Comparative Transcriptome Analysis in Oilseed Rape (Brassica napus) Reveals Distinct Gene Expression Details between Nitrate and Ammonium Nutrition

 ,
,  ,
,
Abstract
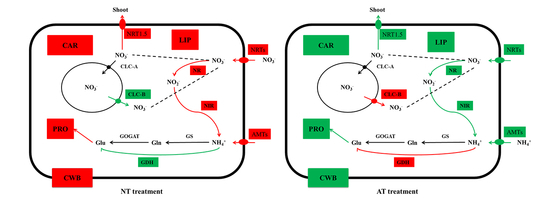
:
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Treatments
2.2. Measurement of Growth-Related Parameters
2.3. RNA Quantification and Qualification
2.4. Bioinformatic Analysis
2.5. Quantitative RT-PCR Analysis
2.6. Statistical Analysis
3. Results
3.1. Effects of Different N Treatments on the Growth of Two Oilseed Rape Cultivars
3.2. Summary of High-Throughput RNA Sequencing Data
3.3. Differentially Expressed Gene Analysis
3.4. Gene Ontology Enrichment Analysis of Differentially Expressed Genes
3.5. Kyoto Encyclopedia of Genes and Genomes Metabolic Pathway Enrichment Analysis
3.6. Genes Involved in Starch and Sucrose Metabolism, Fatty Acid Biosynthesis, Ribosome and Cell Wall Biogenesis
3.7. Genes Related to N Uptake, Transport, and Assimilation
3.8. Validation of Genes Using Quantitative Real-Time PCR
4. Discussion
4.1. Oilseed Rape Seedling Growth Was Inhibited by AT treatment
4.2. N Metabolism of Oilseed Rape Was Inhibited by AT treatment
4.3. Expression Levels of Genes Involved in Cell Wall Biogenesis Decreased with AT treatment
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lu, Y.L.; Xu, Y.C.; Shen, Q.R.; Dong, C.X. Effects of different nitrogen forms on the growth and cytokinin content in xylem sap of tomato (Lycopersicon esculentum Mill.) seedlings. Plant Soil 2009, 315, 67–77. [Google Scholar] [CrossRef]
- Yang, S.Y.; Hao, D.L.; Song, Z.Z.; Yang, G.Z.; Wang, L.; Su, Y.H. RNA-seq analysis of differentially expressed genes in rice under varied nitrogen supplies. Gene 2015, 555, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.; Gerendas, J.; Hardter, R.; Sattelmacher, B. Effect of nitrogen form and root-zone ph on growth and nitrogen uptake of tea (Camellia sinensis) plants. Ann. Bot. 2006, 99, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.Z.; Wang, H.X.; Chen, Y.M.; Li, G.Z. Blueberry physiological ecology research progress in our country. North. Hortic. 2006, 3, 48–49. [Google Scholar]
- Elgharably, A.; Marschner, P.; Rengasamy, P. Wheat growth in a saline sandy loam soil as affected by N form and application rate. Plant Soil 2010, 328, 303–312. [Google Scholar] [CrossRef]
- Liu, N.; Zhang, L.; Meng, X.X.; Neelam, A.; Yang, J.H.; Zhang, M.F. Effect of nitrate/ammonium ratios on growth, root morphology and nutrient elements uptake of watermelon (Citrullus lanatus) seedlings. J. Plant Nutr. 2014, 37, 14. [Google Scholar]
- Hachiya, T.; Watanabe, C.K.; Fujimoto, M.; Ishikawa, T.; Takahara, K.; Kawaiyamada, M.; Uchimiya, H.; Uesono, Y.; Terashima, I.; Noguchi, K. Nitrate addition alleviates ammonium toxicity without lessening ammonium accumulation, organic acid depletion and inorganic cation depletion in Arabidopsis thaliana shoots. Plant Cell Physiol. 2012, 53, 577–591. [Google Scholar] [CrossRef]
