Genome-Wide Association Studies for Methane Production in Dairy Cattle

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Measurement of CH4 Emissions
2.2. DNA Sampling and Genotyping
2.3. Quality Control of Genotypes
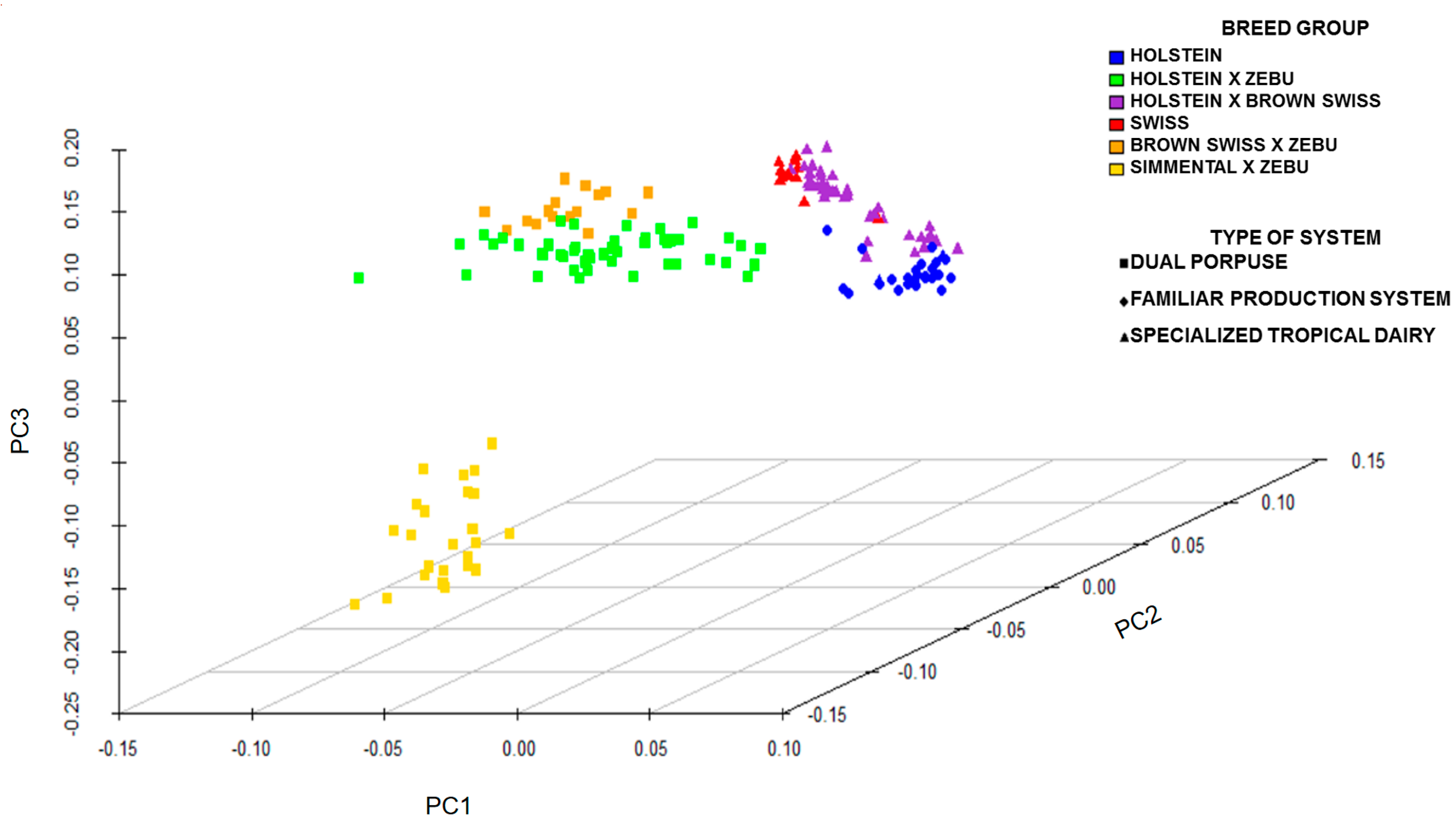
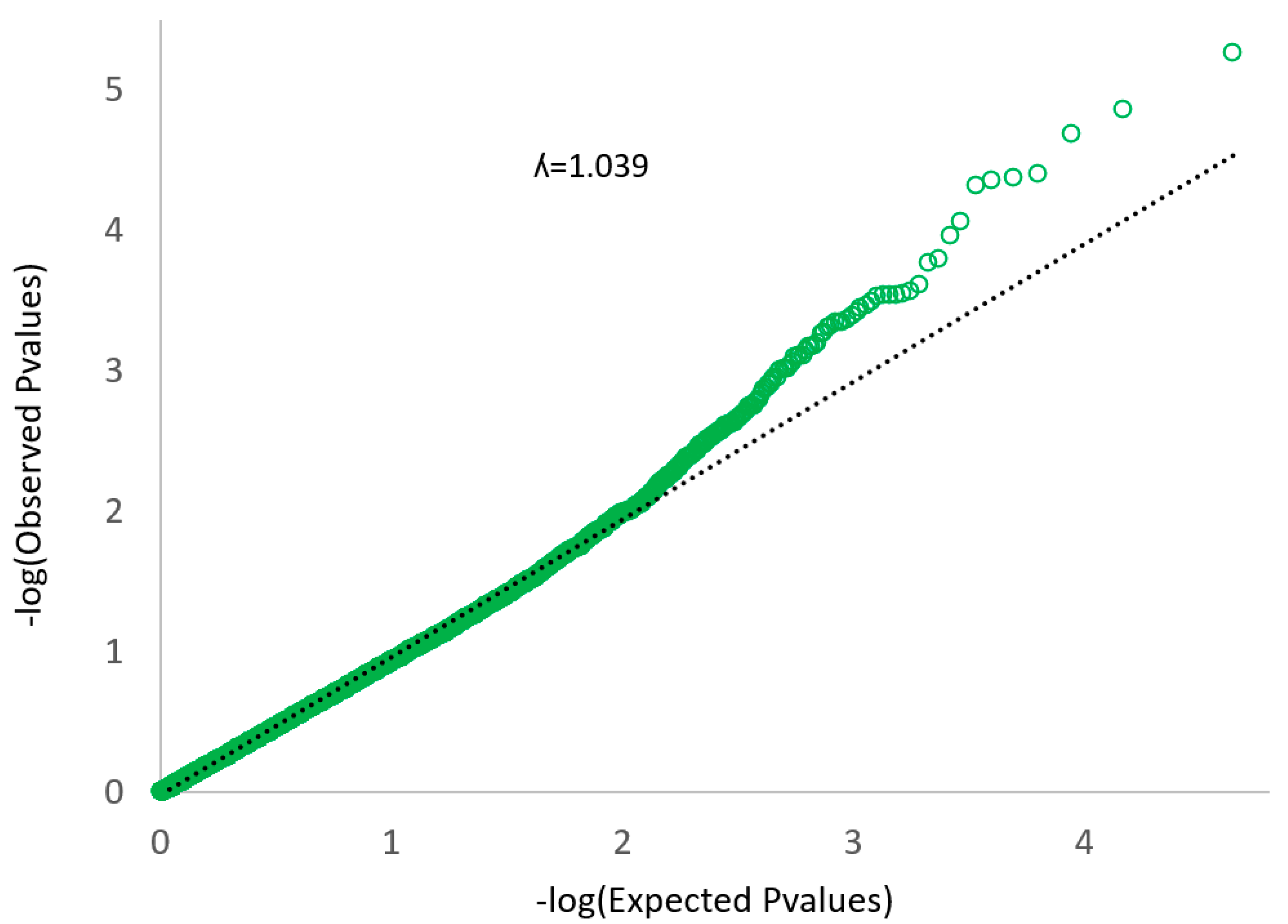
2.4. Statistical Analysis
- yijk = MEIm in the k-th observation in the i-th herd and j-th months in milk.
- μ = overall mean.
- Hi = fixed effect of the i-th herd.
- Mj = fixed effect of the j-th month in milk.
- ε(ij)k = CH4 production during milking residual in the k-th observation in the i-th herd and j-th months in milk.
