MYC Oncogene Contributions to Release of Cell Cycle Brakes



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
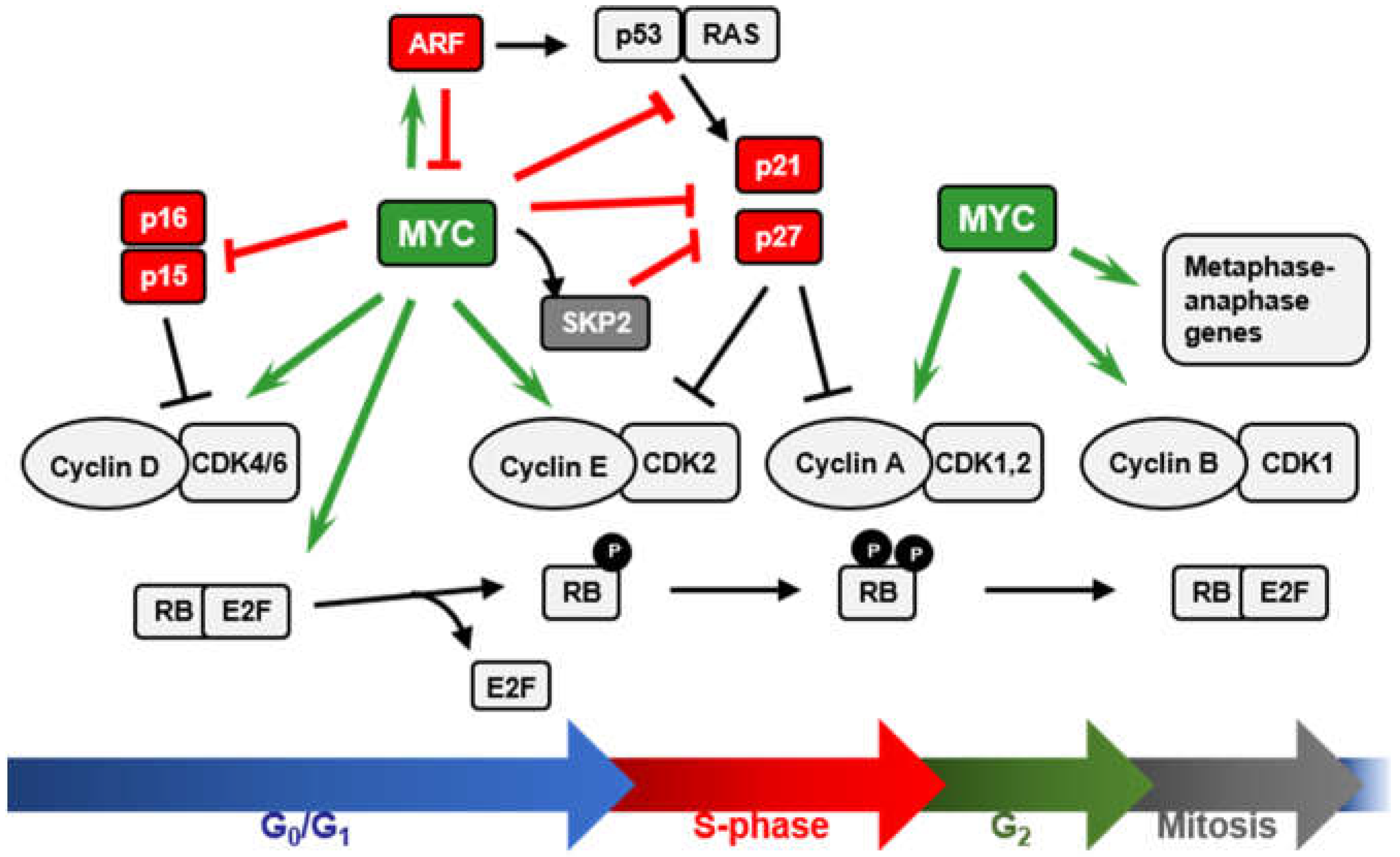
2. MYC and the INK4A/ARF/INK4B Locus
2.1. MYC and p15INK4B Regulation
2.2. MYC Regulation of ARF Expression
2.3. ARF-Mediated Regulation of MYC Activity
3. MYC and p21 Regulation
3.1. MYC-Mediated p21 Repression by Direct Recruitment to Its Core Promoter Region
3.2. MYC-Dependent Switch from Cell-Cycle Arrest to Apoptosis by Inhibiting p53-Dependent Activation of p21 Expression
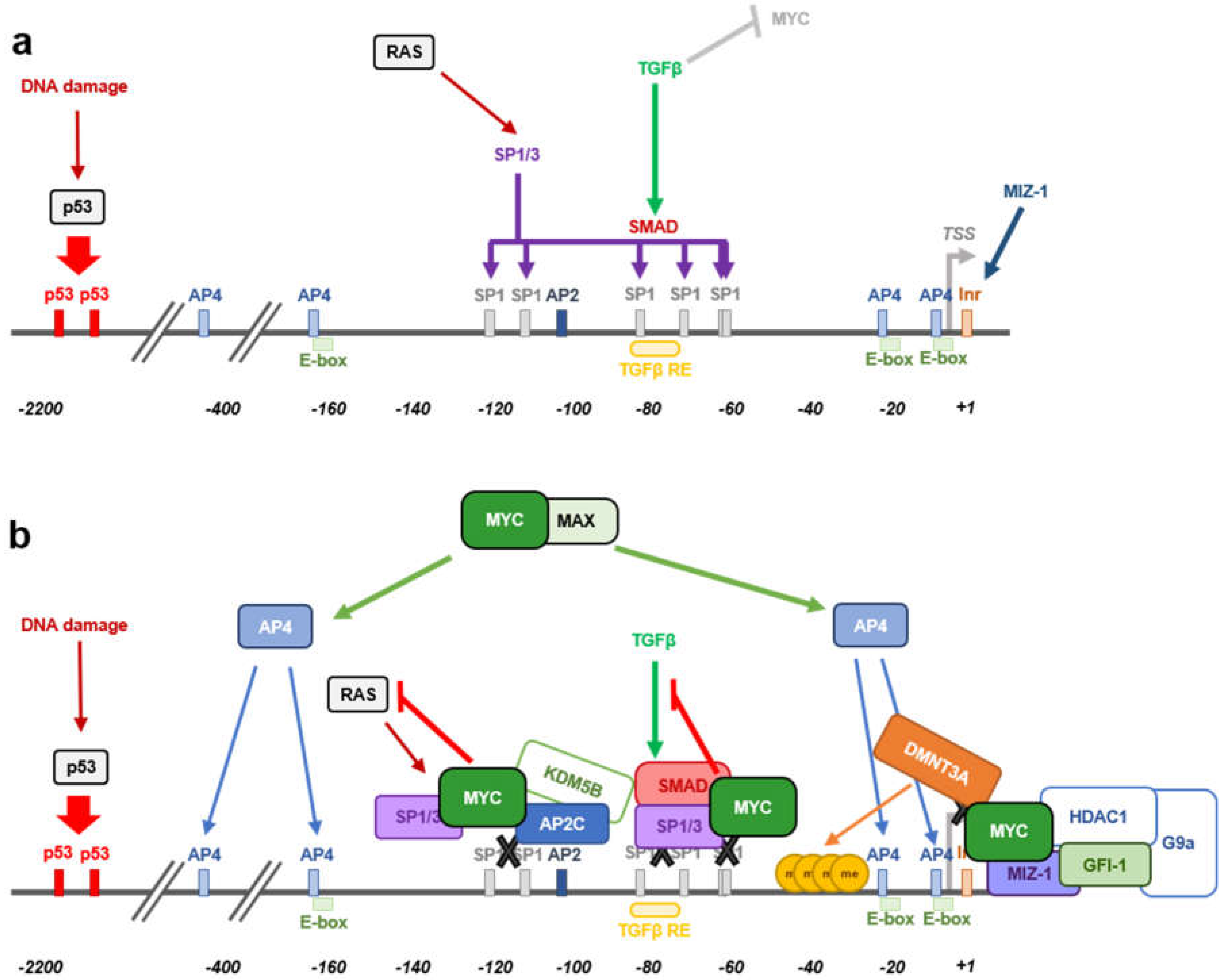
3.3. MYC-Mediated Inhibition of RAS-Induced CDKN1A Expression
3.4. MYC-Indirect Repression of CDKN1A Expression
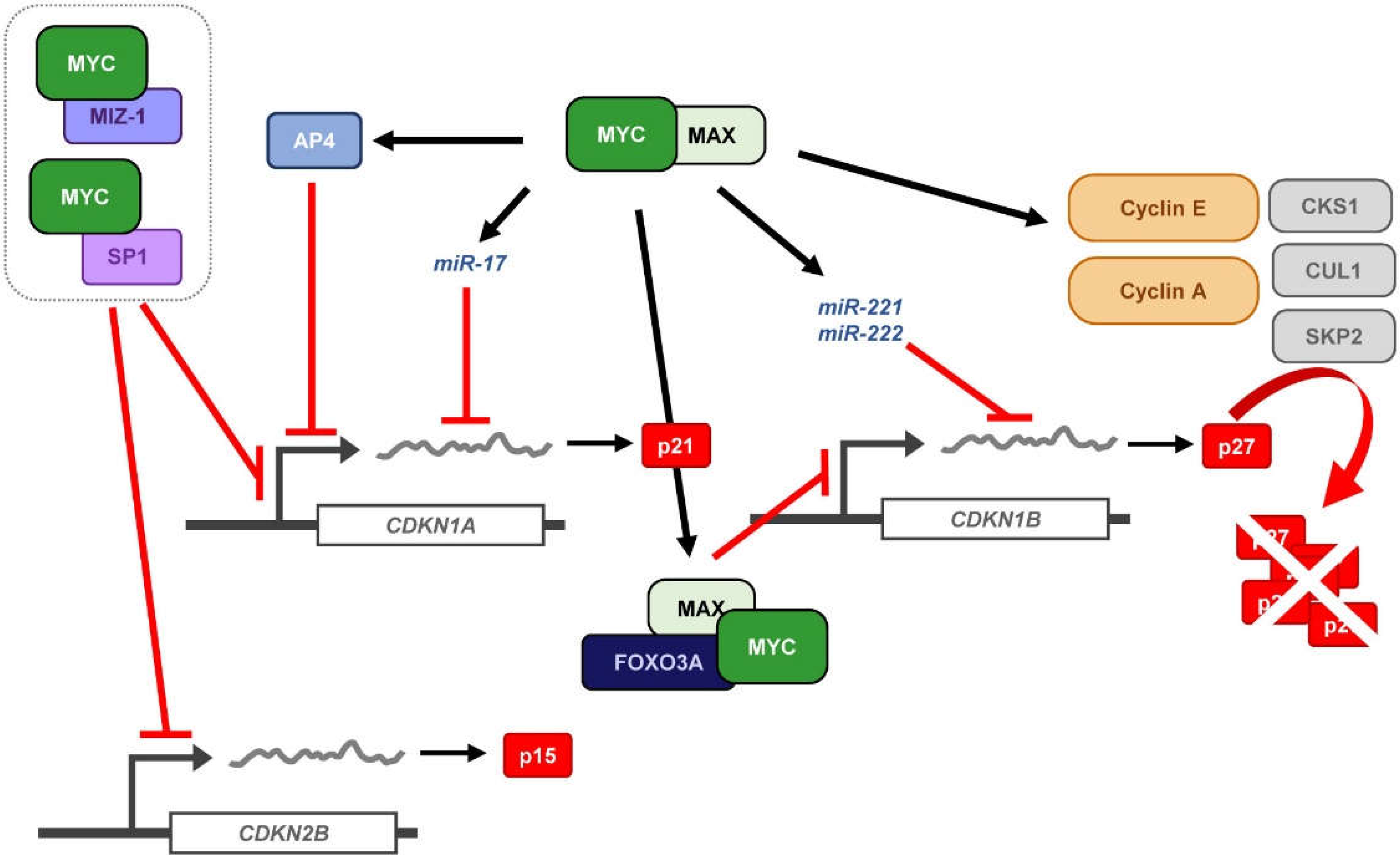
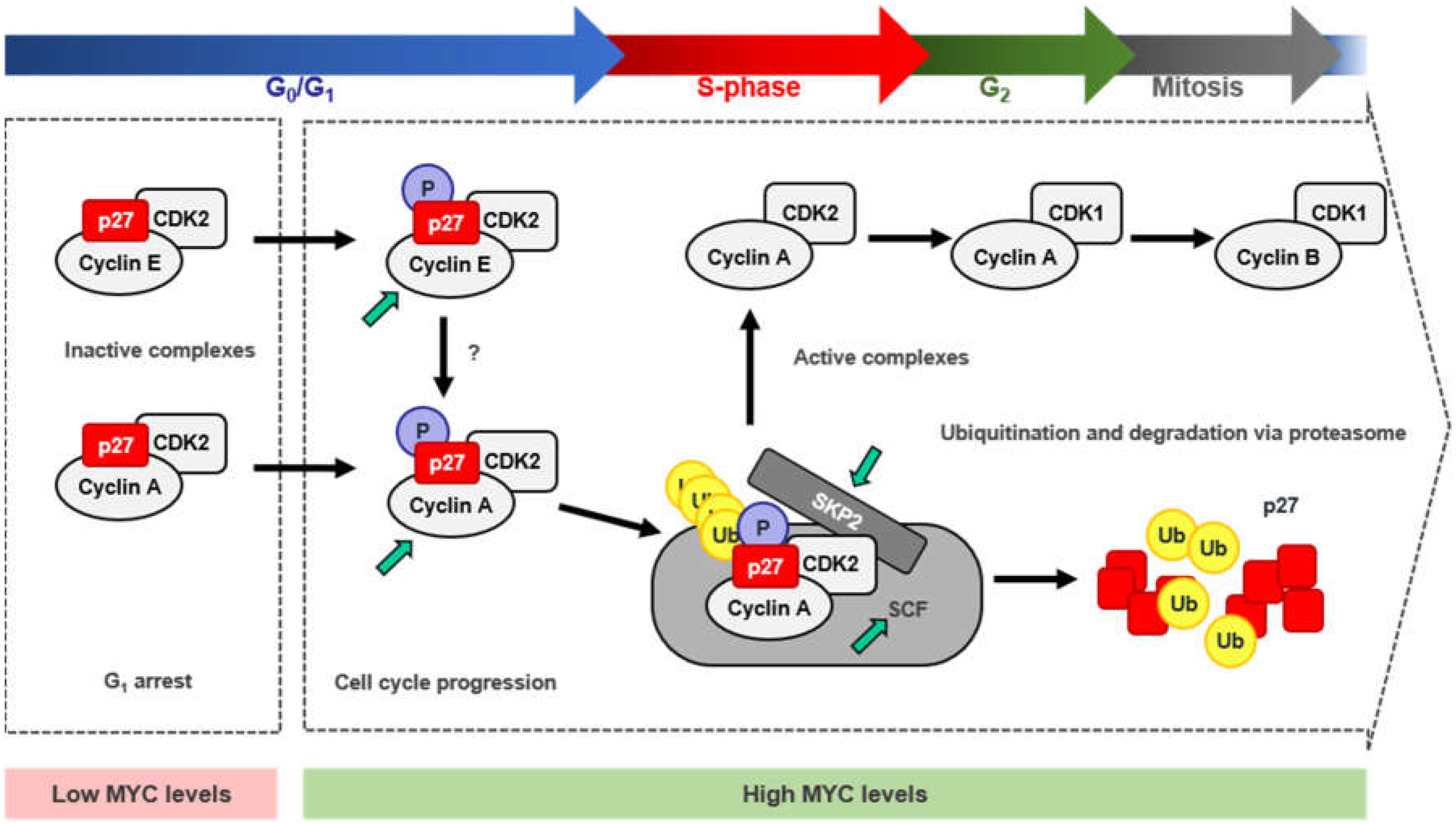
4. MYC and p27 Regulation
4.1. Repression of CDKN1B Expression
4.2. MYC-Induced Repression of p27 Through miRNA Up-Regulation
4.3. Sequestration of p27 by Cyclin D-CDK4/6 Complexes
4.4. Induction of p27 Degradation Through the MYC/CDK2/SKP2 Axis
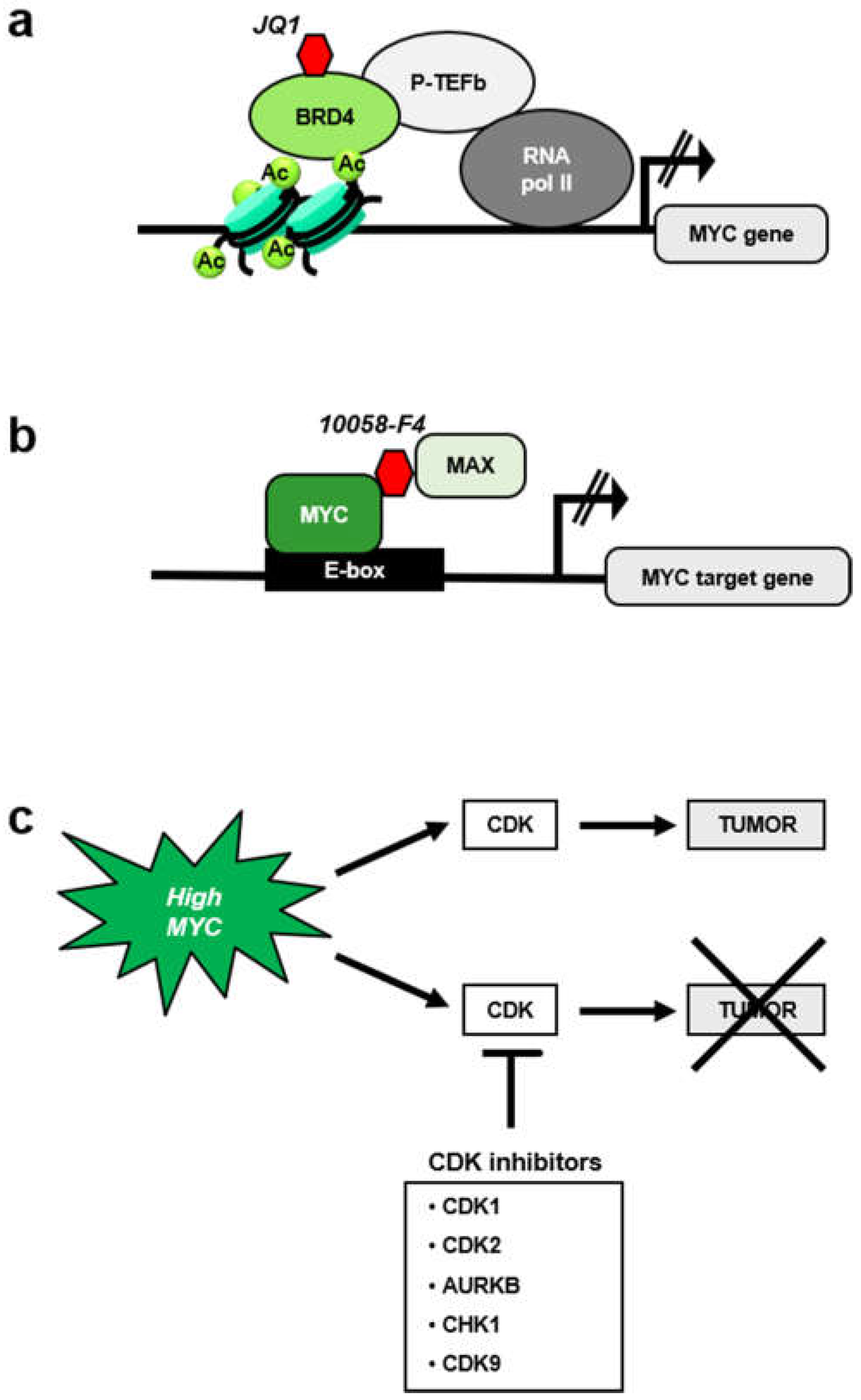
5. MYC-Mediated Synthetic Lethality and the Cell Cycle
5.1. MYC and CDK1 Inhibitors
5.2. MYC and Aurora Kinase Inhibitors
5.3. MYC and CHK1 Inhibitors
5.4. MYC and CDK9 Inhibition
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M.D.; Leon, J. Myc roles in hematopoiesis and leukemia. Genes Cancer 2010, 1, 605–616. [Google Scholar] [CrossRef]
- Vita, M.; Henriksson, M. The Myc oncoprotein as a therapeutic target for human cancer. Semin. Cancer Biol. 2006, 16, 318–330. [Google Scholar] [CrossRef]
- Kalkat, M.; De Melo, J.; Hickman, K.A.; Lourenco, C.; Redel, C.; Resetca, D.; Tamachi, A.; Tu, W.B.; Penn, L.Z. MYC Deregulation in Primary Human Cancers. Genes 2017, 8, 151. [Google Scholar] [CrossRef]
- Schaub, F.X.; Dhankani, V.; Berger, A.C.; Trivedi, M.; Richardson, A.B.; Shaw, R.; Zhao, W.; Zhang, X.; Ventura, A.; Liu, Y.; et al. Pan-cancer Alterations of the MYC Oncogene and Its Proximal Network across the Cancer Genome Atlas. Cell Syst. 2018, 6, 282–300. [Google Scholar] [CrossRef]
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef] [PubMed]
- Eilers, M.; Eisenman, R.N. Myc’s broad reach. Genes Dev. 2008, 22, 2755–2766. [Google Scholar] [CrossRef] [PubMed]
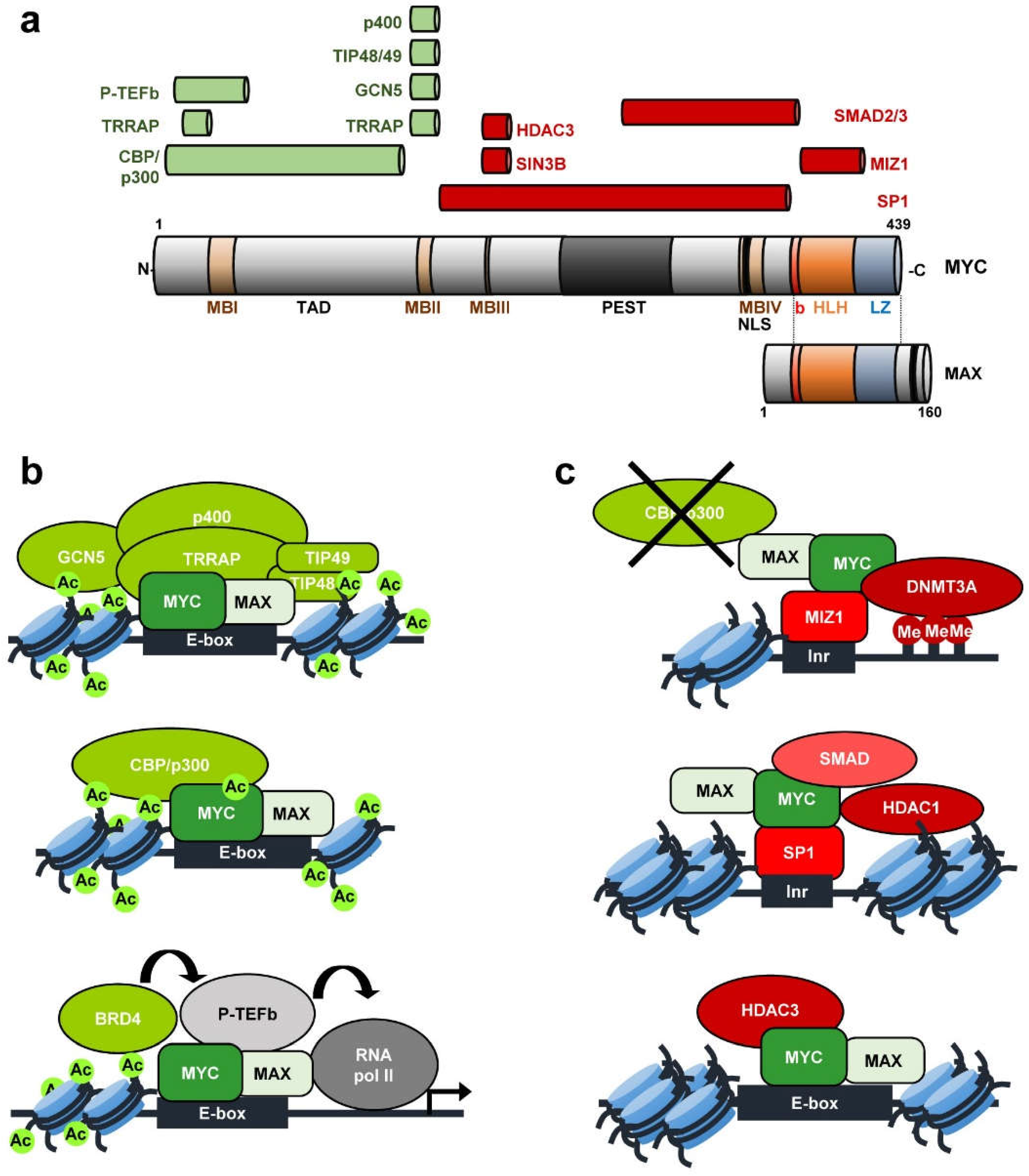
- Luscher, B.; Vervoorts, J. Regulation of gene transcription by the oncoprotein MYC. Gene 2012, 494, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Blackwood, E.M.; Eisenman, R.N. Max: A helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science 1991, 251, 1211–1217. [Google Scholar] [CrossRef]
- Carroll, P.A.; Freie, B.W.; Mathsyaraja, H.; Eisenman, R.N. The MYC transcription factor network: Balancing metabolism, proliferation and oncogenesis. Front. Med. 2018, 12, 412–425. [Google Scholar] [CrossRef]
- Diolaiti, D.; McFerrin, L.; Carroll, P.A.; Eisenman, R.N. Functional interactions among members of the MAX and MLX transcriptional network during oncogenesis. Biochim. Biophys. Acta 2015, 1849, 484–500. [Google Scholar] [CrossRef] [PubMed]
- Van Dang, C.; McMahon, S.B. Emerging Concepts in the Analysis of Transcriptional Targets of the MYC Oncoprotein: Are the Targets Targetable? Genes Cancer 2010, 1, 560–567. [Google Scholar]
- Bretones, G.; Delgado, M.D.; Leon, J. Myc and cell cycle control. Biochim. Biophys. Acta 2015, 1849, 506–516. [Google Scholar] [CrossRef]
- Leon, J.; Ferrandiz, N.; Acosta, J.C.; Delgado, M.D. Inhibition of cell differentiation: A critical mechanism for MYC-mediated carcinogenesis? Cell Cycle 2009, 8, 1148–1157. [Google Scholar] [CrossRef]
- Wolpaw, A.J.; Dang, C.V. MYC-induced metabolic stress and tumorigenesis. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 43–50. [Google Scholar] [CrossRef]
- Conacci-Sorrell, M.; McFerrin, L.; Eisenman, R.N. An overview of MYC and its interactome. Cold Spring Harbor Perspect. Med. 2014, 4, a014357. [Google Scholar] [CrossRef]
- Tu, W.B.; Helander, S.; Pilstal, R.; Hickman, K.A.; Lourenco, C.; Jurisica, I.; Raught, B.; Wallner, B.; Sunnerhagen, M.; Penn, L.Z. Myc and its interactors take shape. Biochim. Biophys. Acta 2015, 1849, 469–483. [Google Scholar] [CrossRef] [PubMed]
- Adhikary, S.; Eilers, M. Transcriptional regulation and transformation by Myc proteins. Nat. Rev. Mol. Cell Biol. 2005, 6, 635–645. [Google Scholar] [CrossRef]
- Garcia-Sanz, P.; Quintanilla, A.; Lafita, M.C.; Moreno-Bueno, G.; Garcia-Gutierrez, L.; Tabor, V.; Varela, I.; Shiio, Y.; Larsson, L.G.; Portillo, F.; et al. Sin3b interacts with Myc and decreases Myc levels. J. Biol. Chem. 2014, 289, 22221–22236. [Google Scholar] [CrossRef] [PubMed]
- Kurland, J.F.; Tansey, W.P. Myc-mediated transcriptional repression by recruitment of histone deacetylase. Cancer Res. 2008, 68, 3624–3629. [Google Scholar] [CrossRef] [PubMed]
- Cowling, V.H.; Chandriani, S.; Whitfield, M.L.; Cole, M.D. A conserved Myc protein domain, MBIV, regulates DNA binding, apoptosis, transformation, and G2 arrest. Mol. Cell. Biol. 2006, 26, 4226–4239. [Google Scholar] [CrossRef] [PubMed]
- Amati, B.; Dalton, S.; Brooks, M.W.; Littlewood, T.D.; Evan, G.I.; Land, H. Transcriptional activation by the human c-Myc oncoprotein in yeast requires interaction with Max. Nature 1992, 359, 423–426. [Google Scholar] [CrossRef]
- Kato, G.J.; Lee, W.M.; Chen, L.L.; Dang, C.V. Max: Functional domains and interaction with c-Myc. Genes Dev. 1992, 6, 81–92. [Google Scholar] [CrossRef]
- Poole, C.J.; van Riggelen, J. MYC-Master Regulator of the Cancer Epigenome and Transcriptome. Genes 2017, 8, 142. [Google Scholar] [CrossRef] [PubMed]
- McMahon, S.B.; Van Buskirk, H.A.; Dugan, K.A.; Copeland, T.D.; Cole, M.D. The novel ATM-related protein TRRAP is an essential cofactor for the c-Myc and E2F oncoproteins. Cell 1998, 94, 363–374. [Google Scholar] [CrossRef]
- Frank, S.R.; Schroeder, M.; Fernandez, P.; Taubert, S.; Amati, B. Binding of c-Myc to chromatin mediates mitogen-induced acetylation of histone H4 and gene activation. Genes Dev. 2001, 15, 2069–2082. [Google Scholar] [CrossRef]
- Nie, Z.; Hu, G.; Wei, G.; Cui, K.; Yamane, A.; Resch, W.; Wang, R.; Green, D.R.; Tessarollo, L.; Casellas, R.; et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 2012, 151, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Loven, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R.A. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012, 151, 56–67. [Google Scholar] [CrossRef]
- Sabo, A.; Kress, T.R.; Pelizzola, M.; de Pretis, S.; Gorski, M.M.; Tesi, A.; Morelli, M.J.; Bora, P.; Doni, M.; Verrecchia, A.; et al. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature 2014, 511, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Wolf, E.; Lin, C.Y.; Eilers, M.; Levens, D.L. Taming of the beast: Shaping Myc-dependent amplification. Trends Cell Biol. 2015, 25, 241–248. [Google Scholar] [CrossRef]
