Development of a Transformation Method for Metschnikowia borealis and other CUG-Serine Yeasts


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Microbial Strains and Growth Conditions
2.2. Codon-Optimization of KanMX and Sh ble
2.3. Yeast DNA Isolation
2.4. Lithium Acetate Transformations
2.5. Electroporation Conditions
2.6. Locating the Insertion Site
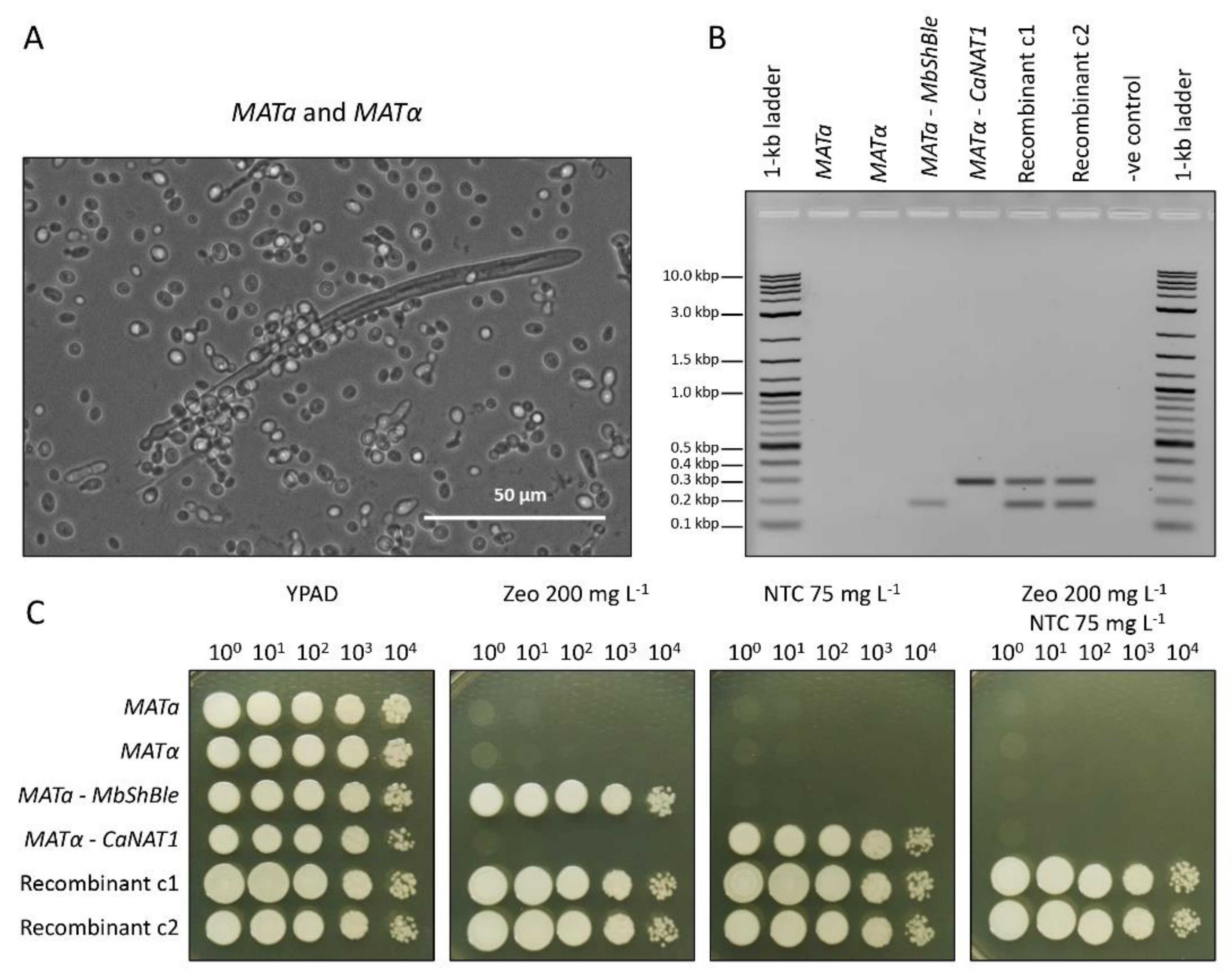
2.7. Mating Experiments
3. Results
3.1. Identification of Antibiotics that Inhibit Growth of M. borealis
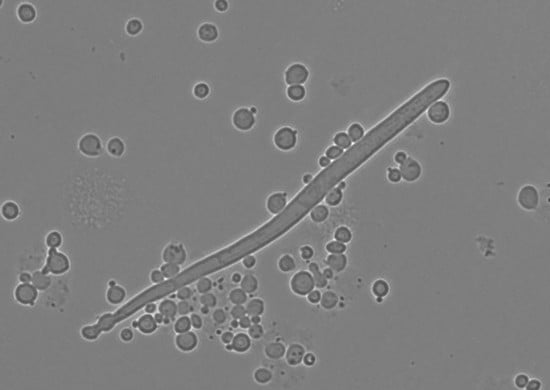
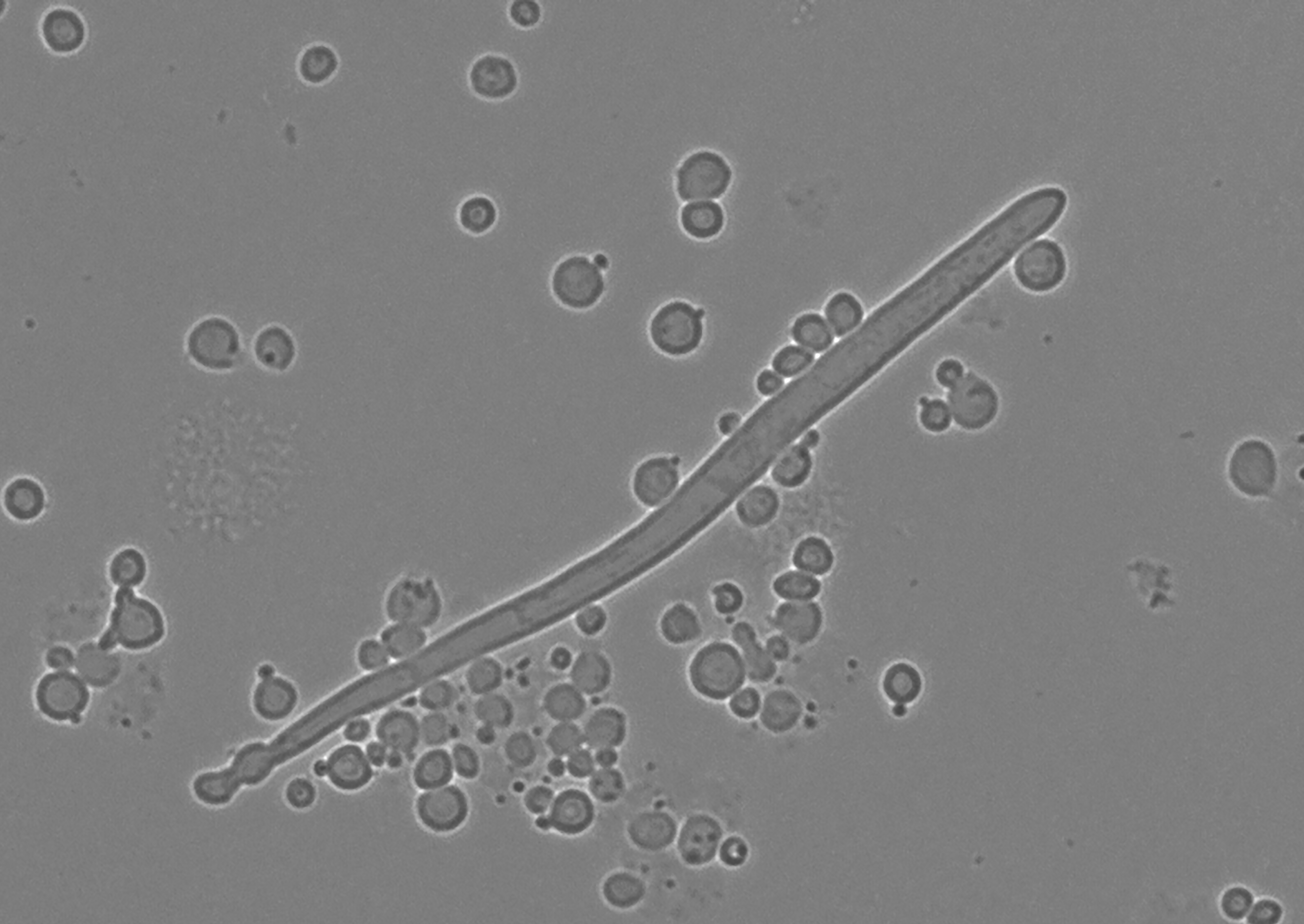
3.2. Growth Rate of M. borealis
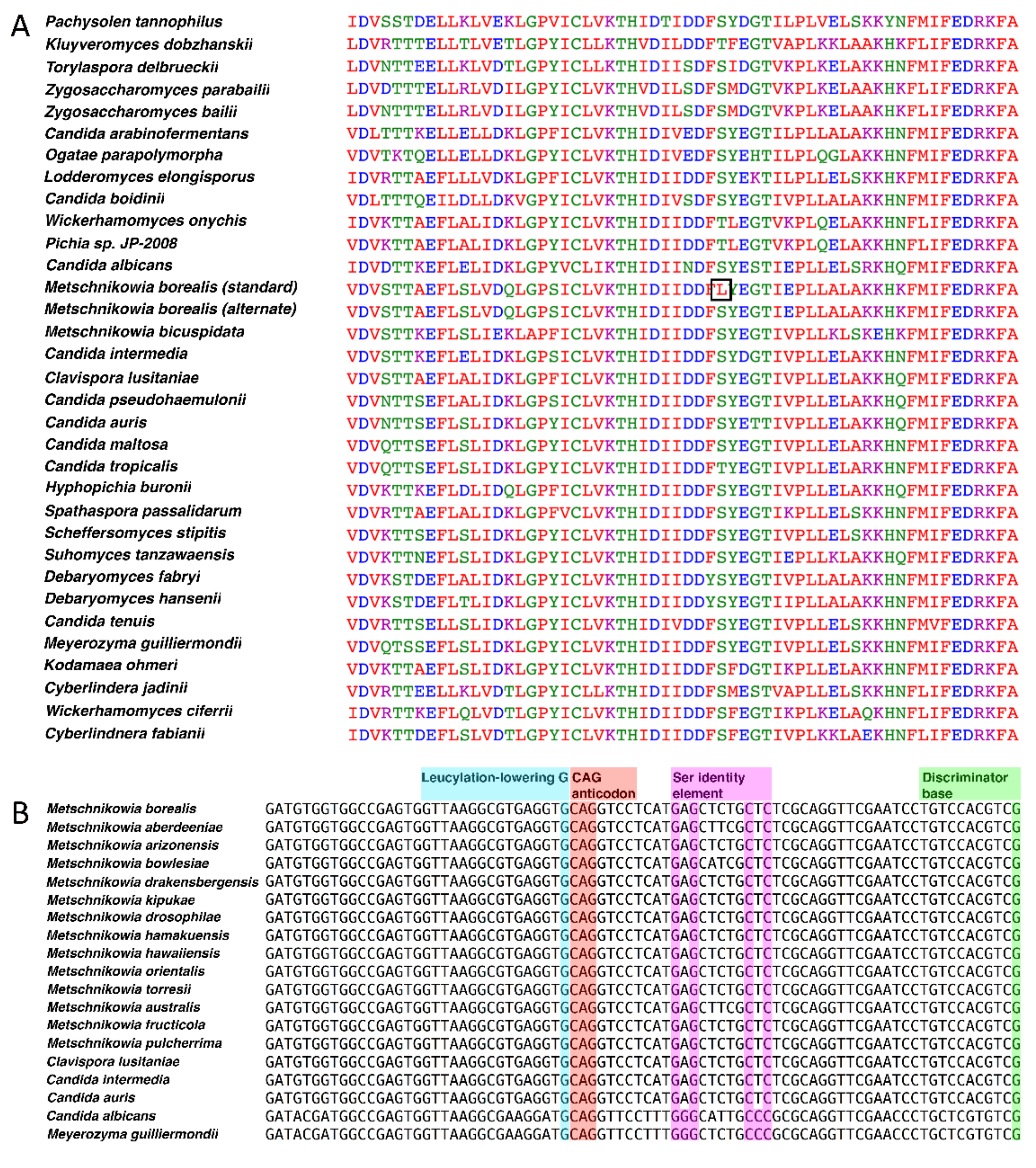
3.3. M. borealis Codon Usage
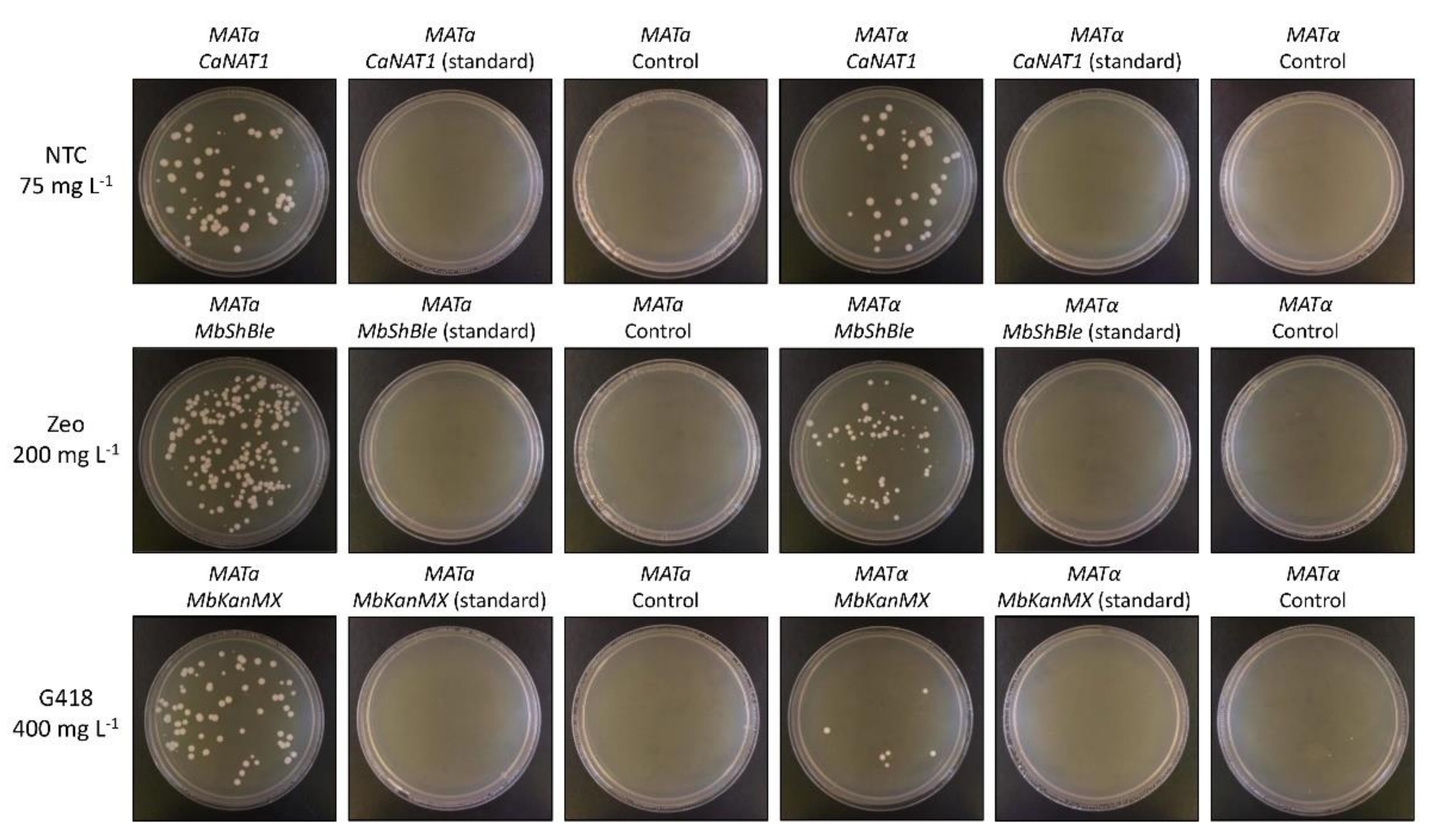
3.4. Transformation of M. borealis with Codon-Optimized Cassettes
3.5. Location of Insertion Site
3.6. Mating Experiments
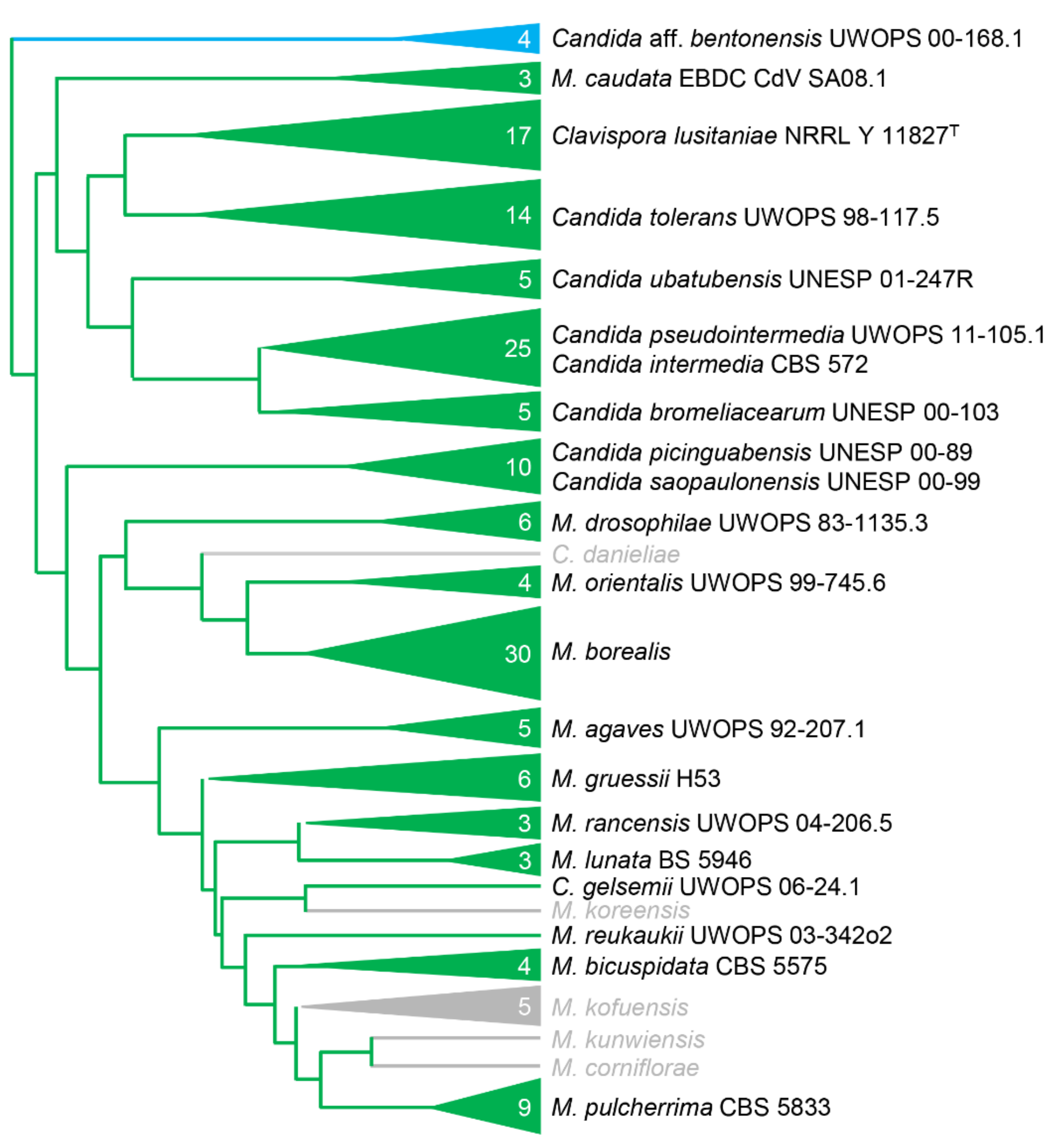
3.7. Transformation of other CUG-Ser Yeasts