2.5. Complete Genome Association
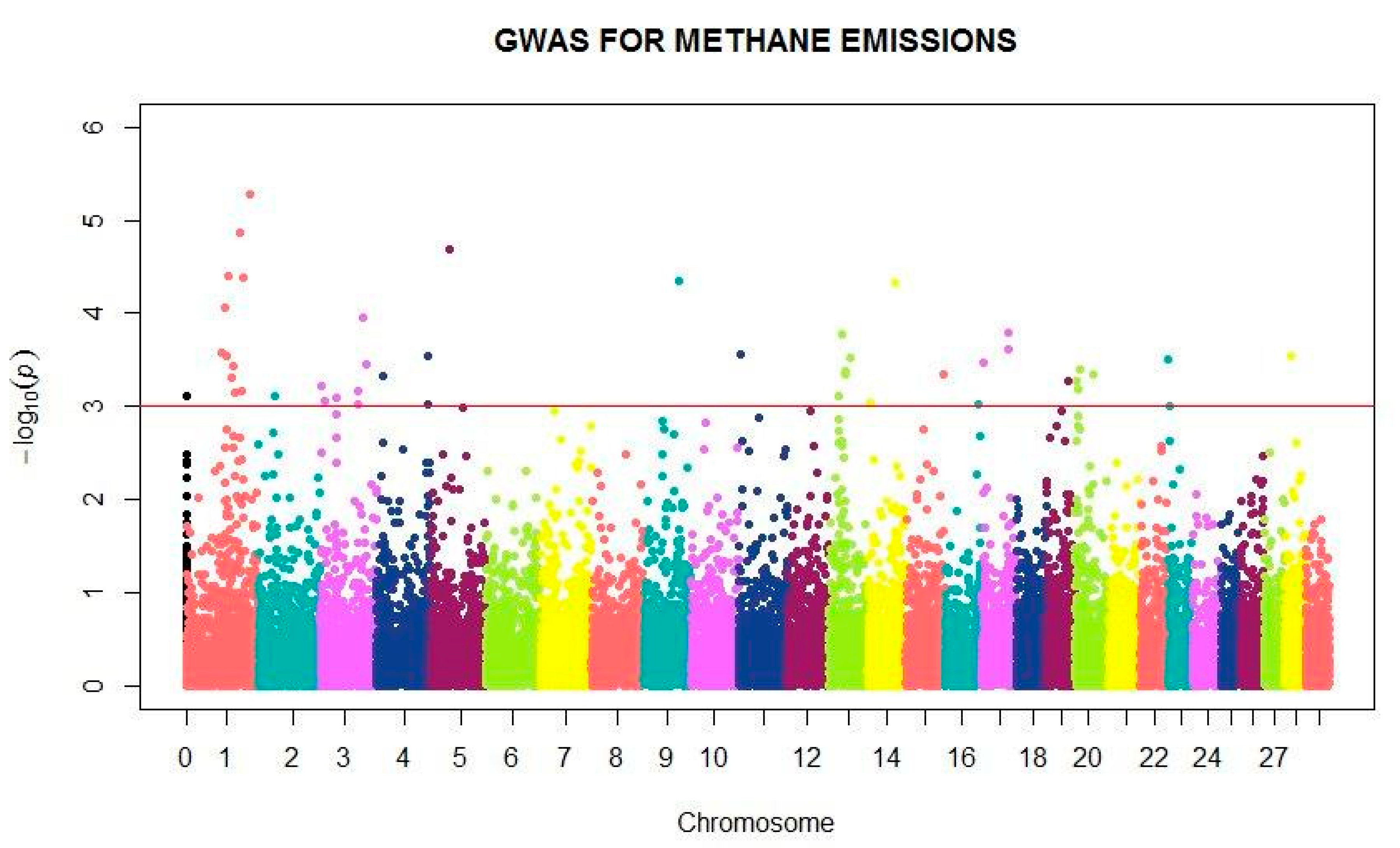
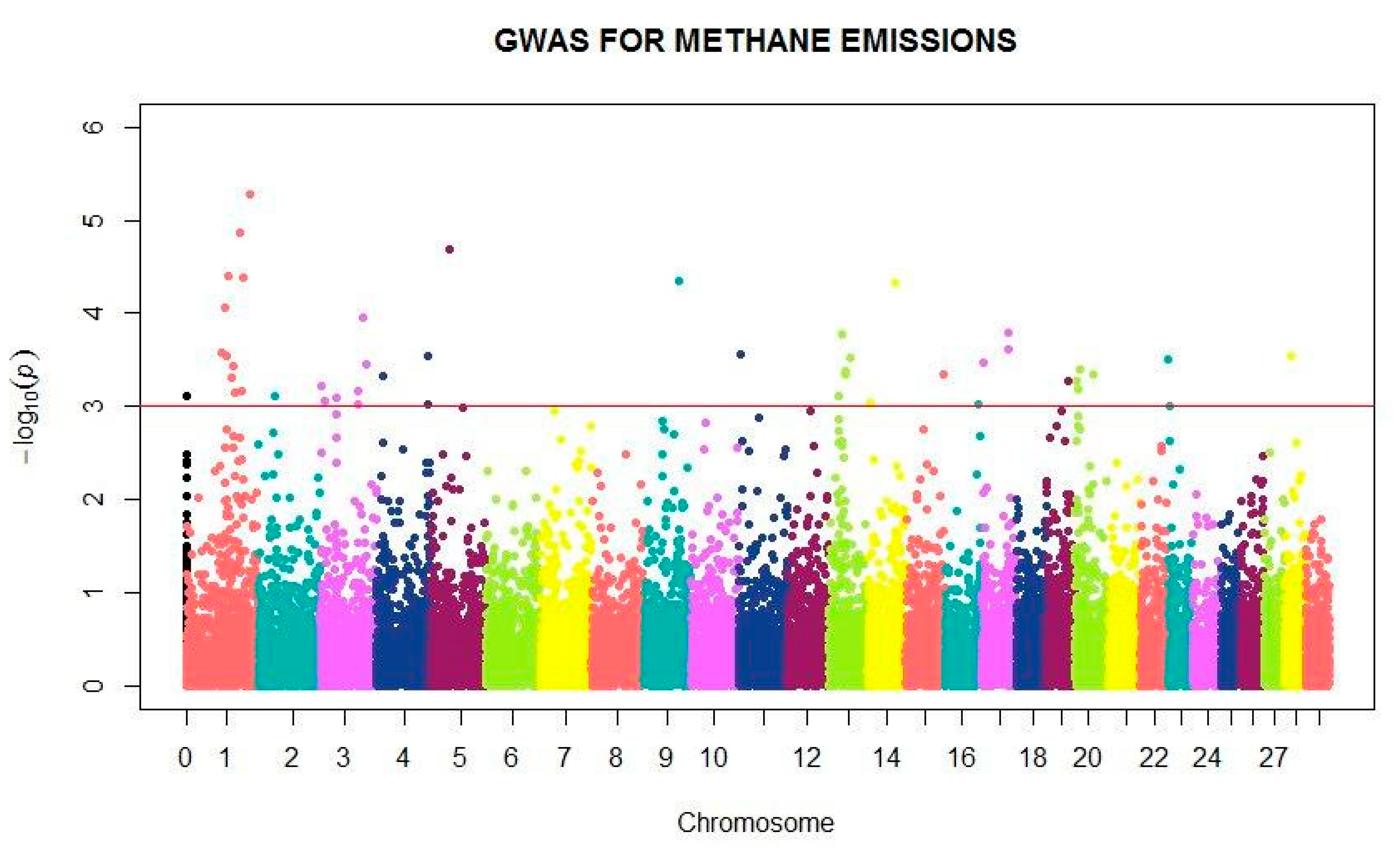
3. Results
4. Discussion
4.1. Markers Associated with Dairy Traits
4.2. Markers Associated with Beef Traits
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- OECD Publishing. OECD-FAO OECD-FAO Agricultural Outlook 2016-2025; OECD: Paris, France, 2016; ISBN 978-92-64-25323-0. [Google Scholar]
- Food and Agriculture Organization of the United Nations. Greenhouse Gas Emissions from the Dairy Sector; FAO: Roma, Italy, 2010. [Google Scholar]
- IPCC. Climate Change 2014: Mitigation of Climate Change; Cambridge University Press: New York, NY, USA, 2014; ISBN 978-11-07-65481-5. [Google Scholar]
- Wall, E.; Simm, G.; Moran, D. Developing breeding schemes to assist mitigation of greenhouse gas emissions. Animal 2010, 4, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Grainger, C.; Clarke, T.; McGinn, S.M.; Auldist, M.J.; Beauchemin, K.A.; Hannah, M.C.; Waghorn, G.C.; Clark, H.; Eckard, R.J. Methane emissions from dairy cows measured using the sulfur hexafluoride (SF6) tracer and chamber techniques. J. Dairy Sci. 2007, 90, 2755–2766. [Google Scholar] [CrossRef] [PubMed]
- Demeyer, D.; Fievez, V. Article de synthèse Ruminants et environnement: La méthanogenèse. Ann. Zootech. 2000, 49, 95–112. [Google Scholar] [CrossRef]
- Demeyer, D.I.; van Nevel, C.J. Methanogenesis, an integrated part of carbohydrate fermentation and its control. In Digestion and Metabolism in the Ruminant I; University of New England Publishing Unit: Armidale, Australia, 1975; pp. 366–382. [Google Scholar]
- Sosa, A.; Galindo, J.; Bocourt, R. Metanogénesis ruminal: Aspectos generales y manipulación para su control. Rev. Cuba. Cienc. Agrícola 2007, 41, 105–114. [Google Scholar]