- Rahl, P.B.; Lin, C.Y.; Seila, A.C.; Flynn, R.A.; McCuine, S.; Burge, C.B.; Sharp, P.A.; Young, R.A. c-Myc regulates transcriptional pause release. Cell 2010, 141, 432–445. [Google Scholar] [CrossRef] [PubMed]
- Rahl, P.B.; Young, R.A. MYC and transcription elongation. Cold Spring Harbor Perspect. Med. 2014, 4, a020990. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; O’Donnell, K.A.; Zeller, K.I.; Nguyen, T.; Osthus, R.C.; Li, F. The c-Myc target gene network. Semin. Cancer Biol. 2006, 16, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Herkert, B.; Eilers, M. Transcriptional repression: The dark side of myc. Genes Cancer 2010, 1, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Staller, P.; Peukert, K.; Kiermaier, A.; Seoane, J.; Lukas, J.; Karsunky, H.; Moroy, T.; Bartek, J.; Massague, J.; Hanel, F.; et al. Repression of p15INK4b expression by Myc through association with Miz-1. Nat. Cell Biol. 2001, 3, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Wiese, K.E.; Walz, S.; von Eyss, B.; Wolf, E.; Athineos, D.; Sansom, O.; Eilers, M. The role of MIZ-1 in MYC-dependent tumorigenesis. Cold Spring Harbor Perspect. Med. 2013, 3, a014290. [Google Scholar] [CrossRef]
- Jiang, G.; Espeseth, A.; Hazuda, D.J.; Margolis, D.M. c-Myc and Sp1 contribute to proviral latency by recruiting histone deacetylase 1 to the human immunodeficiency virus type 1 promoter. J. Virol. 2007, 81, 10914–10923. [Google Scholar] [CrossRef]
- Feng, X.H.; Liang, Y.Y.; Liang, M.; Zhai, W.; Lin, X. Direct interaction of c-Myc with Smad2 and Smad3 to inhibit TGF-β-mediated induction of the CDK inhibitor p15Ink4B. Mol. Cell 2002, 9, 133–143. [Google Scholar] [CrossRef]
- Brenner, C.; Deplus, R.; Didelot, C.; Loriot, A.; Vire, E.; De Smet, C.; Gutierrez, A.; Danovi, D.; Bernard, D.; Boon, T.; et al. Myc represses transcription through recruitment of DNA methyltransferase corepressor. EMBO J. 2005, 24, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Gartel, A.L.; Ye, X.; Goufman, E.; Shianov, P.; Hay, N.; Najmabadi, F.; Tyner, A.L. Myc represses the p21(WAF1/CIP1) promoter and interacts with Sp1/Sp3. Proc. Natl. Acad. Sci. USA 2001, 98, 4510–4515. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Cetinkaya, C.; Munoz-Alonso, M.J.; von der Lehr, N.; Bahram, F.; Beuger, V.; Eilers, M.; Leon, J.; Larsson, L.G. Myc represses differentiation-induced p21CIP1 expression via Miz-1-dependent interaction with the p21 core promoter. Oncogene 2003, 22, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Seoane, J.; Pouponnot, C.; Staller, P.; Schader, M.; Eilers, M.; Massague, J. TGFβ influences Myc, Miz-1 and Smad to control the CDK inhibitor p15INK4b. Nat. Cell Biol. 2001, 3, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. Mammalian cyclin-dependent kinases. Trends Biochem. Sci. 2005, 30, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Harlow, E.; Hunt, T.; Hunter, T.; Lahti, J.M.; Manning, G.; Morgan, D.O.; Tsai, L.H.; Wolgemuth, D.J. Cyclin-dependent kinases: A family portrait. Nat. Cell Biol. 2009, 11, 1275–1276. [Google Scholar] [CrossRef]
- Sherr, C.J. D-type cyclins. Trends Biochem. Sci. 1995, 20, 187–190. [Google Scholar] [CrossRef]
- Dyson, N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998, 12, 2245–2262. [Google Scholar] [CrossRef] [PubMed]
- Nevins, J.R. The Rb/E2F pathway and cancer. Hum. Mol. Genet. 2001, 10, 699–703. [Google Scholar] [CrossRef]
- Sherr, C.J.; Roberts, J.M. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004, 18, 2699–2711. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Besson, A.; Dowdy, S.F.; Roberts, J.M. CDK inhibitors: Cell cycle regulators and beyond. Dev. Cell 2008, 14, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. To cycle or not to cycle: A critical decision in cancer. Nat. Rev. Cancer 2001, 1, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.; Hannon, G.J.; Beach, D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 1993, 366, 704–707. [Google Scholar] [CrossRef] [PubMed]
- Quelle, D.E.; Zindy, F.; Ashmun, R.A.; Sherr, C.J. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell 1995, 83, 993–1000. [Google Scholar] [PubMed]
- Weber, J.D.; Jeffers, J.R.; Rehg, J.E.; Randle, D.H.; Lozano, G.; Roussel, M.F.; Sherr, C.J.; Zambetti, G.P. p53-independent functions of the p19(ARF) tumor suppressor. Genes Dev. 2000, 14, 2358–2365. [Google Scholar] [CrossRef]
- McKeller, R.N.; Fowler, J.L.; Cunningham, J.J.; Warner, N.; Smeyne, R.J.; Zindy, F.; Skapek, S.X. The Arf tumor suppressor gene promotes hyaloid vascular regression during mouse eye development. Proc. Natl. Acad. Sci. USA 2002, 99, 3848–3853. [Google Scholar] [CrossRef]
- Zindy, F.; Eischen, C.M.; Randle, D.H.; Kamijo, T.; Cleveland, J.L.; Sherr, C.J.; Roussel, M.F. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 1998, 12, 2424–2433. [Google Scholar] [CrossRef] [PubMed]
- Gregory, M.A.; Qi, Y.; Hann, S.R. The ARF tumor suppressor: Keeping Myc on a leash. Cell Cycle 2005, 4, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.Y.; Sharpless, N.E. The regulation of INK4/ARF in cancer and aging. Cell 2006, 127, 265–275. [Google Scholar] [CrossRef]
- Serrano, M.; Lee, H.; Chin, L.; Cordon-Cardo, C.; Beach, D.; DePinho, R.A. Role of the INK4a locus in tumor suppression and cell mortality. Cell 1996, 85, 27–37. [Google Scholar] [CrossRef]
- Kamijo, T.; Zindy, F.; Roussel, M.F.; Quelle, D.E.; Downing, J.R.; Ashmun, R.A.; Grosveld, G.; Sherr, C.J. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell 1997, 91, 649–659. [Google Scholar] [CrossRef]
- Zindy, F.; Quelle, D.E.; Roussel, M.F.; Sherr, C.J. Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene 1997, 15, 203–211. [Google Scholar] [CrossRef]
- Land, H.; Parada, L.F.; Weinberg, R.A. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature 1983, 304, 596–602. [Google Scholar] [CrossRef]
- Ruley, H.E. Adenovirus early region 1A enables viral and cellular transforming genes to transform primary cells in culture. Nature 1983, 304, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Reynisdottir, I.; Polyak, K.; Iavarone, A.; Massague, J. Kip/Cip and Ink4 Cdk inhibitors cooperate to induce cell cycle arrest in response to TGF-β. Genes Dev. 1995, 9, 1831–1845. [Google Scholar] [CrossRef] [PubMed]
- Reynisdottir, I.; Massague, J. The subcellular locations of p15(Ink4b) and p27Kip1 coordinate their inhibitory interactions with cdk4 and cdk2. Genes Dev. 1997, 11, 492–503. [Google Scholar] [CrossRef]
- Warner, B.J.; Blain, S.W.; Seoane, J.; Massague, J. Myc downregulation by transforming growth factor β required for activation of the p15(Ink4b) G1 arrest pathway. Mol. Cell. Biol. 1999, 19, 5913–5922. [Google Scholar] [CrossRef] [PubMed]
- Claassen, G.F.; Hann, S.R. Myc-mediated transformation: The repression connection. Oncogene 1999, 18, 2925–2933. [Google Scholar] [CrossRef]
