PVTree: A Sequential Pattern Mining Method for Alignment Independent Phylogeny Reconstruction


Abstract
:1. Introduction
2. Materials and Methods
2.1. Simulated and Real Data
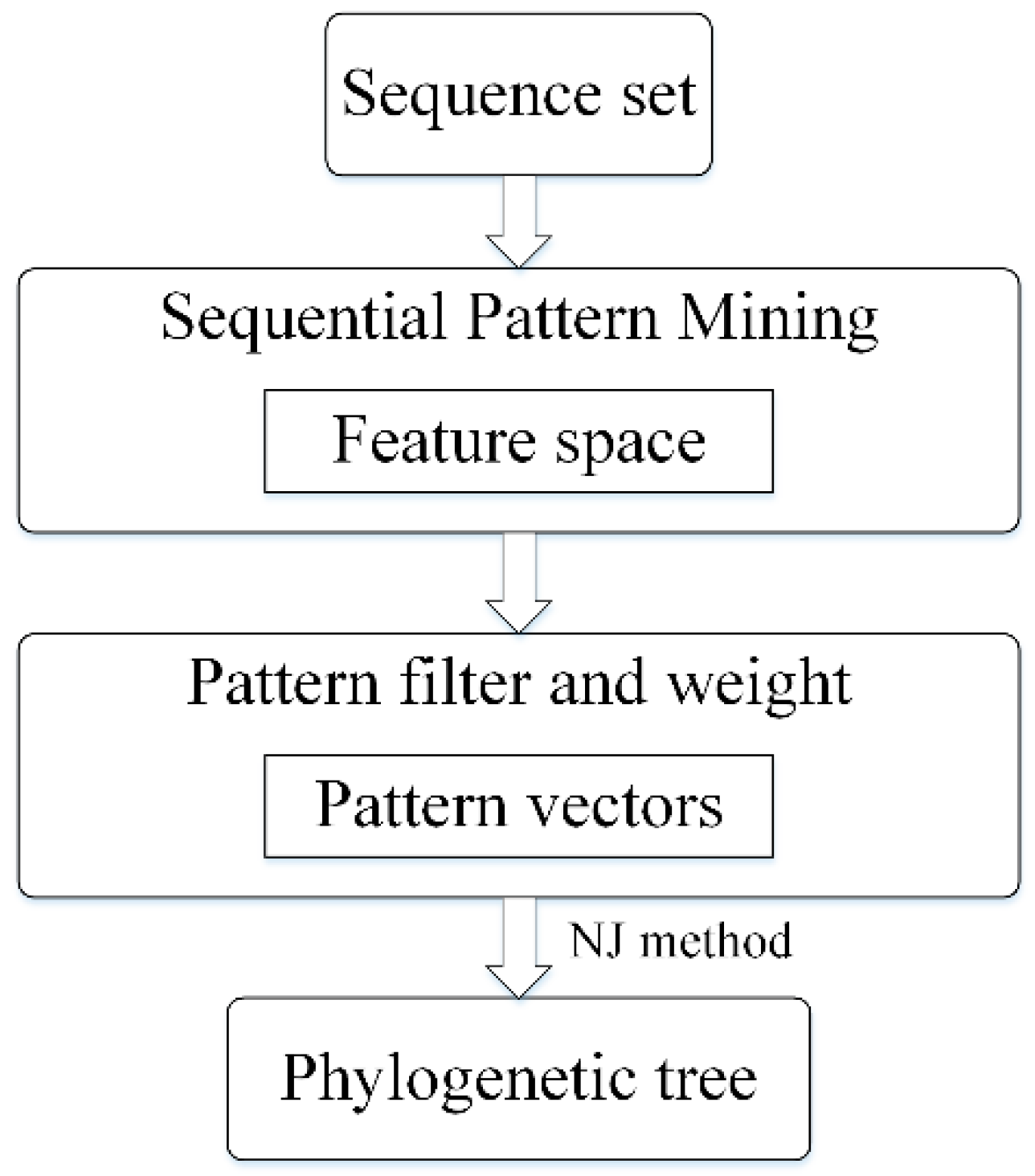
2.2. Sequential Pattern Mining from Unaligned Sequences
| Algorithm 1 Mining Sequential Pattern (Note: min_support is the minimum support threshold, min_mon_wc is the minimal non-wildcard threshold, and last (a) is the last amino acid or nucleotides) |
| 1: Mining (,) |
| 2: if || ≥ then |
| 3: if () ≥ and last () ≠ then |
| 4: if is a closed pattern then |
| 5: report , || |
| 6: for each residue or do |
| 7: : = with or appended to it |
| 8: Mining (,) |
2.3. Converting a Sequence to a Weighted and Non-Redundant Pattern Vector
2.4. Distance Matrix and Tree Construction
2.5. Reference Tree and Tree Comparison
2.6. Code Availability
3. Results
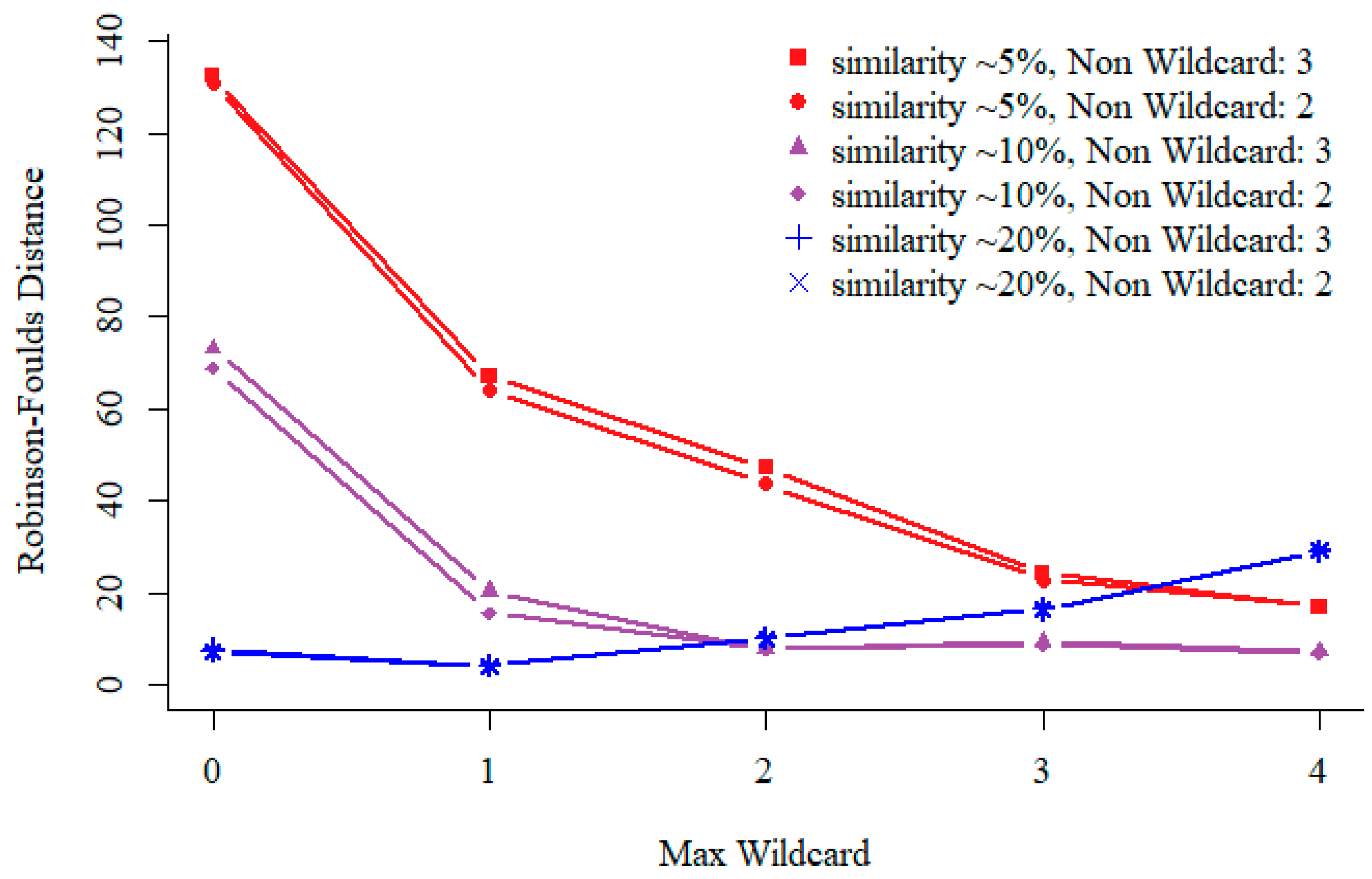
