cell. Biol. 2018, 19, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Burgering, B.M.T.; Kops, G.J.P.L. Cell cycle and death control: Long live Forkheads. Trends Biochem. Sci. 2002, 27, 352–360. [Google Scholar] [CrossRef]
- Puig, O.; Tjian, R. Transcriptional feedback control of Insulin Receptor by dFOXO/FOXO1. Genes Dev. 2005, 19, 2435–2446. [Google Scholar] [CrossRef]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell. Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef]
- Puig, O.; Marr, M.T.; Ruhf, M.L.; Tjian, R. Control of cell number by Drosophila FOXO: Downstream and feedback regulation of the Insulin Receptor pathway. Genes Dev. 2003, 17, 2006–2020. [Google Scholar] [CrossRef]
- Kahn, C.R.; Neville, D.M.; Gorden, P.; Freychet, P.; Roth, J. Insulin Receptor defect in insulin resistance: Studies in the obese-hyperglycemic mouse. Biochem. Biophys. Res. Commun. 1972, 48, 135–142. [Google Scholar] [CrossRef]
- Batista, T.M.; Garcia-Martin, R.; Cai, W.; Konishi, M.; O’Neill, B.T.; Sakaguchi, M.; Kim, J.H.; Jung, D.Y.; Kim, J.K.; Kahn, C.R. Multi-dimensional Transcriptional Remodeling by Physiological Insulin In Vivo. Cell Rep. 2019, 26, 3429–3443. [Google Scholar] [CrossRef]
- Psiachou, H.; Mitton, S.; Alaghband-Zadeh, J.; Hone, J.; Taylor, S.I.; Sinclair, L. Leprechaunism and homozygous nonsense mutation in the Insulin Receptor gene. Lancet 1993, 342, 924. [Google Scholar] [CrossRef]
- Ebina, Y.; Ellis, L.; Jarnagin, K.; Edery, M.; Graf, L.; Clauser, E.; Ou, J.; Masiarz, F.; Kan, Y.W.; Goldfine, I.D.; et al. The human Insulin Receptor cDNA: The structural basis for hormone-activated transmembrane signalling. Cell 1985, 40, 747–758. [Google Scholar] [CrossRef]
- Ullrich, A.; Bell, J.R.; Chen, E.Y.; Herrera, R.; Petruzzelli, L.M.; Dull, T.J.; Gray, A.; Coussens, L.; Lia, Y.C.; Tsubokawa, M.; et al. Human Insulin Receptor and its relationship to the tyrosine kinase family of oncogenes. Nature 1985, 313, 756–761. [Google Scholar] [CrossRef] [PubMed]
- Seino, S.; Seino, M.; Nishi, S.; Bell, G.I. Structure of the human Insulin Receptor gene and characterization of its promoter. Proc. Natl. Acad. Sci. USA 1989, 86, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Malakar, P.; Chartarifsky, L.; Hija, A.; Leibowitz, G.; Glaser, B.; Dor, Y.; Karni, R. Insulin Receptor alternative splicing is regulated by insulin signaling and modulates beta cell survival. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef]
- Moller, D.E.; Yokota, A.; Caro, J.F.; Flier, J.S. Tissue-specific expression of two alternatively spliced Insulin Receptor mRNAs in man. Mol. Endocrinol. 1989, 3, 1263–1269. [Google Scholar] [CrossRef]
- Goldstein, B.J.; Dudley, A.L. The rat Insulin Receptor: Primary structure and conservation of tissue-specific alternative messenger RNA splicing. Mol. Endocrinol. 1990, 4, 235–244. [Google Scholar] [CrossRef]
- Kaminska, D.; Hämäläinen, M.; Cederberg, H.; Käkelä, P.; Venesmaa, S.; Miettinen, P.; Ilves, I.; Herzig, K.-H.; Kolehmainen, M.; Karhunen, L.; et al. Adipose tissue INSR splicing in humans associates with fasting insulin level and is regulated by weight loss. Diabetologia 2014, 57, 347–351. [Google Scholar] [CrossRef]
- Araki, E.; Shimada, F.; Uzawa, H.; Mori, M.; Ebina, Y. Characterization of the promoter region of the human insulin receptor gene. J. Biol. Chem. 1987, 262, 16186–16191. [Google Scholar] [CrossRef]
- Tewari, D.S.; Cook, D.M.; Taub, R. Characterization of the promoter region and 3’ end of the human Insulin Receptor gene. J. Biol. Chem. 1989, 264, 16238–16245. [Google Scholar]
- Mamula, P.W.; Wong, K.Y.; Maddux, B.A.; McDonald, A.R.; Goldfine, I.D. Sequence and analysis of promoter region of human insulin-receptor gene. Diabetes 1988, 37, 1241–1246. [Google Scholar] [CrossRef]