- Mohammed, R.; McGinn, S.M.; Beauchemin, K.A. Prediction of enteric methane output from milk fatty acid concentrations and rumen fermentation parameters in dairy cows fed sunflower, flax, or canola seeds. J. Dairy Sci. 2011, 94, 6057–6068. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, H.; Huhtanen, P. Effects of the Ratio of Ruminal Propionate to Butyrate on Milk Yield and Blood Metabolites in Dairy Cows. J. Dairy Sci. 1996, 79, 851–861. [Google Scholar] [CrossRef]
- Pearson, T.A.; Manolio, T.A. How to Interpret a Genome-wide Association Study. Am. Med. Assoc. 2008, 299, 1335–1344. [Google Scholar] [CrossRef]
- García-Ruiz, A.; Cole, J.B.; VanRaden, P.M.; Wiggans, G.R.; Ruiz-López, F.J.; Tassell, C.P. Van Changes in genetic selection differentials and generation intervals in US Holstein dairy cattle as a result of genomic selection. Proc. Natl. Acad. Sci. USA 2016, 113, E3995–E4004. [Google Scholar] [CrossRef]
- Manzanilla-Pech, C.I.V.; De Haas, Y.; Hayes, B.J.; Veerkamp, R.F.; Khansefid, M.; Donoghue, K.A.; Arthur, P.F.; Pryce, J.E. Genomewide association study of methane emissions in angus beef cattle with validation in dairy cattle. J. Anim. Sci. 2016, 94, 4151–4166. [Google Scholar] [CrossRef]
- FAO. World Agriculture: Towards 2015/2030; Bruinsma, J., Ed.; Earthscan Publications Ltd: London, UK, 2003; ISBN 925-1048-355. [Google Scholar]
- Van Soest, P.J.; Robertson, J.B.; Lewis, B.A. Methods for Dietary Fiber, Neutral Detergent Fiber, and Nonstarch Polysaccharides in Relation to Animal Nutrition. J. Dairy Sci. 1991, 74, 3583–3597. [Google Scholar] [CrossRef]
- Weiss, W.P.; Conrad, H.R.; Pierre, N.R. A theoretically-based model for predicting total digestible nutrient values of forages and concentrates. Anim. Feed Sci. Technol. 1992, 39, 95–110. [Google Scholar] [CrossRef]
- Garnsworthy, P.C.; Craigon, J.; Hernandez-Medrano, J.H.; Saunders, N. On-farm methane measurements during milking correlate with total methane production by individual dairy cows. J. Dairy Sci. 2012, 95, 3166–3180. [Google Scholar] [CrossRef] [PubMed]
- SAS, I. Base SAS® 9.4 Procedures Guide: Stadistical Procedures; SAS Institute Inc.: Cary, NC, USA, 2013. [Google Scholar]