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Deng, J.; Deng, L.; Su, S.; Zhang, M.; Lin, X.; Wei, L.; Minai, A.A.; Hassett, D.J.; Lu, L.J. Investigating the predictability of essential genes across distantly related organisms using an integrative approach. Nucl. Acids Res. 2011, 39, 795–807. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Ou, H.-Y.; Zhang, C.-T. DEG: A database of essential genes. Nucl. Acids Res. 2004, 32, D271–D272. [Google Scholar] [CrossRef] [PubMed]
- De Backer, M.D.; Maes, D.; Vandoninck, S.; Logghe, M.; Contreras, R.; Luyten, W.H. Transformation of Candida albicans by electroporation. Yeast Chichester Engl. 1999, 15, 1609–1618. [Google Scholar] [CrossRef]
- Yildirim, S.; Thompson, M.G.; Jacobs, A.C.; Zurawski, D. V.; Kirkup, B.C. Evaluation of parameters for high efficiency transformation of Acinetobacter baumannii. Sci. Rep. 2016, 6, 22110. [Google Scholar] [CrossRef] [PubMed]
- Lachance, M.-A. Metschnikowia: Half tetrads, a regicide and the fountain of youth. Yeast Chichester Engl. 2016. [Google Scholar] [CrossRef] [PubMed]