- Dang, C.V. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol. Cell. Biol. 1999, 19, 1–11. [Google Scholar] [CrossRef]
- Roy, A.L.; Carruthers, C.; Gutjahr, T.; Roeder, R.G. Direct role for Myc in transcription initiation mediated by interactions with TFII-I. Nature 1993, 365, 359–361. [Google Scholar] [CrossRef]
- Schneider, A.; Peukert, K.; Eilers, M.; Hanel, F. Association of Myc with the zinc-finger protein Miz-1 defines a novel pathway for gene regulation by Myc. Curr. Top. Microbiol. Immunol. 1997, 224, 137–146. [Google Scholar]
- Shrivastava, A.; Saleque, S.; Kalpana, G.V.; Artandi, S.; Goff, S.P.; Calame, K. Inhibition of transcriptional regulator Yin-Yang-1 by association with c-Myc. Science 1993, 262, 1889–1892. [Google Scholar] [CrossRef] [PubMed]
- Evan, G.I.; Wyllie, A.H.; Gilbert, C.S.; Littlewood, T.D.; Land, H.; Brooks, M.; Waters, C.M.; Penn, L.Z.; Hancock, D.C. Induction of apoptosis in fibroblasts by c-Myc protein. Cell 1992, 69, 119–128. [Google Scholar] [CrossRef]
- Hermeking, H.; Eick, D. Mediation of c-Myc-induced apoptosis by p53. Science 1994, 265, 2091–2093. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.J.; Kokontis, J.M.; Hay, N. Myc-mediated apoptosis requires wild-type p53 in a manner independent of cell cycle arrest and the ability of p53 to induce p21waf1/cip1. Genes Dev. 1994, 8, 2817–2830. [Google Scholar] [CrossRef] [PubMed]
- Korgaonkar, C.; Zhao, L.; Modestou, M.; Quelle, D.E. ARF function does not require p53 stabilization or Mdm2 relocalization. Mol. Cell. Biol. 2002, 22, 196–206. [Google Scholar] [CrossRef]
- Weber, J.D.; Taylor, L.J.; Roussel, M.F.; Sherr, C.J.; Bar-Sagi, D. Nucleolar Arf sequesters Mdm2 and activates p53. Nat. Cell Biol. 1999, 1, 20–26. [Google Scholar] [CrossRef]
- Adams, J.M.; Harris, A.W.; Pinkert, C.A.; Corcoran, L.M.; Alexander, W.S.; Cory, S.; Palmiter, R.D.; Brinster, R.L. The c-Myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 1985, 318, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Eischen, C.M.; Weber, J.D.; Roussel, M.F.; Sherr, C.J.; Cleveland, J.L. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 1999, 13, 2658–2669. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, C.A.; McCurrach, M.E.; de Stanchina, E.; Wallace-Brodeur, R.R.; Lowe, S.W. INK4a/ARF mutations accelerate lymphomagenesis and promote chemoresistance by disabling p53. Genes Dev. 1999, 13, 2670–2677. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, A.; Bravo, A.; Jorcano, J.L.; Vidal, M. Sequences 5′ of the bovine keratin 5 gene direct tissue- and cell-type-specific expression of a lacZ gene in the adult and during development. Differ. Res. Biol. Divers. 1994, 58, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Rounbehler, R.J.; Schneider-Broussard, R.; Conti, C.J.; Johnson, D.G. Myc lacks E2F1’s ability to suppress skin carcinogenesis. Oncogene 2001, 20, 5341–5349. [Google Scholar] [CrossRef]
- Russell, J.L.; Powers, J.T.; Rounbehler, R.J.; Rogers, P.M.; Conti, C.J.; Johnson, D.G. ARF differentially modulates apoptosis induced by E2F1 and Myc. Mol. Cell. Biol. 2002, 22, 1360–1368. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, C.; Lee, S.; Paulus-Hock, V.; Loddenkemper, C.; Eilers, M.; Schmitt, C.A. FoxO transcription factors suppress Myc-driven lymphomagenesis via direct activation of Arf. Genes Dev. 2007, 21, 2775–2787. [Google Scholar] [CrossRef] [PubMed]
- Bates, S.; Phillips, A.C.; Clark, P.A.; Stott, F.; Peters, G.; Ludwig, R.L.; Vousden, K.H. p14ARF links the tumour suppressors RB and p53. Nature 1998, 395, 124–125. [Google Scholar] [CrossRef] [PubMed]
- Baudino, T.A.; Maclean, K.H.; Brennan, J.; Parganas, E.; Yang, C.; Aslanian, A.; Lees, J.A.; Sherr, C.J.; Roussel, M.F.; Cleveland, J.L. Myc-mediated proliferation and lymphomagenesis, but not apoptosis, are compromised by E2f1 loss. Mol. Cell 2003, 11, 905–914. [Google Scholar] [CrossRef]
- Meyer, N.; Kim, S.S.; Penn, L.Z. The Oscar-worthy role of Myc in apoptosis. Semin. Cancer Biol. 2006, 16, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Shan, J.; Zhu, W.G.; Qin, J.; Gu, W. Transcription-independent ARF regulation in oncogenic stress-mediated p53 responses. Nature 2010, 464, 624–627. [Google Scholar] [CrossRef]
- Chen, D.; Kon, N.; Zhong, J.; Zhang, P.; Yu, L.; Gu, W. Differential effects on ARF stability by normal versus oncogenic levels of c-Myc expression. Mol. Cell 2013, 51, 46–56. [Google Scholar] [CrossRef]
- Cleveland, J.L.; Sherr, C.J. Antagonism of Myc functions by Arf. Cancer Cell 2004, 6, 309–311. [Google Scholar] [CrossRef]
- Datta, A.; Nag, A.; Pan, W.; Hay, N.; Gartel, A.L.; Colamonici, O.; Mori, Y.; Raychaudhuri, P. Myc-ARF (alternate reading frame) interaction inhibits the functions of Myc. J. Biol. Chem. 2004, 279, 36698–36707. [Google Scholar] [CrossRef]
- Qi, Y.; Gregory, M.A.; Li, Z.; Brousal, J.P.; West, K.; Hann, S.R. p19ARF directly and differentially controls the functions of c-Myc independently of p53. Nature 2004, 431, 712–717. [Google Scholar] [CrossRef]
- Zhang, Q.; Spears, E.; Boone, D.N.; Li, Z.; Gregory, M.A.; Hann, S.R. Domain-specific c-Myc ubiquitylation controls c-Myc transcriptional and apoptotic activity. Proc. Natl. Acad. Sci. USA 2013, 110, 978–983. [Google Scholar] [CrossRef]
- Amente, S.; Gargano, B.; Varrone, F.; Ruggiero, L.; Dominguez-Sola, D.; Lania, L.; Majello, B. p14ARF directly interacts with Myc through the Myc BoxII domain. Cancer Biol. Ther. 2006, 5, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Conzen, S.D.; Gottlob, K.; Kandel, E.S.; Khanduri, P.; Wagner, A.J.; O’Leary, M.; Hay, N. Induction of cell cycle progression and acceleration of apoptosis are two separable functions of c-Myc: Transrepression correlates with acceleration of apoptosis. Mol. Cell. Biol. 2000, 20, 6008–6018. [Google Scholar] [CrossRef] [PubMed]
- Oster, S.K.; Mao, D.Y.; Kennedy, J.; Penn, L.Z. Functional analysis of the N-terminal domain of the Myc oncoprotein. Oncogene 2003, 22, 1998–2010. [Google Scholar] [CrossRef] [PubMed]
- Soucek, L.; Jucker, R.; Panacchia, L.; Ricordy, R.; Tato, F.; Nasi, S. Omomyc, a potential Myc dominant negative, enhances Myc-induced apoptosis. Cancer Res. 2002, 62, 3507–3510. [Google Scholar]
- El-Deiry, W.S. p21(WAF1) Mediates Cell-Cycle Inhibition, Relevant to Cancer Suppression and Therapy. Cancer Res. 2016, 76, 5189–5191. [Google Scholar] [CrossRef] [PubMed]
- Karimian, A.; Ahmadi, Y.; Yousefi, B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair 2016, 42, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Brugarolas, J.; Moberg, K.; Boyd, S.D.; Taya, Y.; Jacks, T.; Lees, J.A. Inhibition of cyclin-dependent kinase 2 by p21 is necessary for retinoblastoma protein-mediated G1 arrest after gamma-irradiation. Proc. Natl. Acad. Sci. USA 1999, 96, 1002–1007. [Google Scholar] [CrossRef] [PubMed]