- Rathke, G.W.; Behrens, T.; Diepenbrock, W. Integrated nitrogen management strategies to improve seed yield, oil content and nitrogen efficiency of winter oilseed rape (Brassica napus L.). A review. Agric. Ecosyst. Environ. 2006, 117, 80–108. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Song, H.X.; Liu, Q.; Rong, X.M.; Peng, J.W.; Xie, G.X.; Zhang, Y.P.; Chen, L.R.; Guan, C.Y.; Gu, J.D. Responses of seed yield and quality to nitrogen application levels in two oilseed rape (Brassica napus L.) varieties differing in nitrogen efficiency. Plant Prod. Sci. 2012, 15, 265–269. [Google Scholar] [CrossRef]
- Kessel, B.; Schierholt, A.; Becker, H.C. Nitrogen use efficiency in a genetically diverse set of winter oilseed rape (Brassica napus L.). Crop Sci. 2012, 52, 2546–2554. [Google Scholar] [CrossRef]
- Falkengrengrerup, U. Interspecies differences in the preference of ammonium and nitrate in vascular plants. Oecologia 1995, 102, 305–311. [Google Scholar] [CrossRef]
- Correa, E.F.; Germon, J.C. Dissimilative nitrate reduction to ammonium in different soils in waterlogged conditions. Develop. Geochem. 1991, 6, 295–308. [Google Scholar]
- Tang, W.J.; Guan, C.Y.; Lin, L.B.; Li, L.P.; Zhang, Z.H.; Wan, F.; Xiao, G.; Li, B.; Liu, Y.X. Effects of nitrate and ammonium supply ratios on growth, physiology and yield of oilseed rape (Brassica napus L.). J. Plant Nutr. Fert. 2018, 24, 1338–1348. [Google Scholar]
- Patterson, K.; Cakmak, T.; Cooper, A.; Lager, I.; Rasmusson, A.G.; Escobar, M.A. Distinct signalling pathways and transcriptome response signatures differentiate ammonium- and nitrate-supplied plants. Plant Cell Environ. 2010, 33, 1486–1501. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.Y.; Wang, F.; Wan, Q.; Ruan, J.Y. Transcriptome analysis using RNA-seq revealed the effects of nitrogen form on major secondary metabolite biosynthesis in tea (Camellia sinensis) plants. Acta Physiol. Plant. 2018, 40, 127. [Google Scholar] [CrossRef]
- Han, Y.L.; Song, H.X.; Liao, Q.; Yu, Y.; Jian, S.F.; Lepo, J.E.; Liu, Q.; Rong, X.M.; Tian, C.; Zeng, J.; et al. Nitrogen use efficiency is mediated by vacuolar nitrate sequestration capacity in roots of Brassica napus. Plant Physiol. 2016, 170, 1684. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Song, H.X.; Liu, Q.; Rong, X.M.; Xie, G.X.; Peng, J.W.; Zhang, Y.P.; Guan, C.Y.; Chen, S.Y. Nitrogen redistribution characteristics of oilseed rape varieties with different nitrogen use efficiencies during later growth period. Commun. Soil Sci. Plant Anal. 2010, 41, 1693–1706. [Google Scholar] [CrossRef]
- Zhang, H.P.; Wu, J.Y.; Tao, S.T.; Wu, T.; Qi, K.J.; Zhang, S.J.; Wang, J.Z.; Huang, W.J.; Wu, J.; Zhang, S.L. Evidence for apoplasmic phloem unloading in pear fruit. Plant Mol. Biol. Rep. 2014, 32, 931–939. [Google Scholar] [CrossRef]
- Chalhoub, B.; Denoeud, F.; Liu, S.Y.; Parkin, I.A.P.; Tang, H.B.; Wang, X.Y.; Chiquet, J.; Belcram, H.; Tong, C.B.; Samans, B.; et al. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript level expression analysis of RNA-seq experiments with hisat, stringtie and ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. Stringtie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [Green Version]