- McKeon, C.; Moncada, V.; Pham, T.; Salvatore, P.; Kadowak, T.; Accili, D.; Taylore, S.I. Structural and functional analysis of the Insulin Receptor promoter. Mol. Endocrinol. 1990, 4, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Gokhale, R.H.; Sonnenschein, A.; Montgomery, K.M.; Ingersoll, A.; Arnosti, D.N. Complex cis-regulatory landscape of the Insulin Receptor gene underlies the broad expression of a central signaling regulator. Development 2016, 143, 3591–3603. [Google Scholar] [CrossRef] [PubMed]
- Moyerbrailean, G.A.; Richards, A.L.; Kurtz, D.; Kalita, C.A.; Davis, G.O.; Harvey, C.T.; Alazizi, A.; Watza, D.; Sorokin, Y.; Hauff, N.; et al. High-throughput allele-specific expression across 250 environmental conditions. Genome Res. 2016, 26, 1627–1638. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Tam, J.W.O.; Tsai, M.J.; Tsai, S.Y. Identification of cis- and trans-acting factors regulating the expression of the human Insulin Receptor gene. J. Biol. Chem. 1992, 267, 4638–4645. [Google Scholar] [PubMed]
- Yoshizato, K.; Shirotani, T.; Furukawa, N.; Taguchi, T.; Motoshima, H.; Toyonaga, T.; Hirashima, Y.; Kawashima, J.; Ebina, Y.; Shichiri, M.; et al. Identification of a cis-acting element and a novel trans-acting factor of the human Insulin Receptor gene in HepG2 and rat liver cells. Biochem. Biophys. Res. Commun. 2001, 280, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.R.; Hug, V. Nuclear protein-binding analysis of a GC-rich Insulin Receptor promoter regulatory region. Diabetes 1993, 42, 66–73. [Google Scholar] [CrossRef]
- Cameron, K.E.; Resnik, J.; Webster, N.J.G. Transcriptional regulation of the human Insulin Receptor promoter. J. Biol. Chem. 1992, 267, 17375–17383. [Google Scholar]
- Araki, E.; Murakami, T.; Shirotani, T.; Kanai, F.; Shinohara, Y.; Shimada, F.; Mori, M.; Shichiri, M.; Ebina, Y. A cluster of four Sp1 binding sites required for efficient expression of the human Insulin Receptor gene. J. Biol. Chem. 1991, 266, 3944–3948. [Google Scholar]
- Brunetti, A.; Manfioletti, G.; Chiefari, E.; Goldfine, I.D.; Foti, D. Transcriptional regulation of human Insulin Receptor gene by the high-mobility group protein HMGI(Y). FASEB J. 2001, 15, 492–500. [Google Scholar] [CrossRef]
- Foti, D.; Chiefari, E.; Fedele, M.; Iuliano, R.; Brunetti, L.; Paonessa, F.; Manfioletti, G.; Barbetti, F.; Brunetti, A.; Croce, C.M.; et al. Lack of the architectural factor HMGA1 causes insulin resistance and diabetes in humans and mice. Nat. Med. 2005, 11, 765–773. [Google Scholar] [CrossRef]
- McKeon, C.; Pham, T. Transactivation of the human Insulin Receptor gene by the CAAT/enchancer binding protein. Biochem. Biophys. Res. Commun. 1991, 174, 721–728. [Google Scholar] [CrossRef]
- Webster, N.J.G.; Resnik, J.L.; Reichart, D.B.; Strauss, B.; Haas, M.; Seely, B.L. Repression of the Insulin Receptor promoter by the tumor suppressor gene product p53: A possible mechanism for receptor overexpression in breast cancer. Cancer Res. 1996, 56, 2781–2788. [Google Scholar] [PubMed]
- Singh, R.; De Aguiar, R.B.; Naik, S.; Mani, S.; Ostadsharif, K.; Wencker, D.; Sotoudeh, M.; Malekzadeh, R.; Sherwin, R.; Mani, A. LRP6 enhances glucose metabolism by promoting TCF7L2-dependent Insulin Receptor expression and ICF receptor stabilization in humans. Cell Metab. 2013, 17, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Costa, V.; Foti, D.; Paonessa, F.; Chiefari, E.; Palaia, L.; Brunetti, G.; Gulletta, E.; Fusco, A.; Brunetti, A. The Insulin Receptor: A new anticancer target for peroxisome proliferator-activated receptor-γ (PPARγ) and thiazolidinedione- PPARγ agonists. Endocr.-Relat. Cancer 2008, 15, 325–335. [Google Scholar] [CrossRef]