- Gartel, A.L.; Serfas, M.S.; Tyner, A.L. p21—Negative regulator of the cell cycle. Proc. Soc. Exp. Biol. Med. Soc. Exp. Biol. Med. 1996, 213, 138–149. [Google Scholar] [CrossRef]
- Bunz, F.; Dutriaux, A.; Lengauer, C.; Waldman, T.; Zhou, S.; Brown, J.P.; Sedivy, J.M.; Kinzler, K.W.; Vogelstein, B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998, 282, 1497–1501. [Google Scholar] [CrossRef] [PubMed]
- Waldman, T.; Kinzler, K.W.; Vogelstein, B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res. 1995, 55, 5187–5190. [Google Scholar] [PubMed]
- Perez-Roger, I.; Kim, S.H.; Griffiths, B.; Sewing, A.; Land, H. Cyclins D1 and D2 mediate Myc-induced proliferation via sequestration of p27Kip1 and p21Cip1. EMBO J. 1999, 18, 5310–5320. [Google Scholar] [CrossRef] [PubMed]
- Sewing, A.; Wiseman, B.; Lloyd, A.C.; Land, H. High-intensity Raf signal causes cell cycle arrest mediated by p21Cip1. Mol. Cell. Biol. 1997, 17, 5588–5597. [Google Scholar] [CrossRef]
- Coller, H.A.; Grandori, C.; Tamayo, P.; Colbert, T.; Lander, E.S.; Eisenman, R.N.; Golub, T.R. Expression analysis with oligonucleotide microarrays reveals that MYC regulates genes involved in growth, cell cycle, signaling, and adhesion. Proc. Natl. Acad. Sci. USA 2000, 97, 3260–3265. [Google Scholar] [CrossRef] [PubMed]
- Sowa, Y.; Orita, T.; Hiranabe-Minamikawa, S.; Nakano, K.; Mizuno, T.; Nomura, H.; Sakai, T. Histone deacetylase inhibitor activates the p21/WAF1/Cip1 gene promoter through the Sp1 sites. Ann. N. Y. Acad. Sci. 1999, 886, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Claassen, G.F.; Hann, S.R. A role for transcriptional repression of p21CIP1 by c-Myc in overcoming transforming growth factor β -induced cell-cycle arrest. Proc. Natl. Acad. Sci. USA 2000, 97, 9498–9503. [Google Scholar] [CrossRef]
- Vaque, J.P.; Navascues, J.; Shiio, Y.; Laiho, M.; Ajenjo, N.; Mauleon, I.; Matallanas, D.; Crespo, P.; Leon, J. Myc antagonizes Ras-mediated growth arrest in leukemia cells through the inhibition of the Ras-ERK-p21Cip1 pathway. J. Biol. Chem. 2005, 280, 1112–1122. [Google Scholar] [CrossRef]
- Peukert, K.; Staller, P.; Schneider, A.; Carmichael, G.; Hanel, F.; Eilers, M. An alternative pathway for gene regulation by Myc. EMBO J. 1997, 16, 5672–5686. [Google Scholar] [CrossRef]
- Liu, Q.; Basu, S.; Qiu, Y.; Tang, F.; Dong, F. A role of Miz-1 in Gfi-1-mediated transcriptional repression of CDKN1A. Oncogene 2010, 29, 2843–2852. [Google Scholar] [CrossRef]
- Hock, H.; Hamblen, M.J.; Rooke, H.M.; Traver, D.; Bronson, R.T.; Cameron, S.; Orkin, S.H. Intrinsic requirement for zinc finger transcription factor Gfi-1 in neutrophil differentiation. Immunity 2003, 18, 109–120. [Google Scholar] [CrossRef]
- Karsunky, H.; Zeng, H.; Schmidt, T.; Zevnik, B.; Kluge, R.; Schmid, K.W.; Duhrsen, U.; Moroy, T. Inflammatory reactions and severe neutropenia in mice lacking the transcriptional repressor Gfi1. Nat. Genet. 2002, 30, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Jankovic, D.; Grinberg, A.; Guo, L.; Paul, W.E. Gfi-1 plays an important role in IL-2-mediated Th2 cell expansion. Proc. Natl. Acad. Sci. USA 2006, 103, 18214–18219. [Google Scholar] [CrossRef] [PubMed]
- Kazanjian, A.; Wallis, D.; Au, N.; Nigam, R.; Venken, K.J.; Cagle, P.T.; Dickey, B.F.; Bellen, H.J.; Gilks, C.B.; Grimes, H.L. Growth factor independence-1 is expressed in primary human neuroendocrine lung carcinomas and mediates the differentiation of murine pulmonary neuroendocrine cells. Cancer Res. 2004, 64, 6874–6882. [Google Scholar] [CrossRef] [PubMed]
- Shroyer, N.F.; Wallis, D.; Venken, K.J.; Bellen, H.J.; Zoghbi, H.Y. Gfi1 functions downstream of Math1 to control intestinal secretory cell subtype allocation and differentiation. Genes Dev. 2005, 19, 2412–2417. [Google Scholar] [CrossRef] [PubMed]
- Wallis, D.; Hamblen, M.; Zhou, Y.; Venken, K.J.; Schumacher, A.; Grimes, H.L.; Zoghbi, H.Y.; Orkin, S.H.; Bellen, H.J. The zinc finger transcription factor Gfi1, implicated in lymphomagenesis, is required for inner ear hair cell differentiation and survival. Development 2003, 130, 221–232. [Google Scholar] [CrossRef]
- Schmidt, T.; Karsunky, H.; Gau, E.; Zevnik, B.; Elsasser, H.P.; Moroy, T. Zinc finger protein GFI-1 has low oncogenic potential but cooperates strongly with pim and myc genes in T-cell lymphomagenesis. Oncogene 1998, 17, 2661–2667. [Google Scholar] [CrossRef]
- Zornig, M.; Schmidt, T.; Karsunky, H.; Grzeschiczek, A.; Moroy, T. Zinc finger protein GFI-1 cooperates with myc and pim-1 in T-cell lymphomagenesis by reducing the requirements for IL-2. Oncogene 1996, 12, 1789–1801. [Google Scholar]
- Duan, Z.; Horwitz, M. Targets of the transcriptional repressor oncoprotein Gfi-1. Proc. Natl. Acad. Sci. USA 2003, 100, 5932–5937. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.; Zarebski, A.; Montoya-Durango, D.; Grimes, H.L.; Horwitz, M. Gfi1 coordinates epigenetic repression of p21Cip/WAF1 by recruitment of histone lysine methyltransferase G9a and histone deacetylase 1. Mol. Cell. Biol. 2005, 25, 10338–10351. [Google Scholar] [CrossRef]
- Wong, P.P.; Miranda, F.; Chan, K.V.; Berlato, C.; Hurst, H.C.; Scibetta, A.G. Histone demethylase KDM5B collaborates with TFAP2C and Myc to repress the cell cycle inhibitor p21cip (CDKN1A). Mol. Cell. Biol. 2012, 32, 1633–1644. [Google Scholar] [CrossRef]
- Ferrandiz, N.; Martin-Perez, J.; Blanco, R.; Donertas, D.; Weber, A.; Eilers, M.; Dotto, P.; Delgado, M.D.; Leon, J. HCT116 cells deficient in p21Waf1 are hypersensitive to tyrosine kinase inhibitors and adriamycin through a mechanism unrelated to p21 and dependent on p53. DNA Repair 2009, 8, 390–399. [Google Scholar] [CrossRef]
- Seoane, J.; Le, H.V.; Massague, J. Myc suppression of the p21Cip1 Cdk inhibitor influences the outcome of the p53 response to DNA damage. Nature 2002, 419, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Ceballos, E.; Delgado, M.D.; Gutierrez, P.; Richard, C.; Muller, D.; Eilers, M.; Ehinger, M.; Gullberg, U.; Leon, J. c-Myc antagonizes the effect of p53 on apoptosis and p21WAF1 transactivation in K562 leukemia cells. Oncogene 2000, 19, 2194–2204. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.W.; Barradas, M.; Stone, J.C.; van Aelst, L.; Serrano, M.; Lowe, S.W. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev. 1998, 12, 3008–3019. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Delgado, M.D.; Vaque, J.P.; Arozarena, I.; Lopez-Ilasaca, M.A.; Martinez, C.; Crespo, P.; Leon, J. H-, K- and N-Ras inhibit myeloid leukemia cell proliferation by a p21WAF1-dependent mechanism. Oncogene 2000, 19, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Kivinen, L.; Tsubari, M.; Haapajarvi, T.; Datto, M.B.; Wang, X.F.; Laiho, M. Ras induces p21Cip1/Waf1 cyclin kinase inhibitor transcriptionally through Sp1-binding sites. Oncogene 1999, 18, 6252–6261. [Google Scholar] [CrossRef]