- Boyle, E.I.; Weng, S.; Gollub, J.; Jin, H.; Botstein, D.; Cherry, J.M.; Sherlock, G. Go:: Termfinder-open source software for accessing gene ontology information and finding significantly enriched gene ontology terms associated with a list of genes. Bioinformatics 2004, 20, 3710–3715. [Google Scholar] [CrossRef]
- An, X.; Chen, J.; Zhang, J.Y.; Liao, Y.W.; Dai, L.J.; Wang, B.; Liu, L.J.; Peng, D.X. Transcriptome profiling and identification of transcription factors in ramie (Boehmeria nivea L. gaud) in response to peg treatment, using illumina paired-end sequencing technology. Int. J. Mol. Sci. 2015, 16, 3493–3511. [Google Scholar] [CrossRef]
- Gao, Y.X.; Li, Y.; Yang, X.X.; Li, H.J.; Shen, Q.R.; Guo, S.W. Ammonium nutrition increases water absorption in rice seedlings (Oryza sativa L.) under water stress. Plant Soil 2010, 331, 193–201. [Google Scholar] [CrossRef]
- Martinoia, E.; Heck, U.; Wiemken, A. Vacuoles as storage compartments for nitrate in barley leaves. Nature 1981, 289, 292–294. [Google Scholar] [CrossRef]
- Martinoia, E.; Massonneau, A.; Frangne, N. Transport processes of solutes across the vacuolar membrane of higher plants. Plant Cell Physiol. 2000, 41, 1175–1186. [Google Scholar] [CrossRef]
- Jian, S.F.; Liao, Q.; Song, H.X.; Liu, Q.; Lepo, J.E.; Guan, C.Y.; Zhang, J.H.; Ismail, A.M.; Zhang, Z.H. NRT1.1-related NH4+ toxicity is associated with a disturbed balance between NH4+ uptake and assimilation. Plant Physiol. 2018. [Google Scholar] [CrossRef]
- Moll, R.H.; Kamprath, E.J.; Jackson, W.A. Analysis and interpretation of factors which contribute to efficiency of nitrogen utilization. Agron. J. 1982, 74, 562–568. [Google Scholar] [CrossRef]
- Sinha, S.K.; Sevanthi, V.A.M.; Chaudhary, S.; Tyagi, P.; Venkadesan, S.; Rani, M.; Mandal, P.K. Transcriptome Analysis of two rice varieties contrasting for nitrogen use efficiency under chronic N starvation reveals differences in chloroplast and starch metabolism-related genes. Genes 2018, 9, 206. [Google Scholar] [CrossRef]
- Hua, Y.P.; Zhou, T.; Liao, Q.; Song, H.X.; Guan, C.Y.; Zhang, Z.H. Genomics-assisted identification and characterization of the genetic variants underlying differential nitrogen use efficiencies in allotetraploid rapeseed genotypes. G3 2018, 8, 2757–2771. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Zhou, T.; Liao, Q.; Yao, J.Y.; Liang, G.H.; Song, H.X.; Guan, C.Y.; Hua, Y.P. Integrated physiologic, genomic and transcriptomic strategies involving the adaptation of allotetraploid rapeseed to nitrogen limitation. BMC Plant Biol. 2018, 18, 322. [Google Scholar] [CrossRef]
- Wittgenstein, N.J.V.; Le, C.H.; Hawkins, B.J.; Ehlting, J. Evolutionary classification of ammonium, nitrate, and peptide transporters in land plants. BMC Evol. Biol. 2014, 14, 11. [Google Scholar] [CrossRef]
- Amarasinghe, B.H.R.R.; Bruxelles, G.L.D.; Braddon, M.; Onyeocha, I.; Forde, B.G.; Udvardi, M.K. Regulation of GmNRT2 expression and nitrate transport activity in roots of soybean (Glycine max). Planta 1998, 206, 44–52. [Google Scholar] [CrossRef]