- Pechlivanis, S.; Pardini, B.; Bermejo, J.L.; Wagner, K.; Naccarati, A.; Vodickova, L.; Novotny, J.; Hemminki, K.; Vodicka, P.; Forsti, A. Insulin pathway related genes and risk of colorectal cancer: INSR promoter polymorphism shows a protective effect. Endocr.-Relat. Cancer 2007, 14, 733–740. [Google Scholar] [CrossRef]
- Haruta, T.; Imamura, T.; Iwanishi, M.; Egawa, K.; Goji, K.; Kobayashi, M. Amplification and analysis of promoter region of Insulin Receptor gene in a patient with leprechaunism associated with severe insulin resistance. Metabolism 1995, 44, 430–437. [Google Scholar] [CrossRef]
- Kaplan, R.E.W.; Maxwell, C.S.; Codd, N.K.; Baugh, L.R. Pervasive positive and negative feedback regulation of insulin-like signaling in Caenorhabditis elegans. Genetics 2019, 211, 349–361. [Google Scholar] [CrossRef]
- Petruzzelli, L.; Herrera, R.; Garcia-Arenas, R.; Rosen, O.M. Acquisition of insulin-dependent protein tyrosine kinase activity during Drosophila embryogenesis. J. Biol. Chem. 1985, 260, 16072–16075. [Google Scholar]
- Petruzzelli, L.; Herrera, R.; Arenas-Garcia, R.; Fernandez, R.; Birnbaum, M.J.; Rosen, O.M. Isolation of a Drosophila genomic sequence homologous to the kinase domain of the human Insulin Receptor and detection of the phosphorylated Drosophila receptor with an anti-peptide antibody. Proc. Natl. Acad. Sci. USA 1986, 83, 4710–4714. [Google Scholar] [CrossRef]
- Nishida, Y.; Hata, M.; Nishizuka, Y.; Rutter, W.J.; Ebina, Y. Cloning of a Drosophila cDNA encoding a polypeptide similar to the human Insulin Receptor precursor. Biochem. Biophys. Res. Commun. 1986, 141, 474–481. [Google Scholar] [CrossRef]
- Fernandez, R.; Tabarini, D.; Azpiazu, N.; Frasch, M.; Schlessinger, J. The Drosophila Insulin Receptor homolog: A gene essential for embryonic development encodes two receptor isoforms with different signaling potential. EMBO J. 1995, 14, 3373–3384. [Google Scholar] [CrossRef] [PubMed]
- The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Casas-Tinto, S.; Marr, M.T.; Andreu, P.; Puig, O. Characterization of the Drosophila Insulin Receptor promoter. Biochim. Biophys. Acta 2007, 1769, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, R.S.; Rosen, O.M. Tissue localization of Drosophila melanogaster Insulin Receptor transcripts during development. Mol. Cell. Biol. 1988, 8, 1638–1647. [Google Scholar] [CrossRef]
- Gavin, J.R.; Roth, J.; Neville, D.M.; De Meyts, P.; Buellt, D.N. Insulin-dependent regulation of Insulin Receptor concentrations: A direct demonstration in cell culture. Proc. Nat. Acad. Sci. USA 1974, 71, 84–88. [Google Scholar] [CrossRef]
- Orengo, D.J.; Aguadé, M.; Juan, E. Characterization of dFOXO binding sites upstream of the Insulin Receptor P2 promoter across the Drosophila phylogeny. PLoS ONE 2017, 12, 1–16. [Google Scholar] [CrossRef]
- Gammeltoft, S. Insulin Receptors: Binding kinetics and structure-function relationship of insulin. Physiol. Rev. 1984, 64, 1321–1378. [Google Scholar] [CrossRef]
- Watanabe, M.; Hayasaki, H.; Tamayama, T.; Shimada, M. Histologic distribution of insulin and glucagon receptors. Braz. J. Med. Biol. Res. 1998, 31, 243–256. [Google Scholar] [CrossRef][Green Version]
- Carpentier, J.L.; Dayer, J.M.; Lang, U.; Silverman, R.; Orci, L.; Gorden, P. Down-regulation and recycling of Insulin Receptors. Effect of monensin on IM-9 lymphocytes and U-937 monocyte-like cells. J. Biol. Chem. 1984, 259, 14190–14195. [Google Scholar]
- Tewari, M.; Tewari, D.S.; Taub, R. Posttranscriptional mechanisms account for differences in steady state levels of Insulin Receptor messenger RNA in different cells. Mol. Endocrinol. 1991, 5, 653–660. [Google Scholar] [CrossRef]