3.1. Non-Wildcard and Max-Wildcard
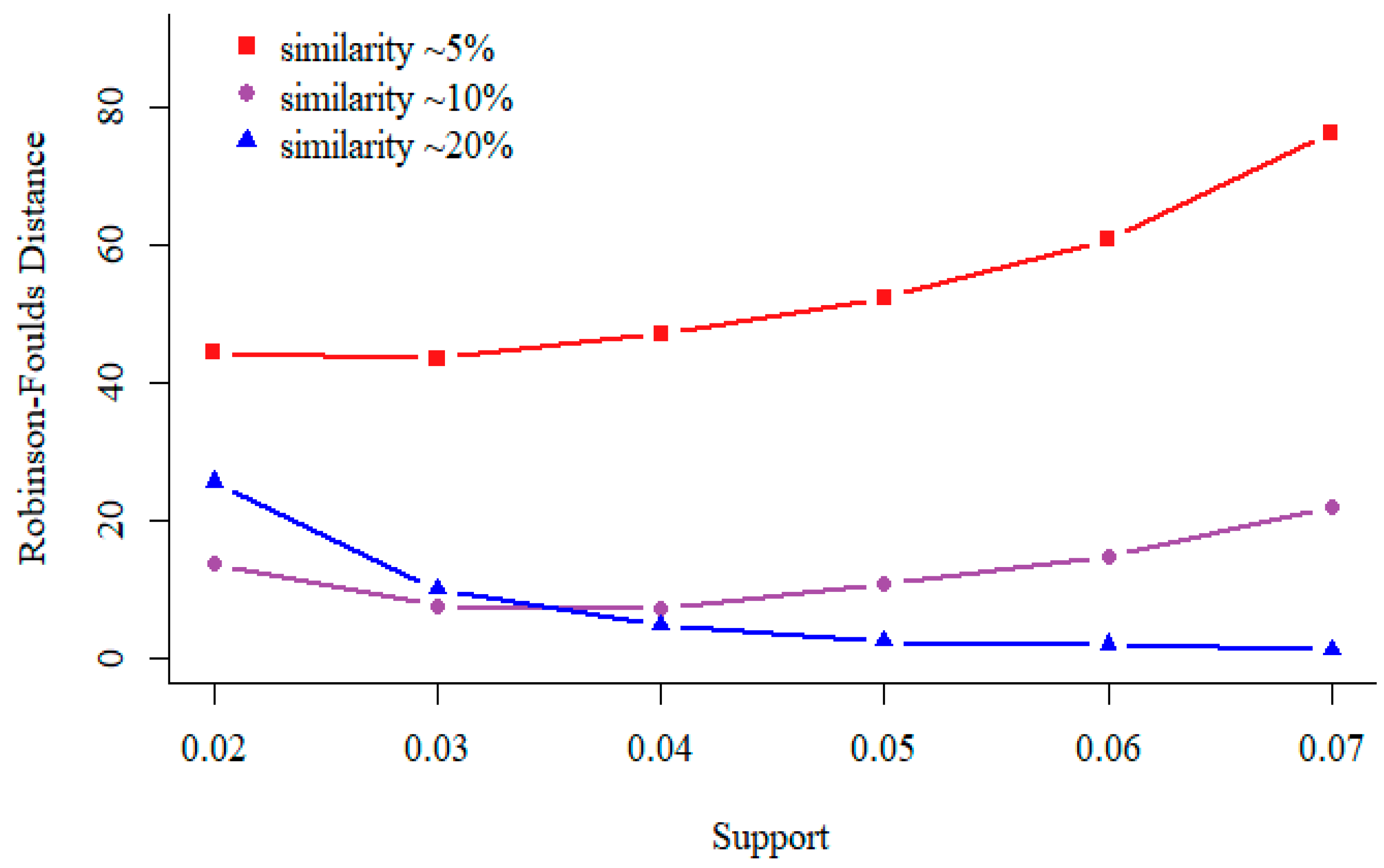
3.2. Support in Sequential Pattern Mining
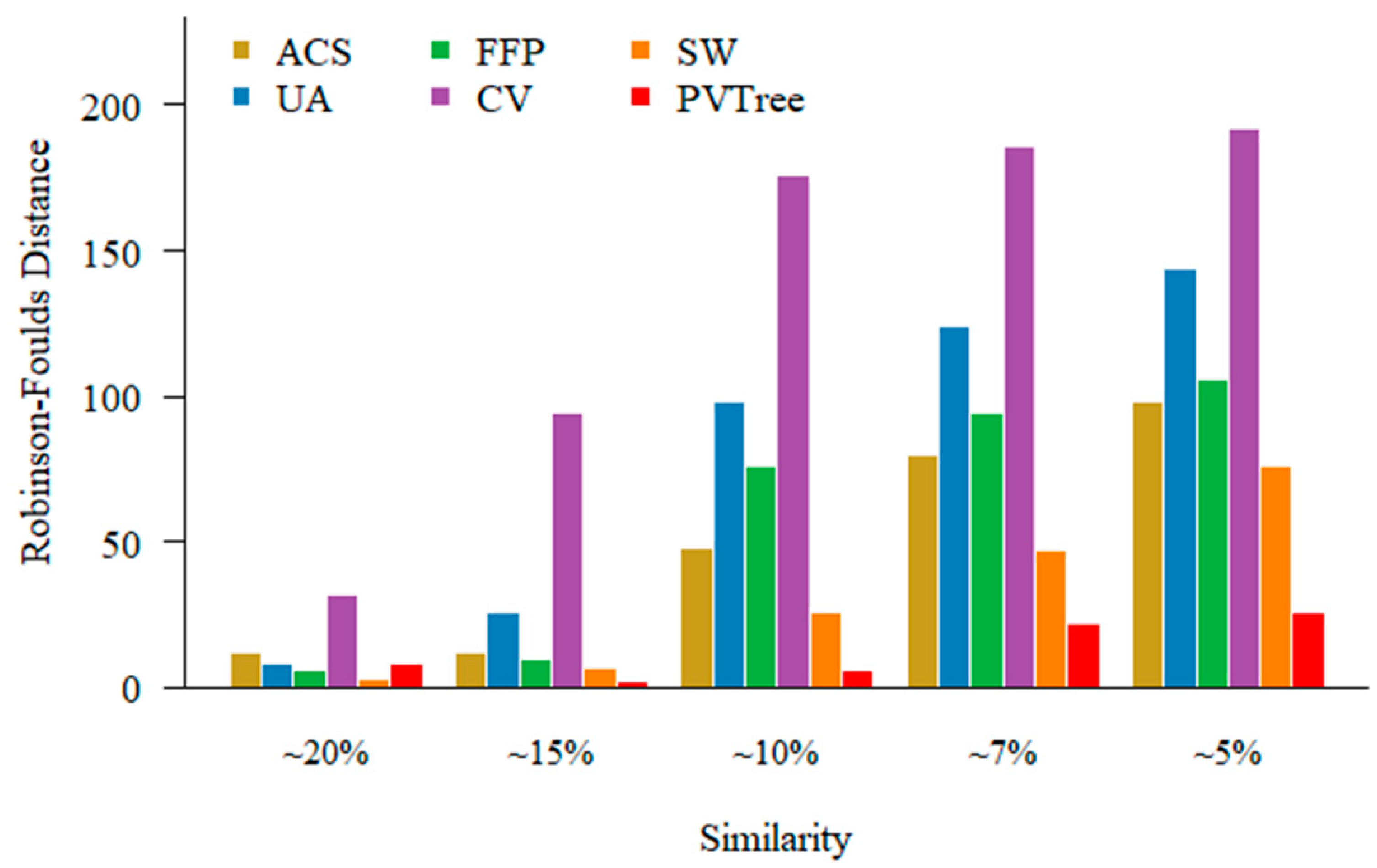
3.3. Sequence Similarity and Performance
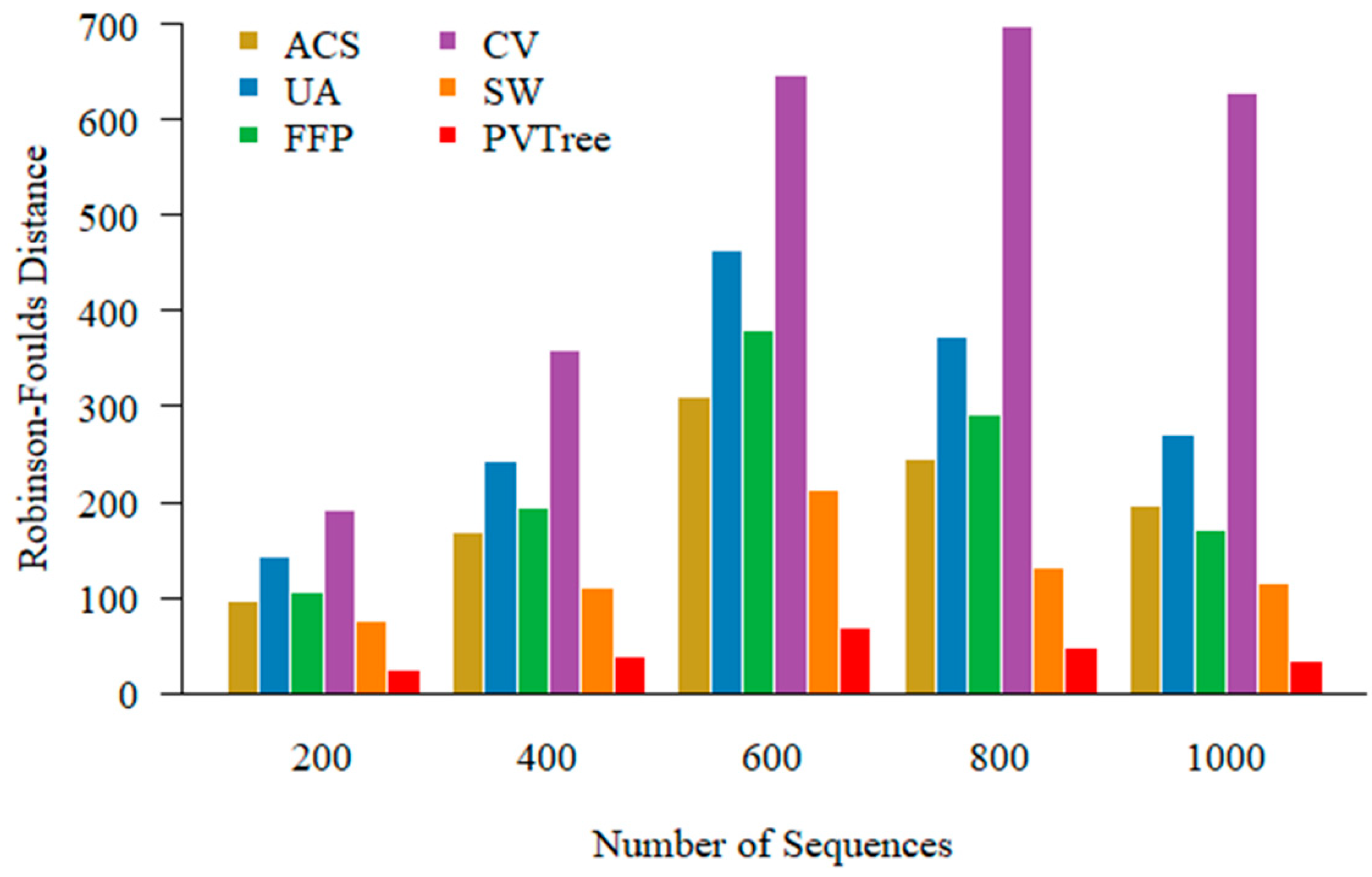
3.4. Numbers of the Sequences and Performance
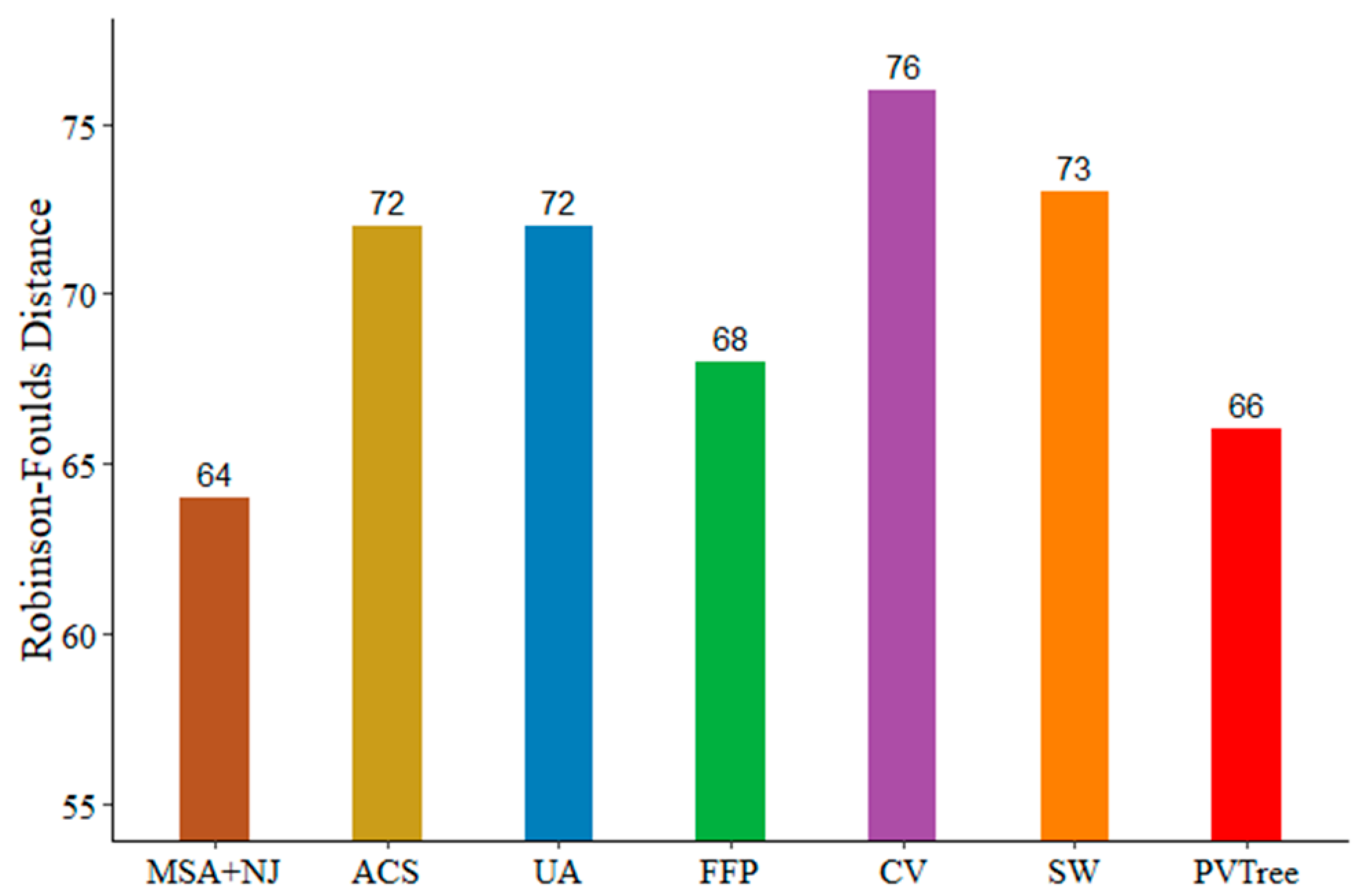
3.5. BALIBASE Data Sets
3.6. Runtime
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [PubMed]
- Löytynoja, A.; Goldman, N. An algorithm for progressive multiple alignment of sequences with insertions. Proc. Nat. Acad. Sci. USA 2005, 102, 10557–10562. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Gascuel, O.A. simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. What is dynamic programming? Nat. Biotechnol. 2004, 22, 909–910. [Google Scholar] [CrossRef]
- Wong, K.M.; Suchard, M.A.; Huelsenbeck, J.P. Alignment uncertainty and genomic analysis. Science 2008, 319, 473–476. [Google Scholar] [CrossRef]