- Okamoto, M. Regulation of NRT1 and NRT2 gene families of Arabidopsis thaliana: Responses to nitrate provision. Plant Cell Physiol. 2003, 44, 304–317. [Google Scholar] [CrossRef]
- Kumar, A.; Silim, S.S.; Okamoto, M.; Siddiqi, M.Y.; Glass, A.D.M. Differential expression of three members of the AMT1 gene family encoding putative high-affinity NH4+ transporters in roots of Oryza sativa subspecies indica. Plant Cell Environ. 2010, 26, 907–914. [Google Scholar] [CrossRef]
- Lin, S.H.; Kuo, H.F.; Canivenc, G.; Lin, C.S.; Lepetit, M.; Hsu, P.K.; Tillard, P.; Lin, H.L.; Wang, Y.Y.; Tsai, C.B.; et al. Mutation of the Arabidopsis NRT1.5 nitrate transporter causes defective root-to-shoot nitrate transport. Plant Cell 2008, 20, 2514–2528. [Google Scholar] [CrossRef]
- Li, J.Y.; Fu, Y.L.; Pike, S.M.; Bao, J.; Tian, W.; Zhang, Y.; Chen, C.Z.; Zhang, Y.; Li, H.M.; Huang, J.; et al. The Arabidopsis nitrate transporter NRT1.8 functions in nitrate removal from the xylem sap and mediates cadmium tolerance. Plant Cell 2010, 22, 1633–1646. [Google Scholar] [CrossRef]
- Tang, Y.F.; Sun, X.C.; Hu, C.X.; Tan, Q.L.; Zhao, X.H. Genotypic differences in nitrate uptake, translocation and assimilation of two Chinese cabbage cultivars [Brassica campestris L. ssp. Chinensis (L.)]. Plant Physiol. Bioch. 2013, 70, 14–20. [Google Scholar] [CrossRef]
- Angeli, A.D.; Monachello, D.; Ephritikhine, G.; Frachisse, J.M.; Thomine, S.; Gambale, F.; Barbier-Brygoo, H. The nitrate/proton antiporer AtCLC a mediates nitrate accumulation in plant vacuoles. Nature 2006, 442, 939–942. [Google Scholar] [CrossRef]
- Fechtbartenbach, J.V.D.; Bogner, M.; Dynowski, M.; Ludewig, U. CLC-b mediated NO3−/H+ exchange across the tonoplast of Arabidopsis vacuoles. Plant Cell Physiol. 2010, 51, 960–968. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, J.Q.; Crawford, N.M. Identification and characterization of a chlorate-resistant mutant of Arabidopsis thaliana with mutations in both nitrate reductase structural genes NIA1 and NIA2. Mol. Gen. Genet. MGG 1993, 239, 289–297. [Google Scholar] [PubMed]
- Hoff, T.; Stummann, B.M.; Henningsen, K.W. Structure, function and regulation of nitrate reductase in higher plants. Physilol. Plantarum 1992, 84, 616–624. [Google Scholar] [CrossRef]
- Fukuoka, H.; Ogawa, T.; Minami, H.; Yano, H.; Ohkawa, Y. Development stage-specific and nitrate-independent regulation of nitrate reductase gene expression in rapeseed. Plant Physiol. 1996, 111, 39–47. [Google Scholar] [CrossRef]
- Tercé-Laforgue, T.; Dubois, F.; Ferrario-Méry, S.; Crecenzo, M.P.D.; Sangwan, R.; Hirel, B. Glutamatedehydrogenase of tobacco (Nicotiana tabacum L.) is mainly induced in the cytosol of phloem companion cells whenammonia is provided either externally or released during photorespiration. Plant Physiol. 2004, 136, 4308–4317. [Google Scholar] [CrossRef]
- Boussama, N.; Ouariti, O.; Suzuki, A.; Ghorbel, M.H. Cd-stress on nitrogen assimilation. J. Plant Physiol. 1994, 155, 310–317. [Google Scholar] [CrossRef]
- Bittsánszkya, A.; Pilinszkya, K.; Gyulaib, G.; Komivesa, T. Overcoming ammonium toxicity. Plant Sci. 2015, 231, 184–190. [Google Scholar] [CrossRef]