- Mermod, N.; Williams, T.J.; Tjian, R. Enhancer binding factors AP-4 and AP-1 act in concert to activate SV40 late transcription in vitro. Nature 1988, 332, 557–561. [Google Scholar] [CrossRef]
- Jung, P.; Menssen, A.; Mayr, D.; Hermeking, H. AP4 encodes a c-MYC-inducible repressor of p21. Proc. Natl. Acad. Sci. USA 2008, 105, 15046–15051. [Google Scholar] [CrossRef]
- Hu, Y.F.; Luscher, B.; Admon, A.; Mermod, N.; Tjian, R. Transcription factor AP-4 contains multiple dimerization domains that regulate dimer specificity. Genes Dev. 1990, 4, 1741–1752. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Okamoto, T. Transcriptional repression of human immunodeficiency virus type 1 by AP-4. J. Biol. Chem. 2006, 281, 12495–12505. [Google Scholar] [CrossRef]
- Kim, M.Y.; Jeong, B.C.; Lee, J.H.; Kee, H.J.; Kook, H.; Kim, N.S.; Kim, Y.H.; Kim, J.K.; Ahn, K.Y.; Kim, K.K. A repressor complex, AP4 transcription factor and geminin, negatively regulates expression of target genes in nonneuronal cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13074–13079. [Google Scholar] [CrossRef] [PubMed]
- Jung, P.; Hermeking, H. The c-MYC-AP4-p21 cascade. Cell Cycle 2009, 8, 982–989. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.H.; Garcia-Martinez, L.F.; Paulssen, E.J.; Gaynor, R.B. Role of flanking E box motifs in human immunodeficiency virus type 1 TATA element function. J. Virol. 1994, 68, 7188–7199. [Google Scholar] [PubMed]
- Ivanovska, I.; Ball, A.S.; Diaz, R.L.; Magnus, J.F.; Kibukawa, M.; Schelter, J.M.; Kobayashi, S.V.; Lim, L.; Burchard, J.; Jackson, A.L.; et al. MicroRNAs in the miR-106b family regulate p21/CDKN1A and promote cell cycle progression. Mol. Cell. Biol. 2008, 28, 2167–2174. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Yu, J.; Han, T.S.; Park, S.Y.; Namkoong, B.; Kim, D.H.; Hur, K.; Yoo, M.W.; Lee, H.J.; Yang, H.K.; et al. Functional links between clustered microRNAs: Suppression of cell-cycle inhibitors by microRNA clusters in gastric cancer. Nucleic Acids Res. 2009, 37, 1672–1681. [Google Scholar] [CrossRef] [PubMed]
- Petrocca, F.; Visone, R.; Onelli, M.R.; Shah, M.H.; Nicoloso, M.S.; de Martino, I.; Iliopoulos, D.; Pilozzi, E.; Liu, C.G.; Negrini, M.; et al. E2F1-regulated microRNAs impair TGFβ-dependent cell-cycle arrest and apoptosis in gastric cancer. Cancer Cell 2008, 13, 272–286. [Google Scholar] [CrossRef]
- Diosdado, B.; van de Wiel, M.A.; Terhaar Sive Droste, J.S.; Mongera, S.; Postma, C.; Meijerink, W.J.; Carvalho, B.; Meijer, G.A. MiR-17-92 cluster is associated with 13q gain and c-myc expression during colorectal adenoma to adenocarcinoma progression. Br. J. Cancer 2009, 101, 707–714. [Google Scholar] [CrossRef]
- O’Donnell, K.A.; Wentzel, E.A.; Zeller, K.I.; Dang, C.V.; Mendell, J.T. c-Myc-regulated microRNAs modulate E2F1 expression. Nature 2005, 435, 839–843. [Google Scholar] [CrossRef]
- Tagawa, H.; Karube, K.; Tsuzuki, S.; Ohshima, K.; Seto, M. Synergistic action of the microRNA-17 polycistron and Myc in aggressive cancer development. Cancer Sci. 2007, 98, 1482–1490. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, M.; Zhu, H.; Zhang, W.; He, S.; Hu, C.; Quan, L.; Bai, J.; Xu, N. Suppression of p21 by c-Myc through members of miR-17 family at the post-transcriptional level. Int. J. Oncol. 2010, 37, 1315–1321. [Google Scholar]
- Bachs, O.; Gallastegui, E.; Orlando, S.; Bigas, A.; Morante-Redolat, J.M.; Serratosa, J.; Farinas, I.; Aligue, R.; Pujol, M.J. Role of p27Kip1 as a transcriptional regulator. Oncotarget 2018, 9, 26259–26278. [Google Scholar] [CrossRef] [PubMed]
- Mateyak, M.K.; Obaya, A.J.; Sedivy, J.M. c-Myc regulates cyclin D-Cdk4 and -Cdk6 activity but affects cell cycle progression at multiple independent points. Mol. Cell. Biol. 1999, 19, 4672–4683. [Google Scholar] [CrossRef]
- Lutz, W.; Leon, J.; Eilers, M. Contributions of Myc to tumorigenesis. Biochim. Biophys. Acta 2002, 1602, 61–71. [Google Scholar] [CrossRef]
- Martins, C.P.; Berns, A. Loss of p27Kip1 but not p21Cip1 decreases survival and synergizes with MYC in murine lymphomagenesis. EMBO J. 2002, 21, 3739–3748. [Google Scholar] [CrossRef]
- Hnit, S.S.; Xie, C.; Yao, M.; Holst, J.; Bensoussan, A.; De Souza, P.; Li, Z.; Dong, Q. p27Kip1 signaling: Transcriptional and post-translational regulation. Int. J. Biochem. Cell Biol. 2015, 68, 9–14. [Google Scholar] [CrossRef]
- Wu, M.; Bellas, R.E.; Shen, J.; Yang, W.; Sonenshein, G.E. Increased p27Kip1 cyclin-dependent kinase inhibitor gene expression following anti-IgM treatment promotes apoptosis of WEHI 231 B cells. J. Immunol. 1999, 163, 6530–6535. [Google Scholar] [PubMed]
- Wu, M.; Arsura, M.; Bellas, R.E.; FitzGerald, M.J.; Lee, H.; Schauer, S.L.; Sherr, D.H.; Sonenshein, G.E. Inhibition of c-myc expression induces apoptosis of WEHI 231 murine B cells. Mol. Cell. Biol. 1996, 16, 5015–5025. [Google Scholar] [CrossRef] [PubMed]
- Schauer, S.L.; Wang, Z.; Sonenshein, G.E.; Rothstein, T.L. Maintenance of nuclear factor-κβ/Rel and c-myc expression during CD40 ligand rescue of WEHI 231 early B cells from receptor-mediated apoptosis through modulation of I kappa B proteins. J. Immunol. 1996, 157, 81–86. [Google Scholar]
- Yang, W.; Shen, J.; Wu, M.; Arsura, M.; FitzGerald, M.; Suldan, Z.; Kim, D.W.; Hofmann, C.S.; Pianetti, S.; Romieu-Mourez, R.; et al. Repression of transcription of the p27Kip1 cyclin-dependent kinase inhibitor gene by c-Myc. Oncogene 2001, 20, 1688–1702. [Google Scholar] [CrossRef]
- Lee, L.A.; Dolde, C.; Barrett, J.; Wu, C.S.; Dang, C.V. A link between c-Myc-mediated transcriptional repression and neoplastic transformation. J. Clin. Investig. 1996, 97, 1687–1695. [Google Scholar] [CrossRef]
- Li, L.H.; Nerlov, C.; Prendergast, G.; MacGregor, D.; Ziff, E.B. c-Myc represses transcription in vivo by a novel mechanism dependent on the initiator element and Myc box II. EMBO J. 1994, 13, 4070–4079. [Google Scholar] [CrossRef]
- Chandramohan, V.; Mineva, N.D.; Burke, B.; Jeay, S.; Wu, M.; Shen, J.; Yang, W.; Hann, S.R.; Sonenshein, G.E. c-Myc represses FOXO3a-mediated transcription of the gene encoding the p27Kip1 cyclin dependent kinase inhibitor. J. Cell. Biochem. 2008, 104, 2091–2106. [Google Scholar] [CrossRef] [PubMed]
- Chandramohan, V.; Jeay, S.; Pianetti, S.; Sonenshein, G.E. Reciprocal control of Forkhead box O 3a and c-Myc via the phosphatidylinositol 3-kinase pathway coordinately regulates p27Kip1 levels. J. Immunol. 2004, 172, 5522–5527. [Google Scholar] [CrossRef]
- Giglio, S.; Cirombella, R.; Amodeo, R.; Portaro, L.; Lavra, L.; Vecchione, A. MicroRNA miR-24 promotes cell proliferation by targeting the CDKs inhibitors p27Kip1 and p16INK4a. J. Cell. Physiol. 2013, 228, 2015–2023. [Google Scholar] [CrossRef] [PubMed]