- Slager, S.L.; Iturria, S.J. Genome-wide linkage analysis of systolic blood pressure: A comparison of two approaches to phenotype definition. BMC Genet. 2003, 4, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Golden Helix Inc. SNP Genome-Wide Association Tutorial; Golden Helix, Inc.: Bozeman, MT, USA, 2017. [Google Scholar]
- Ehret, G.B. Genome-wide association studies: Contribution of genomics to understanding blood pressure and essential hypertension. Curr. Hypertens. Rep. 2010, 12, 17–25. [Google Scholar] [CrossRef]
- Bell, M.; Saunders, N.; Wilcox, R.; Homer, E.; Goodman, J.; Craigon, J.; Garnsworthy, P. Methane emissions among individual dairy cows during milking quantified by eructation peaks or ratio with carbon dioxide. J. Dairy Sci. 2014, 97, 6536–6546. [Google Scholar] [CrossRef]
- Beauchemin, K.A.; McAllister, T.A.; McGinn, S.M. Dietary mitigation of enteric methane from cattle. CAB Rev. Perspect. Agric. Vet. Sci. Nutr. Nat. Resour. 2009, 4, 1–19. [Google Scholar] [CrossRef]
- Barendse, W. Haplotype analysis improved evidence for candidate genes for intramuscular fat percentage from a genome wide association study of cattle. PLoS ONE 2011, 6, e29601. [Google Scholar] [CrossRef]
- Bouwman, A.C.; Bovenhuis, H.; Visker, M.H.; Am Van Arendonk, J. Genome-wide association of milk fatty acids in Dutch dairy cattle. BMC Genet. 2011, 12, 1–12. [Google Scholar] [CrossRef]
- Bouwman, A.C.; Visker, M.H.; Am Van Arendonk, J.; Bovenhuis, H. Genomic regions associated with bovine milk fatty acids in both summer and winter milk samples. BMC Genet. 2012, 13, 1–13. [Google Scholar] [CrossRef]
- Peters, S.O.; Kizilkaya, K.; Garrick, D.J.; Fernando, R.L.; Reecy, J.M.; Weaber, R.L.; Silver, G.A.; Thomas, M.G. Bayesian genome-wide association analysis of growth and yearling ultrasound measures of carcass traits in brangus heifers. J. Anim. Sci. 2012, 90, 3398–3409. [Google Scholar] [CrossRef] [PubMed]