- Min, K.H.; Yang, W.M.; Lee, W. Saturated fatty acids-induced miR-424–5p aggravates insulin resistance via targeting Insulin Receptor in hepatocytes. Biochem. Biophys. Res. Commun. 2018, 503, 1587–1593. [Google Scholar] [CrossRef] [PubMed]
- Spriggs, K.A.; Cobbold, L.C.; Ridley, S.H.; Coldwell, M.; Bottley, A.; Bushell, M.; Willis, A.E.; Siddle, K. The human Insulin Receptor mRNA contains a functional internal ribosome entry segment. Nucleic Acids Res. 2009, 37, 5881–5893. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, B.J.; Muller-Wieland, D.; Kahn, R.C. Variation in Insulin Receptor messenger ribonucleic acid expression in human and rodent tissues. Mol. Endocrinol. 1987, 1, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Norgren, S.; Arner, P.; Luthman, H. Insulin Receptor ribonucleic acid levels and alternative splicing in human liver, muscle, and adipose tissue: Tissue specificity and relation to insulin action. J. Clin. Endocrinol. Metab. 1994, 78, 757–762. [Google Scholar] [PubMed]
- Pezzino, V.; Papa, V.; Trischitta, V.; Brunetti, A.; Goodman, P.A.; Treutelaar, M.K.; Williams, J.A.; Maddux, B.A.; Goldfine, I.D. Human Insulin Receptor radioimmunoassay: Applicability to insulin-resistant states. Am. J. Physiol. 1989, 257, 451–457. [Google Scholar] [CrossRef]
- Carithers, L.J.; Ardlie, K.; Barcus, M.; Branton, P.A.; Britton, A.; Buia, S.A.; Compton, C.C.; DeLuca, D.S.; Peter-Demchok, J.; Gelfand, E.T.; et al. A novel approach to high-quality postmortem tissue procurement: The GTEx project. Biopreserv. Biobank. 2015, 13, 311–317. [Google Scholar] [CrossRef]
- Serrano, R.; Villar, M.; Martínez, C.; Carrascosa, J.M.; Gallardo, N.; Andrés, A. Differential gene expression of Insulin Receptor isoforms A and B and Insulin Receptor substrates 1, 2 and 3 in rat tissues: Modulation by aging and differentiation in rat adipose tissue. J. Mol. Endocrinol. 2005, 34, 153–161. [Google Scholar] [CrossRef]
- Halfon, M.S. Studying transcriptional enhancers: The founder fallacy, validation creep, and other biases. Trends Genet. 2019, 35, 93–103. [Google Scholar] [CrossRef]
- Bar, R.S.; Gorden, P.; Roth, J.; Kahn, C.R.; De Meyts, P. Fluctuations in the affinity and concentration of Insulin Receptors on circulating monocytes of obese patients: Effects of starvation, refeeding, and dieting. J. Clin. Investig. 1976, 58, 1123–1135. [Google Scholar] [CrossRef]
- Olefsky, J.M. Decreased insulin binding to adipocytes and circulating monocytes from obese subjects. J. Clin. Investig. 1976, 57, 1165–1172. [Google Scholar] [CrossRef]
- Spanheimer, R.G.; Bar, R.S.; Ginsberg, B.H.; Peacock, M.L.; Martino, I. Comparison of insulin binding to cells of fed and fasted obese patients: Results in erythrocytes and monocytes. J. Clin. Endocrinol. Metab. 1982, 54, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Briata, P.; Gherzi, R.; Adezati, L.; Cordera, R. Effect of two different glucose concentrations on Insulin Receptor mRNA levels in human hepatoma HepG2 cells. Biochem. Biophys. Res. Commun. 1989, 3, 1415–1420. [Google Scholar] [CrossRef]
- Briata, P.; Gherzi, R. Multifactorial control of Insulin Receptor gene expression in human cell lines. Biochem. Biophys. Res. Commun. 1990, 3, 1184–1190. [Google Scholar] [CrossRef]
- Mamula, P.W.; McDonald, A.R.; Brunetti, A.; Okabayashi, Y.; Wong, K.Y.; Maddux, B.A.; Logsdon, C.; Goldfine, I.D. Regulating insulin-receptor-gene expression by differentiation and hormones. Diabetes Care 1990, 13, 288–301. [Google Scholar] [CrossRef]
- Ujvari, D.; Jakson, I.; Babayeva, S.; Salamon, D.; Rethi, B.; Gidlöf, S.; Hirscberg, A.L. Dysregulation of in vitro decidualization of human endometrial stromal cells by insulin via transcriptional inhibition of forkhead box protein O1. PLoS ONE 2017, 12, 1–18. [Google Scholar] [CrossRef]