- González-Pombo, P.; Pérez, G.; Carrau, F.; Guisán, J.M.; Batista-Viera, F.; Brena, B.M. One-step purification and characterization of an intracellular beta-glucosidase from Metschnikowia pulcherrima. Biotechnol. Lett. 2008, 30, 1469–1475. [Google Scholar] [CrossRef] [PubMed]
- Macwilliam, I.C. A survey of the antibiotic powers of yeasts. J. Gen. Microbiol. 1959, 21, 410–414. [Google Scholar] [CrossRef]
- Marinoni, G.; Piškur, J.; Research, M.L.-F. Ascospores of large-spored Metschnikowia species are genuine meiotic products of these yeasts. FEMS Yeast Res. 2003, 3, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Nigro, F.; Sialer, M.M.F.; Gallitelli, D. Transformation of Metschnikowia pulcherrima 320, biocontrol agent of storage rot, with the Green Fluorescent Protein gene. J. Plant Pathol. 1999, 81, 205–208. [Google Scholar]
- Lachance, M.-A.; Hurtado, E.; Hsiang, T. A stable phylogeny of the large-spored Metschnikowia clade. Yeast Chichester Engl. 2016, 33, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Kawai, S.; Hashimoto, W.; Murata, K. Transformation of Saccharomyces cerevisiae and other fungi. Bioeng. Bugs 2010, 1, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Lachance, M.-A.; Rosa, C.A.; Starmer, W.T.; Schlag-Edler, B.; Baker, J.S.F.; Bowles, J.M. Metschnikowia continentalis var. borealis, Metschnikowia continentalis var. continentalis, and Metschnikowia hibisci, new heterothallic haploid yeasts from ephemeral flowers and associated insects. Can. J. Microbiol. 1998, 44, 279–288. [Google Scholar] [CrossRef]
- Puigbò, P.; Guzmán, E.; Romeu, A.; Garcia-Vallvé, S. OPTIMIZER: A web server for optimizing the codon usage of DNA sequences. Nucl. Acids Res. 2007, 35, W126–W131. [Google Scholar] [CrossRef] [PubMed]
- Gietz, R.D. Yeast transformation by the LiAc/SS carrier DNA/PEG method. Methods Mol. Biol. Clifton 2014, 1205, 1–12. [Google Scholar] [CrossRef]