- Zhong, C.; Cao, X.C.; Hu, J.J.; Zhu, L.F.; Zhang, J.H.; Huang, J.L.; Jin, Q.Y. Nitrogen metabolism in adaptation of photosynthesis to water stress in rice grown under different nitrogen levels. Front. Plant Sci. 2017, 8, 1709. [Google Scholar] [CrossRef]
- Byrt, C.S.; Munns, R.; Burton, R.A.; Gilliham, M.; Wege, S. Root cell wall solutions for crop plants in saline soils. Plant Sci. 2018, 269, 47–55. [Google Scholar] [CrossRef]
- Irshad, M.; Canut, H.; Borderies, G.; Pont-Lezica, R.; Jamet, E. A new picture of cell wall protein dynamics in elongating cells of Arabidopsis thaliana: Confirmed actors and newcomers. BMC Plant Biol. 2008, 8, 94. [Google Scholar] [CrossRef] [PubMed]
- Richmond, T.A.; Somerville, C.R. The cellulose synthase superfamily. Plant Physiol. 2000, 124, 495–498. [Google Scholar] [CrossRef] [PubMed]
- Hématy, K.; Höfte, H. Cellulose and cell elongation. Plant Cell Monogr. 2006, 5, 33–56. [Google Scholar]
- Doblin, M.S.; Kurek, I.D.; Delmer, D.P. Cellulose biosynthesis in plants: From genes to rosettes. A review. Plant Cell Physiol. 2002, 43, 1407. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Nutrient Concentration (mmol/L) | ||||||
|---|---|---|---|---|---|---|---|
| KNO3 | (NH4)2SO4 | K2CO3 | CaCl2 | MgSO4 | KH2PO4 | Fe-EDTA | |
| NT | 15 | 0 | 0 | 2 | 2 | 1 | 0.2 |
| NT + AT | 11.25 | 1.875 | 1.875 | 2 | 2 | 1 | 0.2 |
| AT | 0 | 7.5 | 7.5 | 2 | 2 | 1 | 0.2 |
| Comparison Groups | SSM | FAB | RIB | CWB | ||||
|---|---|---|---|---|---|---|---|---|
| Up | Down | Up | Down | Up | Down | Up | Down | |
| CKvsNT(15) | 158 | 20 | 28 | 1 | 481 | 1 | 8 | 0 |
| CKvsNT(814) | 142 | 13 | 21 | 1 | 566 | 1 | 6 | 0 |
| CKvsAT(15) | 18 | 20 | 3 | 1 | 6 | 29 | 0 | 0 |
| CKvsAT(814) | 4 | 8 | 0 | 2 | 0 | 9 | 0 | 0 |
| NTvsAT(15) | 19 | 161 | 0 | 32 | 2 | 613 | 0 | 9 |
| NTvsAT(814) | 16 | 162 | 0 | 22 | 4 | 631 | 0 | 5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, W.; He, X.; Qian, L.; Wang, F.; Zhang, Z.; Sun, C.; Lin, L.; Guan, C. Comparative Transcriptome Analysis in Oilseed Rape (Brassica napus) Reveals Distinct Gene Expression Details between Nitrate and Ammonium Nutrition. Genes 2019, 10, 391. https://doi.org/10.3390/genes10050391
Tang W, He X, Qian L, Wang F, Zhang Z, Sun C, Lin L, Guan C. Comparative Transcriptome Analysis in Oilseed Rape (Brassica napus) Reveals Distinct Gene Expression Details between Nitrate and Ammonium Nutrition. Genes. 2019; 10(5):391. https://doi.org/10.3390/genes10050391
Chicago/Turabian StyleTang, Weijie, Xin He, Lunwen Qian, Feng Wang, Zhenhua Zhang, Chao Sun, Liangbin Lin, and Chunyun Guan. 2019. "Comparative Transcriptome Analysis in Oilseed Rape (Brassica napus) Reveals Distinct Gene Expression Details between Nitrate and Ammonium Nutrition" Genes 10, no. 5: 391. https://doi.org/10.3390/genes10050391
APA StyleTang, W., He, X., Qian, L., Wang, F., Zhang, Z., Sun, C., Lin, L., & Guan, C. (2019). Comparative Transcriptome Analysis in Oilseed Rape (Brassica napus) Reveals Distinct Gene Expression Details between Nitrate and Ammonium Nutrition. Genes, 10(5), 391. https://doi.org/10.3390/genes10050391