- Rouiller, D.G.; McKeon, C.; Taylor, S.I.; Gorden, P. Hormonal regulation of Insulin Receptor gene expression. Hydrocortisone and insulin act by different mechanisms. J. Biol. Chem. 1988, 263, 13185–13190. [Google Scholar]
- Westermeier, F.; Salomón, C.; González, M.; Puebla, C.; Guzmán-Gutiérrez, E.; Cifuentes, F.; Leiva, A.; Casamello, P.; Sobrevia, L. Insulin restores gestational diabetes mellitus-Reduced adenosine transport involving differential expression of Insulin Receptor isoforms in human umbilical vein endothelium. Diabetes 2011, 60, 1677–1687. [Google Scholar] [CrossRef][Green Version]
- Lane, M.D.; Reed, B.C.; Clements, P.R. Insulin Receptor synthesis and turnover in differentiating 3T3-L1 preadipocytes. Proc. Natl. Acad. Sci. USA 1981, 66, 523–542. [Google Scholar]
- Kasuga, M.; Kahn, C.R.; Hedo, J.A.; Van Obberghen, E.; Yamada, K.M. Insulin-induced receptor loss in cultured human lymphocytes is due to accelerated receptor degradation. Proc. Natl. Acad. Sci. USA 1981, 78, 6917–6921. [Google Scholar] [CrossRef]
- Ronnetts, G.V.; Knutsong, V.P.; Lane, M.D. Insulin-induced sown-regulation of Insulin Receptors in 3T3-Ll adipocytes. J. Biol. Chem. 1982, 257, 4285–4291. [Google Scholar]
- Hatada, E.N.; McClain, D.A.; Potter, E.; Ullrich, A.; Olefsky, J.M. Effects of growth and insulin treatment on the levels of Insulin Receptors and their mRNA in Hep G2 cells. J. Biol. Chem. 1989, 264, 6741–6747. [Google Scholar] [PubMed]
- Ducluzeau, P.H.; Perretti, N.; Laville, M.; Andreelli, F.; Vega, N.; Riou, J.P.; Vidal, H. Regulation by insulin of gene expression in human skeletal muscle and adipose tissue: Evidence for specific defects in type 2 diabetes. Diabetes 2001, 50, 1134–1142. [Google Scholar] [CrossRef] [PubMed]
- Kaizer, E.C.; Glaser, C.L.; Chaussabel, D.; Banchereau, J.; Pascual, V.; White, P.C. Gene expression in peripheral blood mononuclear cells from children with diabetes. J. Clin. Endocrinol. Metab. 2007, 92, 3705–3711. [Google Scholar] [CrossRef] [PubMed]
- Folli, F.; Okada, T.; Perego, C.; Gunton, J.; Liew, C.W.; Akiyama, M.; D’Amico, A.; Rosa, S.L.; Placidi, C.; Lupi, R.; et al. Altered Insulin Receptor signalling and β-cell cycle dynamics in type 2 diabetes mellitus. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Thompson, B.D.; de Gregorio, M.; Sommercorn, J. Insulin regulation of multuple ribonucleic acid species in human skeletal muscle in insulin-sensitive and insulin-resistant subjects. J. Clin. Endocrinol. Metab. 1996, 81, 519–523. [Google Scholar]
- Sánchez, J.; Priego, T.; Picó, C.; Ahrens, W.; De Henauw, S.; Fraterman, A.; Marild, S.; Molnar, D.; Moreno, L.A.; Peplies, J.; et al. Blood cells as a source of transcriptional biomarkers of childhood obesity and its related metabolic alterations: Results of the IDEFICS study. J. Clin. Endocrinol. Metab. 2012, 97, 648–652. [Google Scholar] [CrossRef]
- Sbraccia, P.; Giaccari, A.; D’Amado, S.; Caiola, L.; Morviducci, L.; Zorretta, D.; Maroccia, E.; Buongiorno, A.; Tamburrano, G. Expression of the two Insulin Receptor isoforms is not altered in the skeletal muscle and liver of diabetic rats. Metabolism 1998, 47, 129–132. [Google Scholar] [CrossRef]
- Capiotti, K.M.; Junior, R.A.; Kist, L.W.; Bogo, M.R.; Bonan, C.D.; Da Silva, R.S. Persistent impaired glucose metabolism in a zebrafish hyperglycemia model. Comp. Biochem. Physiol. Part. B 2014, 171, 58–65. [Google Scholar] [CrossRef]
- Musselman, L.P.; Fink, J.L.; Narzinski, K.; Ramachandran, P.V.; Hathiramani, S.S.; Cagan, R.L.; Baranski, T.J. A high-sugar diet produces obesity and insulin resistance in wild-type Drosophila. Dis. Model. Mech. 2011, 4, 842–849. [Google Scholar] [CrossRef]