- Saatchi, M.; Garrick, D.J.; Tait Jr, R.G.; Mayes, M.S.; Drewnoski, M.; Schoonmaker, J.; Diaz, C.; Beitz, D.C.; Reecy, J.M. Genome-wide association and prediction of direct genomic breeding values for composition of fatty acids in Angus beef cattle a. BMC Genom. 2013, 14, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Saatchi, M.; Beever, J.E.; Decker, J.E.; Faulkner, D.B.; Freetly, H.C.; Hansen, S.L.; Yampara-Iquise, H.; Johnson, K.A.; Kachman, S.D.; Kerley, M.S.; et al. QTLs associated with dry matter intake, metabolic mid-test weight, growth and feed efficiency have little overlap across 4 beef cattle studies. BMC Genom. 2014, 15, 1–14. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, P.; Cesar, A.; Nascimento, M.L.; Chaves, A.S.; Tizioto, P.C.; Tullio, R.R.; Lanna, D.; Rosa, A.N.; Sonstegard, T.S.; Mourao, G.B.; et al. Identification of genomic regions associated with feed efficiency in Nelore cattle. BMC Genet. 2014, 15, 1–10. [Google Scholar] [CrossRef]
- Den Berg, S.; Vandenplas, J.; Eeuwijk, F.A.; Lopes, M.S.; Veerkamp, R.F. Significance testing and genomic inflation factor using high-density genotypes or whole-genome sequence data. J. Anim. Breed. Genet. 2019, 418–429. [Google Scholar] [CrossRef]
- Kaler, A.S.; Purcell, L.C. Estimation of a significance threshold for genome-wide association studies. BMC Genom. 2019, 20, 618. [Google Scholar] [CrossRef]
- Chilliard, Y.; Martin, C.; Roual, J.; Doreau, M. Milk fatty acids in dairy cows fed whole crude linseed, extruded linseed, or linseed oil, and their relationship with methane output. J. Dairy Sci. 2009, 92, 5199–5211. [Google Scholar] [CrossRef]
- Dijkstra, J.; van Zijderveld, S.; Apajalahti, J.; Bannink, A.; Gerrits, W.; Newbold, J. Relationships between methane production and milk fatty acid profiles in dairy cattle. Anim. Feed Sci. Technol. 2011, 166–167, 590–595. [Google Scholar] [CrossRef]