- Froyd, C.A.; Kapoor, S.; Dietrich, F.; Rusche, L.N. The deacetylase Sir2 from the yeast Clavispora lusitaniae lacks the evolutionarily conserved capacity to generate subtelomeric heterochromatin. PLoS Genet. 2013, 9, e1003935. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinform. Oxf. Engl. 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mühlhausen, S.; Kollmar, M. Molecular phylogeny of sequenced saccharomycetes reveals polyphyly of the alternative yeast codon usage. Genome Biol. Evol. 2014, 6, 3222–3237. [Google Scholar] [CrossRef] [PubMed]
- Riley, R.; Haridas, S.; Wolfe, K.H.; Lopes, M.R.; Hittinger, C.T.; Göker, M.; Salamov, A.A.; Wisecaver, J.H.; Long, T.M.; Calvey, C.H.; et al. Comparative genomics of biotechnologically important yeasts. Proc. Natl. Acad. Sci. USA 2016, 113, 9882–9887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krassowski, T.; Coughlan, A.Y.; Shen, X.-X.; Zhou, X.; Kominek, J.; Opulente, D.A.; Riley, R.; Grigoriev, I. V.; Maheshwari, N.; Shields, D.C.; et al. Evolutionary instability of CUG-Leu in the genetic code of budding yeasts. Nat. Commun. 2018, 9, 1887. [Google Scholar] [CrossRef] [PubMed]