- Piening, B.D.; Zhou, W.; Contrepois, K.; Röst, H.; Urban, G.G.J.; Mishra, T.; Hanson, B.M.; Bautista, E.J.; Leopold, S.; Yeh, C.Y.; et al. Integrative personal omics profiles during periods of weight gain and loss. Cell Syst. 2018, 6, 157–170. [Google Scholar] [CrossRef]
- McDonald, A.R.; Goldfine, I.D. Glucocorticoid regulation of Insulin Receptor gene transcription in IM-9 cultured lymphocytes. J. Clin. Investig. 1988, 81, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.M.; Rogers, S.D.; Ng, K.W.; Best, J.D. Dexamethasone modulates Insulin Receptor expression and subcellular distribution of the glucose transporter GLUT 1 in UMR 106-01, a clonal osteogenic sarcoma cell line. J. Mol. Endocrinol. 1996, 17, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Norgren, S.; Li, L.S.; Luthman, H. Regulation of human Insulin Receptor RNA splicing in HepG2 cells: Effects of glucocorticoid and low glucose concentration. Biochem. Bioph. Res. Commun. 1994, 119, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Leal, M.A.; Aller, P.; Mas, A.; Del, M.; Carranza, C.; Calle, C. Tissue-specific changes in Insulin Receptor mRNA concentrations in dexamethasone-treated and adrenaiectomized Rats. Endocr. J. 1994, 41, 737–741. [Google Scholar] [CrossRef]
- Sesti, G.; Marini, M.A.; Briata, P.; Tullio, A.N.; Montemurro, A.; Borboni, P.; Pirro, R.D.; Gherzi, R.; Lauro, R. Androgens increase Insulin Receptor mRNA levels, insulin binding, and insulin responsiveness in HEp-2 larynx carcinoma cells. Mol. Cell. Endocrinol. 1992, 86, 111–118. [Google Scholar] [CrossRef]
- Hah, N.; Murakami, S.; Nagari, A.; Danko, C.G.; Kraus, W.L. Enhancer transcripts mark active estrogen receptor binding sites. Genome Res. 2013, 23, 1210–1223. [Google Scholar] [CrossRef]
- Xie, P.; Liu, M.L.; Gu, Y.P.; Lu, J.; Xu, X.; Zeng, W.M.; Song, H.P. Oestrogen improves glucose metabolism and insulin signal transduction in HepG2 cells. Clin. Exp. Pharmacol. Physiol. 2003, 30, 643–648. [Google Scholar] [CrossRef]
- Shibasaki, Y.; Sakura, H.; Odawara, M.; Shibuya, M.; Kanazawa, Y.; Akanuma, Y.; Takaku, F.; Kasuga, M. Glucocorticoids increase insulin binding and the amount of insulin-receptor mRNA in human cultured lymphocytes. Biochem. J. 1988, 249, 715–719. [Google Scholar] [CrossRef]
- García-Arencibia, M.; Dávila, N.; Campión, J.; Carranza, M.C.; Calle, C. Identification of two functional estrogen response elements complexed with AP-1-like sites in the human Insulin Receptor gene promoter. J. Steroid Biochem. Mol. Biol. 2005, 94, 1–14. [Google Scholar] [CrossRef]
- Maestro, B.; Campión, J.; Dávila, N.; Calle, C. Stimulation by 1,25-dihydroxyvitamin D3 of Insulin Receptor expression and insulin responsiveness for glucose transport in U-937 human promonocytic cells. Endocr. J. 2000, 47, 383–391. [Google Scholar] [CrossRef]
- Maestro, B.; Dávila, N.; Carranza, M.C.; Calle, C. Identification of a Vitamin D response element in the human Insulin Receptor gene promoter. J. Steroid Biochem. Mol. Biol. 2003, 84, 223–230. [Google Scholar] [CrossRef]
- Takano, M.; Lu, Z.; Goto, T.; Fusi, L.; Higham, J.; Francis, J.; Withey, A.; Hardt, J.; Cloke, B.; Stavropoulou, A.V.; et al. Transcriptional cross talk between the forkhead transcription factor forkhead box O1A and the progesterone receptor coordinates cell cycle regulation and differentiation in human endometrial stromal cells. Mol. Endocrinol. 2007, 21, 2334–2349. [Google Scholar] [CrossRef] [PubMed]