- Gillies, J.K.; Lorimer, I.A. Regulation of p27Kip1 by miRNA 221/222 in glioblastoma. Cell Cycle 2007, 6, 2005–2009. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.M.; McKenna, M.M.; Walsh, C.P.; McKenna, D.J. miR-24 regulates CDKN1B/p27 expression in prostate cancer. Prostate 2016, 76, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Le Sage, C.; Nagel, R.; Egan, D.A.; Schrier, M.; Mesman, E.; Mangiola, A.; Anile, C.; Maira, G.; Mercatelli, N.; Ciafre, S.A.; et al. Regulation of the p27Kip1 tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. EMBO J. 2007, 26, 3699–3708. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Mori, S.; Nevins, J.R. Myc-induced microRNAs integrate Myc-mediated cell proliferation and cell fate. Cancer Res. 2010, 70, 4820–4828. [Google Scholar] [CrossRef]
- Pineau, P.; Volinia, S.; McJunkin, K.; Marchio, A.; Battiston, C.; Terris, B.; Mazzaferro, V.; Lowe, S.W.; Croce, C.M.; Dejean, A. miR-221 overexpression contributes to liver tumorigenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 264–269. [Google Scholar] [CrossRef]
- Koff, A. How to decrease p27Kip1 levels during tumor development. Cancer Cell 2006, 9, 75–76. [Google Scholar] [CrossRef]
- James, M.K.; Ray, A.; Leznova, D.; Blain, S.W. Differential modification of p27Kip1 controls its cyclin d-cdk4 inhibitory activity. Mol. Cell. Biol. 2008, 28, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; James, M.K.; Larochelle, S.; Fisher, R.P.; Blain, S.W. p27Kip1 inhibits cyclin d-cyclin-dependent kinase 4 by two independent modes. Mol. Cell. Biol. 2009, 29, 986–999. [Google Scholar] [CrossRef] [PubMed]
- Daksis, J.I.; Lu, R.Y.; Facchini, L.M.; Marhin, W.W.; Penn, L.J. Myc induces cyclin D1 expression in the absence of de novo protein synthesis and links mitogen-stimulated signal transduction to the cell cycle. Oncogene 1994, 9, 3635–3645. [Google Scholar] [PubMed]
- Philipp, A.; Schneider, A.; Vasrik, I.; Finke, K.; Xiong, Y.; Beach, D.; Alitalo, K.; Eilers, M. Repression of cyclin D1: A novel function of MYC. Mol. Cell. Biol. 1994, 14, 4032–4043. [Google Scholar] [CrossRef] [PubMed]
- Solomon, D.L.; Philipp, A.; Land, H.; Eilers, M. Expression of cyclin D1 mRNA is not upregulated by Myc in rat fibroblasts. Oncogene 1995, 11, 1893–1897. [Google Scholar]
- Yu, Q.; Ciemerych, M.A.; Sicinski, P. Ras and Myc can drive oncogenic cell proliferation through individual D-cyclins. Oncogene 2005, 24, 7114–7119. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, C.; Dittrich, O.; Kiermaier, A.; Dohmann, K.; Menkel, A.; Eilers, M.; Luscher, B. Regulation of cyclin D2 gene expression by the Myc/Max/Mad network: Myc-dependent TRRAP recruitment and histone acetylation at the cyclin D2 promoter. Genes Dev. 2001, 15, 2042–2047. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, C.; Thieke, K.; Maier, A.; Saffrich, R.; Hanley-Hyde, J.; Ansorge, W.; Reed, S.; Sicinski, P.; Bartek, J.; Eilers, M. Direct induction of cyclin D2 by Myc contributes to cell cycle progression and sequestration of p27. EMBO J. 1999, 18, 5321–5333. [Google Scholar] [CrossRef]
- Hermeking, H.; Rago, C.; Schuhmacher, M.; Li, Q.; Barrett, J.F.; Obaya, A.J.; O’Connell, B.C.; Mateyak, M.K.; Tam, W.; Kohlhuber, F.; et al. Identification of CDK4 as a target of c-MYC. Proc. Natl. Acad. Sci. USA 2000, 97, 2229–2234. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Van Calcar, S.; Qu, C.; Cavenee, W.K.; Zhang, M.Q.; Ren, B. A global transcriptional regulatory role for c-Myc in Burkitt’s lymphoma cells. Proc. Natl. Acad. Sci. USA 2003, 100, 8164–8169. [Google Scholar] [CrossRef] [PubMed]
- Yap, C.S.; Peterson, A.L.; Castellani, G.; Sedivy, J.M.; Neretti, N. Kinetic profiling of the c-Myc transcriptome and bioinformatic analysis of repressed gene promoters. Cell Cycle 2011, 10, 2184–2196. [Google Scholar] [CrossRef] [PubMed]
- Kossatz, U.; Dietrich, N.; Zender, L.; Buer, J.; Manns, M.P.; Malek, N.P. Skp2-dependent degradation of p27kip1 is essential for cell cycle progression. Genes Dev. 2004, 18, 2602–2607. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.; Nagahama, H.; Minamishima, Y.A.; Miyake, S.; Ishida, N.; Hatakeyama, S.; Kitagawa, M.; Iemura, S.; Natsume, T.; Nakayama, K.I. Skp2-mediated degradation of p27 regulates progression into mitosis. Dev. Cell 2004, 6, 661–672. [Google Scholar] [CrossRef]
- Tsvetkov, L.M.; Yeh, K.H.; Lee, S.J.; Sun, H.; Zhang, H. p27Kip1 ubiquitination and degradation is regulated by the SCF(Skp2) complex through phosphorylated Thr187 in p27. Curr. Biol. 1999, 9, 661–664. [Google Scholar] [CrossRef]
- Kitagawa, K.; Kitagawa, M. The SCF-type E3 Ubiquitin Ligases as Cancer Targets. Curr. Cancer Drug Targets 2016, 16, 119–129. [Google Scholar] [CrossRef]
- Nakayama, K.I.; Nakayama, K. Regulation of the cell cycle by SCF-type ubiquitin ligases. Semin. Cell Dev. Biol. 2005, 16, 323–333. [Google Scholar] [CrossRef]
- Bretones, G.; Acosta, J.C.; Caraballo, J.M.; Ferrándiz, N.; Gómez-Casares, M.T.; Albajar, M.; Blanco, R.; Ruiz, P.; Hung, W.C.; Albero, M.P.; et al. SKP2 oncogene is a direct MYC target gene and MYC down-regulates p27KIP1 through SKP2 in human leukemia cells. J. Biol. Chem. 2011, 286, 9815–9825. [Google Scholar] [CrossRef]
- Muller, D.; Bouchard, C.; Rudolph, B.; Steiner, P.; Stuckmann, I.; Saffrich, R.; Ansorge, W.; Huttner, W.; Eilers, M. Cdk2-dependent phosphorylation of p27 facilitates its Myc-induced release from cyclin E/cdk2 complexes. Oncogene 1997, 15, 2561–2576. [Google Scholar] [CrossRef]
- Montagnoli, A.; Fiore, F.; Eytan, E.; Carrano, A.C.; Draetta, G.F.; Hershko, A.; Pagano, M. Ubiquitination of p27 is regulated by Cdk-dependent phosphorylation and trimeric complex formation. Genes Dev. 1999, 13, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Berthet, C.; Aleem, E.; Coppola, V.; Tessarollo, L.; Kaldis, P. Cdk2 knockout mice are viable. Curr. Biol. 2003, 13, 1775–1785. [Google Scholar] [CrossRef]
- Garcia-Gutierrez, L.; Bretones, G.; Leon, J. MYC stimulates cell-cycle progression through the activation of CDK1 and phosphorylation of p27. (manuscript in preparation).
- Steiner, P.; Philipp, A.; Lukas, J.; Godden-Kent, D.; Pagano, M.; Mittnacht, S.; Bartek, J.; Eilers, M. Identification of a Myc-dependent step during the formation of active G1 cyclin-cdk complexes. EMBO J. 1995, 14, 4814–4826. [Google Scholar] [CrossRef] [PubMed]
- Berns, K.; Hijmans, E.M.; Bernards, R. Repression of c-Myc responsive genes in cycling cells causes G1 arrest through reduction of cyclin E/CDK2 kinase activity. Oncogene 1997, 15, 1347–1356. [Google Scholar] [CrossRef]
- Ohtani, K.; DeGregori, J.; Nevins, J.R. Regulation of the cyclin E gene by transcription factor E2F1. Proc. Natl. Acad. Sci. USA 1995, 92, 12146–12150. [Google Scholar] [CrossRef] [PubMed]
- Ohtsubo, M.; Theodoras, A.M.; Schumacher, J.; Roberts, J.M.; Pagano, M. Human cyclin E, a nuclear protein essential for the G1-to-S phase transition. Mol. Cell. Biol. 1995, 15, 2612–2624. [Google Scholar] [CrossRef] [PubMed]