- Sugita, T.; Nakase, T. Non-universal usage of the leucine CUG codon and the molecular phylogeny of the genus Candida. Syst. Appl. Microbiol. 1999, 22, 79–86. [Google Scholar] [CrossRef]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucl. Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Guo, W.; Köhler, J.R. CaNAT1, a heterologous dominant selectable marker for transformation of Candida albicans and other pathogenic Candida species. Infect. Immunity 2005, 73, 1239–1242. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gordon, Z.B.; Soltysiak, M.P.M.; Leichthammer, C.; Therrien, J.A.; Meaney, R.S.; Lauzon, C.; Adams, M.; Lee, D.K.; Janakirama, P.; Lachance, M.-A.; et al. Development of a Transformation Method for Metschnikowia borealis and other CUG-Serine Yeasts. Genes 2019, 10, 78. https://doi.org/10.3390/genes10020078
Gordon ZB, Soltysiak MPM, Leichthammer C, Therrien JA, Meaney RS, Lauzon C, Adams M, Lee DK, Janakirama P, Lachance M-A, et al. Development of a Transformation Method for Metschnikowia borealis and other CUG-Serine Yeasts. Genes. 2019; 10(2):78. https://doi.org/10.3390/genes10020078
Chicago/Turabian StyleGordon, Zachary B., Maximillian P.M. Soltysiak, Christopher Leichthammer, Jasmine A. Therrien, Rebecca S. Meaney, Carolyn Lauzon, Matthew Adams, Dong Kyung Lee, Preetam Janakirama, Marc-André Lachance, and et al. 2019. "Development of a Transformation Method for Metschnikowia borealis and other CUG-Serine Yeasts" Genes 10, no. 2: 78. https://doi.org/10.3390/genes10020078
APA StyleGordon, Z. B., Soltysiak, M. P. M., Leichthammer, C., Therrien, J. A., Meaney, R. S., Lauzon, C., Adams, M., Lee, D. K., Janakirama, P., Lachance, M.-A., & Karas, B. J. (2019). Development of a Transformation Method for Metschnikowia borealis and other CUG-Serine Yeasts. Genes, 10(2), 78. https://doi.org/10.3390/genes10020078