- Goldfine, I.D.; Papa, V.; Vigneri, R.; Siiteri, P.; Rosenthal, S. Progestin regulation of insulin and insulin-like growth factor I receptors in cultured human breast cancer cells. Breast Cancer Res. Treat. 1992, 22, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Kong, W.J.; Shan, Y.Q.; Song, D.Q.; Li, Y.; Wang, Y.M.; Jiang, J.D. Protein kinase D activation stimulates the transcription of the Insulin Receptor gene. Mol. Cell. Endocrinol. 2010, 330, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Kriauciunas, K.M.; Goldstein, B.J.; Lipes, M.A.; Kahn, C.R. Modulation of expression of insulin and IGF-I receptor by Epstein-Barr Virus and its gene products LMP and EBNA-2 in lymphocyte cell lines. J. Cell. Physiol. 1993, 154, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.P.; Carroll, J.M.; Carroll, P.A.; DeFilippis, V.R.; Lagunoff, M.; Moses, A.V.; Roberts, C.T.; Fruh, K. The Insulin Receptor is essential for virus-induced tumorigenesis of Kaposi’s sarcoma. Oncogene 2007, 26, 1995–2005. [Google Scholar] [CrossRef]
- Belfiore, A.; Frasca, F.; Pandini, G.; Sciacca, L.; Vigneri, R. Insulin Receptor isoforms and Insulin Receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr. Rev. 2009, 30, 586–623. [Google Scholar] [CrossRef]
- Vella, V.; Milluzzo, A.; Scalisi, N.M.; Vigneri, P.; Sciacca, L. Insulin Receptor isoforms in cancer. Int. J. Mol. Sci. 2018, 19, 3615. [Google Scholar] [CrossRef]
- Wang, Y.; Song, F.; Zhang, B.; Zhang, L.; Xu, J.; Li, D.; Choudhary, M.N.K.; Li, Y.; Hu, M.; Hardison, R.; et al. The 3D Genome Browser: A web-based browser for visualizing 3D genome organization and long-range chromatin interactions. Genome Biol. 2018, 19, 1–12. [Google Scholar] [CrossRef]
- Dekker, J.; Misteli, T. Long-range chromatin interactions. Cold Spring Harb. Perspect. Biol. 2015, 7, 1–22. [Google Scholar] [CrossRef]
- Shlyueva, D.; Stampfel, G.; Stark, A. Transcriptional enhancers: From properties to genome-wide predictions. Nat. Rev. Genet. 2014, 15, 272–286. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Schneider, R.; Myers, F.A.; Thorne, A.W.; Crane-Robinson, C.; Kouzarides, T. Spatial distribution of di- and tri-methyl lysine 36 of histone H3 at active genes. J. Biol. Chem. 2005, 280, 17732–17736. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.S.; Nicetto, D.; Zaert, K.S. H3K9me3-dependent heterochromatin: Barrier to cell fate changes. Trends Genet. 2016, 32, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Shu, W.; Chen, H.; Bo, X.; Wang, S. Genome-wide analysis of the relationships between DNaseI HS, histone modifications and gene expression reveals distinct modes of chromatin domains. Nucleic Acids Res. 2011, 39, 7428–7443. [Google Scholar] [CrossRef] [PubMed]
- Iwafuchi-Doi, M.; Zaret, K.S. Cell fate control by pioneer transcription factors. Development. 2016, 143, 1833–1837. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yu, S.; Dhiman, V.K.; Brunetti, T.; Eckart, H.; White, K.P. Functional assessment of human enhancer activities using whole-genome STARR-sequencing. Genome Biol. 2017, 18, 1–13. [Google Scholar] [CrossRef]
- Tobi, E.W.; Goeman, J.J.; Monajemi, R.; Gu, H.; Putter, H.; Zhang, Y.; Slieker, R.C.; Stok, A.P.; Thijssen, P.E.; Müller, F.; et al. DNA methylation signatures link prenatal famine exposure to growth and metabolism. Nat. Commun. 2014, 5, 5592, Erratum in Nat. Commun. 2015, 6, 7740. [Google Scholar] [CrossRef]
- Roadmap Epigenomics Consortium. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–329. [Google Scholar] [CrossRef]
- Kent, J.W.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef]
- Paaby, A.B.; Blacket, M.J.; Hoffmann, A.A.; Schmidt, P.S. Identification of a candidate adaptive polymorphism for Drosophila life history by parallel independent clines on two continents. Mol. Ecol. 2010, 19, 760–774. [Google Scholar] [CrossRef]
- Riddle, M.R.; Aspiras, A.C.; Gaudenz, K.; Peuß, R.; Sung, J.Y.; Martineau, B.; Peavey, M.; Box, A.C.; Tabin, J.A.; McGaugh, S.; et al. Insulin resistance in cavefish as an adaptation to a nutrient-limited environment. Nature 2018, 555, 647–651. [Google Scholar] [CrossRef] [PubMed]