- Nelesen, S.; Liu, K.; Zhao, D.; Linder, C.R.; Warnow, T. The effect of the guide tree on multiple sequence alignments and subsequent phylogenetic analyses. In Proceedings of the Pacific Symposium on Biocomputing, Kohala Coast, HI, USA, 4–8 January 2008; p. 25. [Google Scholar]
- Rost, B. Twilight zone of protein sequence alignments. Protein Eng. 1999, 12, 85–94. [Google Scholar] [CrossRef] [Green Version]
- Zielezinski, A.; Vinga, S.; Almeida, J.; Karlowski, W.M. Alignment-free sequence comparison: Benefits, applications, and tools. Genome Biol. 2017, 18, 186. [Google Scholar] [CrossRef]
- Sims, G.E.; Jun, S.R.; Wu, G.A.; Kim, S.H. Alignment-Free Genome Comparison with Feature Frequency Profiles (FFP) and Optimal Resolutions. Proc. Natl. Acad. Sci. USA 2009, 106, 2677–2682. [Google Scholar] [CrossRef]
- Qi, J.; Luo, H.; Hao, B. CVTree: A phylogenetic tree reconstruction tool based on whole genomes. Nucleic Acids Res. 2004, 32, 45–47. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [PubMed]
- Sokal, R.R.; Michener, C.D. A statistical method for evaluating systematic relationships. Univ. Kans. Sci. Bull. 1958, 38, 1409–1438. [Google Scholar]
- Leimeister, C.A.; Boden, M.; Horwege, S.; Lindner, S.; Morgenstern, B. Fast alignment-free sequence comparison using spaced-word frequencies. Bioinformatics 2014, 30, 1991–1999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulitsky, I.; Burstein, D.; Tuller, T.; Chor, B. The average common substring approach to phylogenomic reconstruction. J. Comput. Mol. Cell Biol. 2006, 13, 336–350. [Google Scholar] [CrossRef] [PubMed]
- Comin, M.; Verzotto, D. Alignment-free phylogeny of whole genomes using underlying subwords. Algorithms Mol. Biol. 2012, 7, 34. [Google Scholar] [CrossRef] [Green Version]