- Van Gastelen, S.; Hettinga, K.A.; Dijkstra, J. Relationships between methane emission of Holstein Friesian dairy cows and fatty acids, volatile metabolites and non-volatile metabolites in milk. Animal 2017, 11, 1539–1548. [Google Scholar] [CrossRef]
- Fitzsimons, C.; Kenny, D.A.; Deighton, M.H.; Fahey, A.G.; McGee, M. Methane emissions, body composition, and rumen fermentation traits of beef heifers differing in residual feed intake. J. Anim. Sci. 2013, 91, 5789–5800. [Google Scholar] [CrossRef]
- Pickering, N.K.; Oddy, V.H.; Basarab, J.; Cammack, K.; Hayes, B.; Hegarty, R.S.; Lassen, J.; McEwan, J.C.; Miller, S.; Pinares-Patiño, C.S.; et al. Animal board invited review: Genetic possibilities to reduce enteric methane emissions from ruminants. Animal 2015, 9, 1431–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feeding | Herds | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| A | B | C | D | E | F | G | H | I | |
| Concentrate (kg) | 3.5 | 2 | 2 | 11 | 6.6 | 10.4 | 6.3 | 3.5 | 3.5 |
| Corn silage (kg) | 5.2 | ||||||||
| Alfalfa hay (kg) | 1.7 | ||||||||
| Corn stubble (kg) | 5.4 | 8 | 4.8 | 3.3 | |||||
| Saccharum sinense (kg) | 20 | 20 | 20 | ||||||
| Grazing | |||||||||
| Cynodon plectostachyus Vanderyst (kg) | X | X | X | X | |||||
| Sorghum vulgare (kg) | X | ||||||||
| Megathyrsus maximus (kg) | X | X | |||||||
| Brachiaria decumbens Stapf (kg) | X | X | |||||||
| Desmodium ovalifolium (kg) | X | ||||||||
| Andropogon gayanus Kunth (kg) | X | ||||||||
| Pennisetum sp. (kg) | X | ||||||||
| Digitaría decumbens Stend (kg) | X | ||||||||
| Native grasses | X | X | X | X | |||||
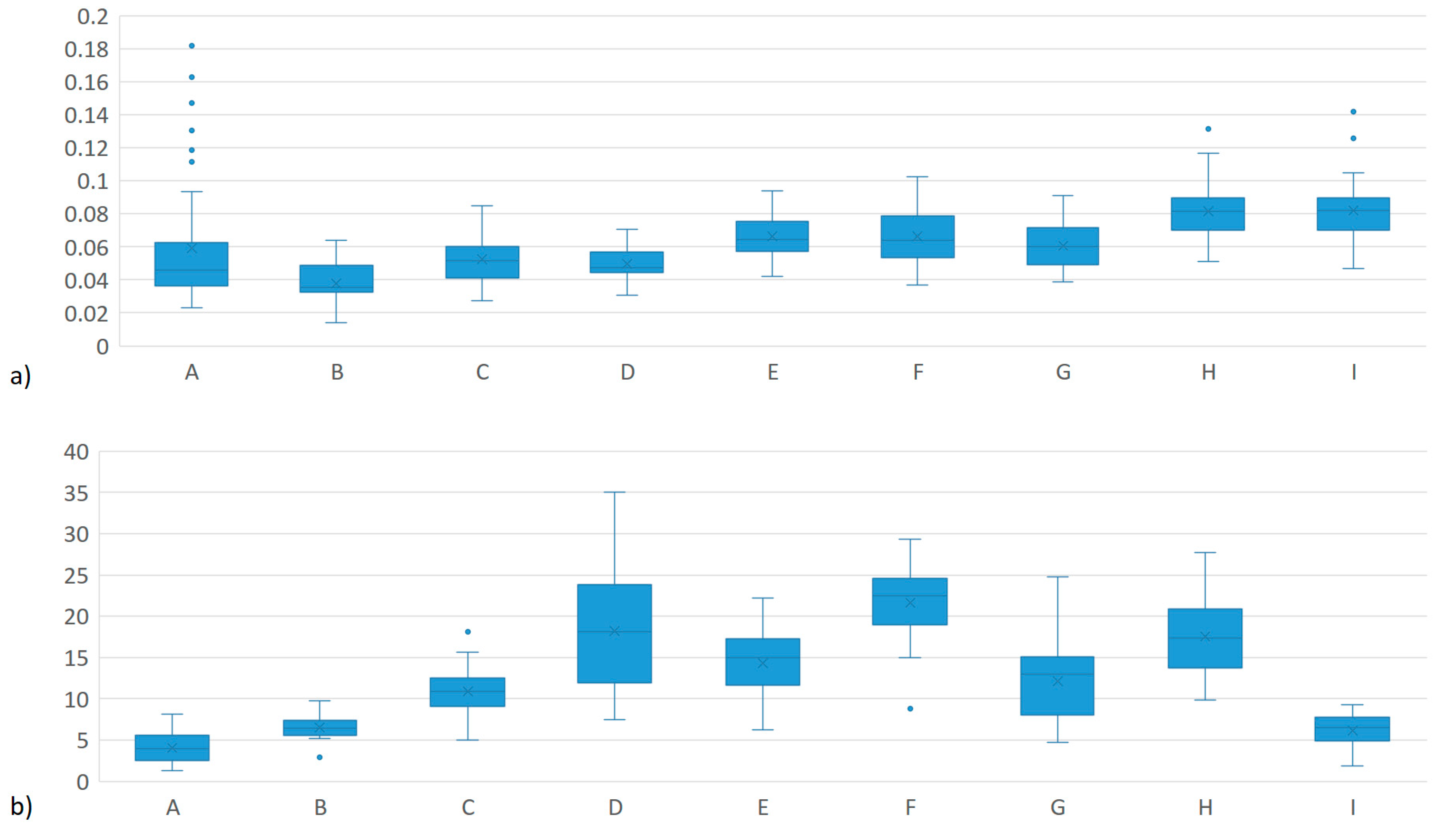
| System | Herd | n | MEIm (mg/L) | MY (kg per day) | ||
|---|---|---|---|---|---|---|
| µ | σ | µ | σ | |||
| Dual Purpose | A | 51 | 0.076 ab | 0.076 | 4.2 g | 2.0 |
| B | 16 | 0.038 bc | 0.012 | 6.5 g | 1.6 | |
| C | 33 | 0.053 abc | 0.015 | 10.9 ef | 2.8 | |
| TOTAL | 100 | 0.062 A | 0.057 | 6.8 A | 3.7 | |
| Familiar Production System | D | 24 | 0.050 abc | 0.010 | 18.2 abc | 7.6 |