- Guirao-Rico, S.; Aguadé, M. Positive selection has driven the evolution of the drosophila Insulin-Like Receptor (InR) at different timescales. Mol. Biol. Evol. 2009, 26, 1723–1732. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Ponce, D.; Aguadé, M.; Rozas, J. Network-level molecular evolutionary analysis of the insulin/TOR signal transduction pathway across 12 Drosophila genomes. Genome Res. 2009, 19, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Ponce, D.; Aguadé, M.; Rozas, J. Comparative genomics of the vertebrate insulin/TOR signal transduction pathway: A network-level analysis of selective pressures. Genome Biol. Evol. 2011, 3, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Shingleton, A.W.; Das, J.; Vinicius, L.; Stern, D.L. The temporal requirements for insulin signaling during development in Drosophila. PLoS Biol. 2005, 3, 1607–1617. [Google Scholar] [CrossRef]
- Emlen, D.J.; Warren, I.A.; Johns, A.; Dworkin, I.; Lavine, L.C. A mechanism of extreme growth and reliable signaling in sexually selected ornaments and weapons. Science 2012, 337, 860–864. [Google Scholar] [CrossRef]
- Green, D.A.; Extavour, C.G. Insulin signalling underlies both plasticity and divergence of a reproductive trait in Drosophila. Proc. R. Soc. B Biol. Sci. 2014, 281. [Google Scholar] [CrossRef]
- Xu, H.J.; Xue, J.; Lu, B.; Zhang, X.C.; Zhuo, J.C.; He, S.F.; Ma, X.-F.; Jiang, Y.-Q.; Fan, H.-W.; Xu, J.-Y.; et al. Two Insulin Receptors determine alternative wing morphs in planthoppers. Nature 2015, 519, 464–467. [Google Scholar] [CrossRef]
- Lin, X.; Yao, Y.; Wang, B.; Lavine, M.D.; Lavine, L.C. FOXO links wing form polyphenism and wound healing in the brown planthopper, Nilaparvata lugens. Insect Biochem. Mol. Biol. 2016, 70, 24–31. [Google Scholar] [CrossRef]
- Lin, X.; Xu, Y.; Jiang, J.; Lavine, M.; Lavine, L.C. Host quality induces phenotypic plasticity in a wing polyphenic insect. Proc. Natl. Acad. Sci. USA 2018, 115, 7563–7568. [Google Scholar] [CrossRef]
- Radhakrishnan, S.; Literman, R.; Neuwald, J.; Severin, A.; Valenzuela, N. Transcriptomic responses to environmental temperature by turtles with temperaturedependent and genotypic sex determination assessed by RNAseq inform the genetic architecture of embryonic gonadal development. PLoS ONE 2017, 12, 1–34. [Google Scholar] [CrossRef] [PubMed]
- De Meyts, P.; Bianco, A.R.; Roth, J. Site-site interactions among Insulin Receptors. J. Biol. Chem. 1976, 251, 1877–1888. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Cell Line | INSR mRNA Changes | Ref. |
|---|---|---|---|
| Dexamethasone | IM9, UMR 106-01 | increase | [63,81,82] |
| Hydrocortisone | IM9 | increase | [66] |
| Androgen | HEp-2 | increase | [85] |
| Estradiol | MCF-7 | increase | [86] |
| Estradiol | HepG2, IM9 | no change | [87,88] |
| Estradiol | U-937 | decrease (reporter gene) | [89] |
| Vitamin D3 | U-937 | increase (reporter gene) | [90,91] |
| Progesterone | Endometrial stromal cells, T-47D | increase | [92,93] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Payankaulam, S.; Raicu, A.-M.; Arnosti, D.N. Transcriptional Regulation of INSR, the Insulin Receptor Gene. Genes 2019, 10, 984. https://doi.org/10.3390/genes10120984
Payankaulam S, Raicu A-M, Arnosti DN. Transcriptional Regulation of INSR, the Insulin Receptor Gene. Genes. 2019; 10(12):984. https://doi.org/10.3390/genes10120984
Chicago/Turabian StylePayankaulam, Sandhya, Ana-Maria Raicu, and David N. Arnosti. 2019. "Transcriptional Regulation of INSR, the Insulin Receptor Gene" Genes 10, no. 12: 984. https://doi.org/10.3390/genes10120984
APA StylePayankaulam, S., Raicu, A.-M., & Arnosti, D. N. (2019). Transcriptional Regulation of INSR, the Insulin Receptor Gene. Genes, 10(12), 984. https://doi.org/10.3390/genes10120984