- Ye, K.; Kosters, W.A.; Ijzerman, A.P. An efficient, versatile and scalable pattern growth approach to mine frequent patterns in unaligned protein sequences. Bioinformatics 2007, 23, 687–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoye, J.; Evers, D.; Meyer, F. Rose: Generating sequence families. Bioinformatics 1998, 14, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Koehl, P.; Ripp, R.; Poch, O. BAliBASE 3.0: Latest developments of the multiple sequence alignment benchmark. Proteins Struct. Funct. Bioinform. 2005, 61, 127–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Han, J.; Kamber, M.; Pei, J. Data Mining: Concepts and Techniques, 3rd ed.; Morgan Kaufmann: Waltham, MA, USA, 2011; pp. 1–13. [Google Scholar]
- Jones, K.S. A Statistical Interpretation of term specificity and its application in retrieval. J. Doc. 1972, 28, 11–21. [Google Scholar] [CrossRef]
- Lin, J. Divergence measures based on the Shannon entropy. IEEE Trans. Inf. Theor. 1991, 37, 145–151. [Google Scholar] [CrossRef] [Green Version]
- Kullback, S. The Kullback-Leibler Distance. Am. Statist. 1987, 41, 340–341. [Google Scholar]
- Felsenstein, J. PHYLIP-Phylogeny inference package (Version 3.2). Cladistics 1989, 5, 164–166. [Google Scholar]
- Robinson, D.F.; Foulds, L.R. Comparison of phylogenetic trees. Math. Biosci. 1981, 53, 131–147. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Hu, Q.; Guo, M.; Wang, G. HAlign: Fast multiple similar DNA/RNA sequence alignment based on the centre star strategy. Bioinformatics 2015, 31, 2475–2481. [Google Scholar] [CrossRef] [Green Version]
- Zou, Q.; Wan, S.; Zeng, X.; Ma, Z.S. Reconstructing evolutionary trees in parallel for massive sequences. BMC Syst. Biol. 2017, 11, 100. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Runtimes (s) |
|---|---|
| ACS | 1.459 |
| UA | 171.868 |
| FFP (l = 5) | 133.908 |
| CV (k = 3) | 3.455 |
| SW (k = 4) | 1.403 |
| PVTree (2-1) | 5.571 |
| PVTree (2-2) | 12.782 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, Y.; Yang, X.; Lin, J.; Ye, K. PVTree: A Sequential Pattern Mining Method for Alignment Independent Phylogeny Reconstruction. Genes 2019, 10, 73. https://doi.org/10.3390/genes10020073
Kang Y, Yang X, Lin J, Ye K. PVTree: A Sequential Pattern Mining Method for Alignment Independent Phylogeny Reconstruction. Genes. 2019; 10(2):73. https://doi.org/10.3390/genes10020073
Chicago/Turabian StyleKang, Yongyong, Xiaofei Yang, Jiadong Lin, and Kai Ye. 2019. "PVTree: A Sequential Pattern Mining Method for Alignment Independent Phylogeny Reconstruction" Genes 10, no. 2: 73. https://doi.org/10.3390/genes10020073
APA StyleKang, Y., Yang, X., Lin, J., & Ye, K. (2019). PVTree: A Sequential Pattern Mining Method for Alignment Independent Phylogeny Reconstruction. Genes, 10(2), 73. https://doi.org/10.3390/genes10020073