- Mitxelena, J.; Apraiz, A.; Vallejo-Rodriguez, J.; Malumbres, M.; Zubiaga, A.M. E2F7 regulates transcription and maturation of multiple microRNAs to restrain cell proliferation. Nucleic Acids Res. 2016. [Google Scholar] [CrossRef]
- Lolli, G.; Johnson, L.N. CAK-Cyclin-dependent Activating Kinase: A key kinase in cell cycle control and a target for drugs? Cell Cycle 2005, 4, 572–577. [Google Scholar] [CrossRef]
- Morgan, D.O. Principles of CDK regulation. Nature 1995, 374, 131–134. [Google Scholar] [CrossRef]
- Morgan, D.O. Cyclin-dependent kinases: Engines, clocks, and microprocessors. Annu. Rev. Cell Dev. Biol. 1997, 13, 261–291. [Google Scholar] [CrossRef] [PubMed]
- Cowling, V.H.; Cole, M.D. The Myc transactivation domain promotes global phosphorylation of the RNA polymerase II carboxy-terminal domain independently of direct DNA binding. Mol. Cell. Biol. 2007, 27, 2059–2073. [Google Scholar] [CrossRef] [PubMed]
- Obaya, A.J.; Kotenko, I.; Cole, M.D.; Sedivy, J.M. The proto-oncogene c-myc acts through the cyclin-dependent kinase (Cdk) inhibitor p27Kip1 to facilitate the activation of Cdk4/6 and early G1 phase progression. J. Biol. Chem. 2002, 277, 31263–31269. [Google Scholar] [CrossRef] [PubMed]
- Perez-Roger, I.; Solomon, D.L.; Sewing, A.; Land, H. Myc activation of cyclin E/Cdk2 kinase involves induction of cyclin E gene transcription and inhibition of p27Kip1 binding to newly formed complexes. Oncogene 1997, 14, 2373–2381. [Google Scholar] [CrossRef]
- Vlach, J.; Hennecke, S.; Amati, B. Phosphorylation-dependent degradation of the cyclin-dependent kinase inhibitor p27. EMBO J. 1997, 16, 5334–5344. [Google Scholar] [CrossRef]
- Carrano, A.C.; Eytan, E.; Hershko, A.; Pagano, M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat. Cell Biol. 1999, 1, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Sutterluty, H.; Chatelain, E.; Marti, A.; Wirbelauer, C.; Senften, M.; Muller, U.; Krek, W. p45SKP2 promotes p27Kip1 degradation and induces S phase in quiescent cells. Nat. Cell Biol. 1999, 1, 207–214. [Google Scholar] [CrossRef]
- Hao, B.; Zheng, N.; Schulman, B.A.; Wu, G.; Miller, J.J.; Pagano, M.; Pavletich, N.P. Structural basis of the Cks1-dependent recognition of p27Kip1 by the SCF(Skp2) ubiquitin ligase. Mol. Cell 2005, 20, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.; Gitig, D.M.; Koff, A. Cell-free degradation of p27kip1, a G1 cyclin-dependent kinase inhibitor, is dependent on CDK2 activity and the proteasome. Mol. Cell. Biol. 1999, 19, 1190–1201. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.H.; Nguyen, H.; Halicka, H.D.; Traganos, F.; Koff, A. Noncatalytic requirement for cyclin A-cdk2 in p27 turnover. Mol. Cell. Biol. 2004, 24, 6058–6066. [Google Scholar] [CrossRef] [PubMed]
- Bourne, Y.; Watson, M.H.; Hickey, M.J.; Holmes, W.; Rocque, W.; Reed, S.I.; Tainer, J.A. Crystal structure and mutational analysis of the human CDK2 kinase complex with cell cycle-regulatory protein CksHs1. Cell 1996, 84, 863–874. [Google Scholar] [CrossRef]
- Sitry, D.; Seeliger, M.A.; Ko, T.K.; Ganoth, D.; Breward, S.E.; Itzhaki, L.S.; Pagano, M.; Hershko, A. Three different binding sites of Cks1 are required for p27-ubiquitin ligation. J. Biol. Chem. 2002, 277, 42233–42240. [Google Scholar] [CrossRef] [PubMed]
- Keller, U.B.; Old, J.B.; Dorsey, F.C.; Nilsson, J.A.; Nilsson, L.; MacLean, K.H.; Chung, L.; Yang, C.; Spruck, C.; Boyd, K.; et al. Myc targets Cks1 to provoke the suppression of p27Kip1, proliferation and lymphomagenesis. EMBO J. 2007, 26, 2562–2574. [Google Scholar] [CrossRef]
- O’Hagan, R.C.; Ohh, M.; David, G.; de Alboran, I.M.; Alt, F.W.; Kaelin, W.G., Jr.; DePinho, R.A. Myc-enhanced expression of Cul1 promotes ubiquitin-dependent proteolysis and cell cycle progression. Genes Dev. 2000, 14, 2185–2191. [Google Scholar] [CrossRef] [PubMed]
- Skorski, T.; Nieborowska-Skorska, M.; Campbell, K.; Iozzo, R.V.; Zon, G.; Darzynkiewicz, Z.; Calabretta, B. Leukemia treatment in severe combined immunodeficiency mice by antisense oligodeoxynucleotides targeting cooperating oncogenes. J. Exp. Med. 1995, 182, 1645–1653. [Google Scholar] [CrossRef]
- Skorski, T.; Nieborowska-Skorska, M.; Wlodarski, P.; Zon, G.; Iozzo, R.V.; Calabretta, B. Antisense oligodeoxynucleotide combination therapy of primary chronic myelogenous leukemia blast crisis in SCID mice. Blood 1996, 88, 1005–1012. [Google Scholar] [PubMed]
- Roderick, J.E.; Tesell, J.; Shultz, L.D.; Brehm, M.A.; Greiner, D.L.; Harris, M.H.; Silverman, L.B.; Sallan, S.E.; Gutierrez, A.; Look, A.T.; et al. c-Myc inhibition prevents leukemia initiation in mice and impairs the growth of relapsed and induction failure pediatric T-ALL cells. Blood 2014, 123, 1040–1050. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, I.B. Cancer. Addiction to oncogenes—The Achilles heal of cancer. Science 2002, 297, 63–64. [Google Scholar] [CrossRef] [PubMed]
- Soucek, L.; Helmer-Citterich, M.; Sacco, A.; Jucker, R.; Cesareni, G.; Nasi, S. Design and properties of a Myc derivative that efficiently homodimerizes. Oncogene 1998, 17, 2463–2472. [Google Scholar] [CrossRef] [PubMed]
- Soucek, L.; Whitfield, J.R.; Sodir, N.M.; Masso-Valles, D.; Serrano, E.; Karnezis, A.N.; Swigart, L.B.; Evan, G.I. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev. 2013, 27, 504–513. [Google Scholar] [CrossRef]
- Annibali, D.; Whitfield, J.R.; Favuzzi, E.; Jauset, T.; Serrano, E.; Cuartas, I.; Redondo-Campos, S.; Folch, G.; Gonzalez-Junca, A.; Sodir, N.M.; et al. Myc inhibition is effective against glioma and reveals a role for Myc in proficient mitosis. Nat. Commun. 2014, 5, 4632. [Google Scholar] [CrossRef]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [PubMed]
- Fowler, T.; Ghatak, P.; Price, D.H.; Conaway, R.; Conaway, J.; Chiang, C.M.; Bradner, J.E.; Shilatifard, A.; Roy, A.L. Regulation of MYC expression and differential JQ1 sensitivity in cancer cells. PLoS ONE 2014, 9, e87003. [Google Scholar] [CrossRef] [PubMed]
- Bhadury, J.; Nilsson, L.M.; Muralidharan, S.V.; Green, L.C.; Li, Z.; Gesner, E.M.; Hansen, H.C.; Keller, U.B.; McLure, K.G.; Nilsson, J.A. BET and HDAC inhibitors induce similar genes and biological effects and synergize to kill in Myc-induced murine lymphoma. Proc. Natl. Acad. Sci. USA 2014, 111, E2721–E2730. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S.; Prochownik, E.V. Small-molecule inhibitors of the Myc oncoprotein. Biochim. Biophys. Acta 2015, 1849, 525–543. [Google Scholar] [CrossRef]