| E | 16 | 0.067 abc | 0.013 | 14.3 de | 3.8 | |
| F | 33 | 0.067 abc | 0.017 | 21.6 ab | 4.8 | |
| G | 31 | 0.061 abc | 0.014 | 12.1 def | 4.4 | |
| TOTAL | 104 | 0.062 A | 0.015 | 15.9 C | 6.1 | |
| Specialized Tropical Dairy | H | 38 | 0.082 a | 0.017 | 17.5 bc | 4.7 |
| I | 38 | 0.082 abc | 0.018 | 6.1 g | 2.0 | |
| TOTAL | 76 | 0.082 B | 0.017 | 11.8 B | 6.8 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calderón-Chagoya, R.; Hernandez-Medrano, J.H.H.; Ruiz-López, F.J.J.; Garcia-Ruiz, A.; Vega-Murillo, V.E.E.; Montano-Bermudez, M.; Arechavaleta-Velasco, M.E.E.; Gonzalez-Padilla, E.; Mejia-Melchor, E.I.I.; Saunders, N.; et al. Genome-Wide Association Studies for Methane Production in Dairy Cattle. Genes 2019, 10, 995. https://doi.org/10.3390/genes10120995
Calderón-Chagoya R, Hernandez-Medrano JHH, Ruiz-López FJJ, Garcia-Ruiz A, Vega-Murillo VEE, Montano-Bermudez M, Arechavaleta-Velasco MEE, Gonzalez-Padilla E, Mejia-Melchor EII, Saunders N, et al. Genome-Wide Association Studies for Methane Production in Dairy Cattle. Genes. 2019; 10(12):995. https://doi.org/10.3390/genes10120995
Chicago/Turabian StyleCalderón-Chagoya, R., J.H. H. Hernandez-Medrano, F.J. J. Ruiz-López, A. Garcia-Ruiz, V.E. E. Vega-Murillo, M. Montano-Bermudez, M.E. E. Arechavaleta-Velasco, E. Gonzalez-Padilla, E.I. I. Mejia-Melchor, N. Saunders, and et al. 2019. "Genome-Wide Association Studies for Methane Production in Dairy Cattle" Genes 10, no. 12: 995. https://doi.org/10.3390/genes10120995
APA StyleCalderón-Chagoya, R., Hernandez-Medrano, J. H. H., Ruiz-López, F. J. J., Garcia-Ruiz, A., Vega-Murillo, V. E. E., Montano-Bermudez, M., Arechavaleta-Velasco, M. E. E., Gonzalez-Padilla, E., Mejia-Melchor, E. I. I., Saunders, N., Bonilla-Cardenas, J. A. A., Garnsworthy, P. C. C., & Román-Ponce, S. I. I. (2019). Genome-Wide Association Studies for Methane Production in Dairy Cattle. Genes, 10(12), 995. https://doi.org/10.3390/genes10120995