- Yin, X.; Giap, C.; Lazo, J.S.; Prochownik, E.V. Low molecular weight inhibitors of Myc-Max interaction and function. Oncogene 2003, 22, 6151–6159. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Parise, R.A.; Joseph, E.; Egorin, M.J.; Lazo, J.S.; Prochownik, E.V.; Eiseman, J.L. Efficacy, pharmacokinetics, tisssue distribution, and metabolism of the Myc-Max disruptor, 10058-F4 [Z,E]-5-[4-ethylbenzylidine]-2-thioxothiazolidin-4-one, in mice. Cancer Chemother. Pharmacol. 2009, 63, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Toyoshima, M.; Howie, H.L.; Imakura, M.; Walsh, R.M.; Annis, J.E.; Chang, A.N.; Frazier, J.; Chau, B.N.; Loboda, A.; Linsley, P.S.; et al. Functional genomics identifies therapeutic targets for MYC-driven cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 9545–9550. [Google Scholar] [CrossRef] [PubMed]
- Cermelli, S.; Jang, I.S.; Bernard, B.; Grandori, C. Synthetic lethal screens as a means to understand and treat MYC-driven cancers. Cold Spring Harbor Perspect. Med. 2014, 4, a014209. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, J.E.; Gilbert, C.S.; Porter, A.C. Construction by gene targeting in human cells of a “conditional” CDC2 mutant that rereplicates its DNA. Nat. Genet. 1997, 15, 258–265. [Google Scholar] [CrossRef]
- Santamaria, D.; Barriere, C.; Cerqueira, A.; Hunt, S.; Tardy, C.; Newton, K.; Caceres, J.F.; Dubus, P.; Malumbres, M.; Barbacid, M. Cdk1 is sufficient to drive the mammalian cell cycle. Nature 2007, 448, 811–815. [Google Scholar] [CrossRef]
- Gray, N.; Detivaud, L.; Doerig, C.; Meijer, L. ATP-site directed inhibitors of cyclin-dependent kinases. Curr. Med. Chem. 1999, 6, 859–875. [Google Scholar]
- Cicenas, J.; Kalyan, K.; Sorokinas, A.; Stankunas, E.; Levy, J.; Meskinyte, I.; Stankevicius, V.; Kaupinis, A.; Valius, M. Roscovitine in cancer and other diseases. Ann. Transl. Med. 2015, 3, 135. [Google Scholar] [PubMed]
- Goga, A.; Yang, D.; Tward, A.D.; Morgan, D.O.; Bishop, J.M. Inhibition of CDK1 as a potential therapy for tumors over-expressing MYC. Nat. Med. 2007, 13, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, D.; Kusdra, L.; Huskey, N.E.; Chandriani, S.; Lenburg, M.E.; Gonzalez-Angulo, A.M.; Creasman, K.J.; Bazarov, A.V.; Smyth, J.W.; Davis, S.E.; et al. MYC pathway activation in triple-negative breast cancer is synthetic lethal with CDK inhibition. J. Exp. Med. 2012, 209, 679–696. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.; Ji, F.; Ying, S. CDK1: Beyond cell cycle regulation. Aging 2017, 9, 2465–2466. [Google Scholar] [CrossRef]
- Wade, M.; Wahl, G.M. c-Myc, genome instability, and tumorigenesis: The devil is in the details. Curr. Top. Microbiol. Immunol. 2006, 302, 169–203. [Google Scholar] [PubMed]
- Prochownik, E.V.; Li, Y. The ever expanding role for c-Myc in promoting genomic instability. Cell Cycle 2007, 6, 1024–1029. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, A.; Deb-Basu, D.; Cherry, A.; Turner, S.; Ford, J.; Felsher, D.W. Defective double-strand DNA break repair and chromosomal translocations by MYC overexpression. Proc. Natl. Acad. Sci. USA 2003, 100, 9974–9979. [Google Scholar] [CrossRef]
- Li, Q.; Dang, C.V. c-Myc overexpression uncouples DNA replication from mitosis. Mol. Cell. Biol. 1999, 19, 5339–5351. [Google Scholar] [CrossRef]
- Kuschak, T.I.; Kuschak, B.C.; Taylor, C.L.; Wright, J.A.; Wiener, F.; Mai, S. c-Myc initiates illegitimate replication of the ribonucleotide reductase R2 gene. Oncogene 2002, 21, 909–920. [Google Scholar] [CrossRef]
- Sheen, J.H.; Dickson, R.B. Overexpression of c-Myc alters G1/S arrest following ionizing radiation. Mol. Cell. Biol. 2002, 22, 1819–1833. [Google Scholar] [CrossRef]
- Dominguez-Sola, D.; Ying, C.Y.; Grandori, C.; Ruggiero, L.; Chen, B.; Li, M.; Galloway, D.A.; Gu, W.; Gautier, J.; Dalla-Favera, R. Non-transcriptional control of DNA replication by c-Myc. Nature 2007, 448, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.; Asawachaicharn, N.; Galloway, D.A.; Grandori, C. c-Myc accelerates S-Phase and requires WRN to avoid replication stress. PLoS ONE 2009, 4, e5951. [Google Scholar] [CrossRef]
- Albajar, M.; Gomez-Casares, M.T.; Llorca, J.; Mauleon, I.; Vaque, J.P.; Acosta, J.C.; Bermudez, A.; Donato, N.; Delgado, M.D.; Leon, J. MYC in chronic myeloid leukemia: Induction of aberrant DNA synthesis and association with poor response to imatinib. Mol. Cancer Res. 2011, 9, 564–576. [Google Scholar] [CrossRef]
- Willems, E.; Dedobbeleer, M.; Digregorio, M.; Lombard, A.; Lumapat, P.N.; Rogister, B. The functional diversity of Aurora kinases: A comprehensive review. Cell Div. 2018, 13, 7. [Google Scholar] [CrossRef] [PubMed]
- Den Hollander, J.; Rimpi, S.; Doherty, J.R.; Rudelius, M.; Buck, A.; Hoellein, A.; Kremer, M.; Graf, N.; Scheerer, M.; Hall, M.A.; et al. Aurora kinases A and B are up-regulated by Myc and are essential for maintenance of the malignant state. Blood 2010, 116, 1498–1505. [Google Scholar] [CrossRef] [PubMed]
- Harrington, E.A.; Bebbington, D.; Moore, J.; Rasmussen, R.K.; Ajose-Adeogun, A.O.; Nakayama, T.; Graham, J.A.; Demur, C.; Hercend, T.; Diu-Hercend, A.; et al. VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat. Med. 2004, 10, 262–267. [Google Scholar] [CrossRef]
- Yang, D.; Liu, H.; Goga, A.; Kim, S.; Yuneva, M.; Bishop, J.M. Therapeutic potential of a synthetic lethal interaction between the MYC proto-oncogene and inhibition of aurora-B kinase. Proc. Natl. Acad. Sci. USA 2010, 107, 13836–13841. [Google Scholar] [CrossRef] [PubMed]
- McNeely, S.; Beckmann, R.; Bence Lin, A.K. CHEK again: Revisiting the development of CHK1 inhibitors for cancer therapy. Pharmacol. Ther. 2014, 142, 1–10. [Google Scholar] [CrossRef]
- Thompson, R.; Eastman, A. The cancer therapeutic potential of Chk1 inhibitors: How mechanistic studies impact on clinical trial design. Br. J. Clin. Pharmacol. 2013, 76, 358–369. [Google Scholar] [CrossRef]
- Hoglund, A.; Nilsson, L.M.; Muralidharan, S.V.; Hasvold, L.A.; Merta, P.; Rudelius, M.; Nikolova, V.; Keller, U.; Nilsson, J.A. Therapeutic implications for the induced levels of Chk1 in Myc-expressing cancer cells. Clin. Cancer Res. 2011, 17, 7067–7079. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.H.; Lujambio, A.; Zuber, J.; Tschaharganeh, D.F.; Doran, M.G.; Evans, M.J.; Kitzing, T.; Zhu, N.; de Stanchina, E.; Sawyers, C.L.; et al. CDK9-mediated transcription elongation is required for MYC addiction in hepatocellular carcinoma. Genes Dev. 2014, 28, 1800–1814. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Gutiérrez, L.; Delgado, M.D.; León, J. MYC Oncogene Contributions to Release of Cell Cycle Brakes. Genes 2019, 10, 244. https://doi.org/10.3390/genes10030244
García-Gutiérrez L, Delgado MD, León J. MYC Oncogene Contributions to Release of Cell Cycle Brakes. Genes. 2019; 10(3):244. https://doi.org/10.3390/genes10030244
Chicago/Turabian StyleGarcía-Gutiérrez, Lucía, María Dolores Delgado, and Javier León. 2019. "MYC Oncogene Contributions to Release of Cell Cycle Brakes" Genes 10, no. 3: 244. https://doi.org/10.3390/genes10030244
APA StyleGarcía-Gutiérrez, L., Delgado, M. D., & León, J. (2019). MYC Oncogene Contributions to Release of Cell Cycle Brakes. Genes, 10(3), 244. https://doi.org/10.3390/genes10030244