Effect of Long-Term Farming Practices on Agricultural Soil Microbiome Members Represented by Metagenomically Assembled Genomes (MAGs) and Their Predicted Plant-Beneficial Genes

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Analyzed Soil Samples and Extraction of Total Microbial DNA
2.2. Metagenome Library Preparation, Sequencing and Preprocessing of Sequencing Data
2.3. Taxonomic and Functional Microbiome Analyses Based on Single Read Data
2.4. Metagenome Assembly and Binning
2.5. Functional Analyses of Obtained Metagenomically Assembled Genomes (MAGs)
2.6. Detection of Differentially Abundant Features (DAFs)
2.7. Sequence Submission to a Public Sequence Archive
3. Results and Discussion
3.1. Layout and Soil Cultivation Treatment Regime at the Long-Term Experimental Field Site (LTE-1) Located at Bernburg-Strenzfeld (Saxony-Anhalt, Germany)
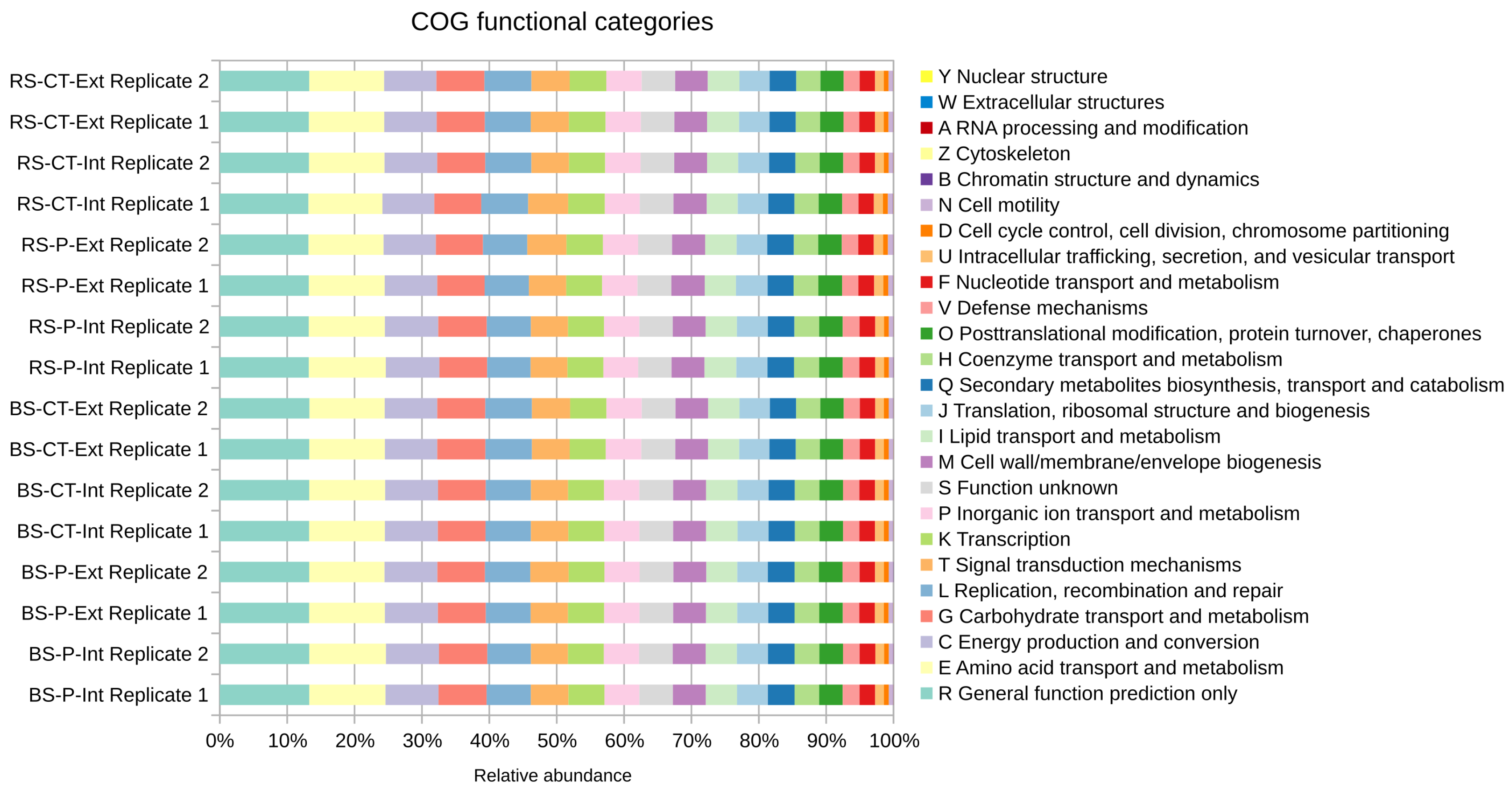
3.2. Unraveling the Soil Microbial Community Composition and Functional Potential of the LTE-1 Field Site Based on Metagenome Single Read Analyses
3.3. Assembly of Metagenomic Reads Yielded Approx. 6.9 Mio. Genes Characterizing the LTE-1 Microbiome
3.4. Binning of Metagenomically Assembled Contigs to Access the Most Prominent Genomes of the LTE-1 Soil Microbiome
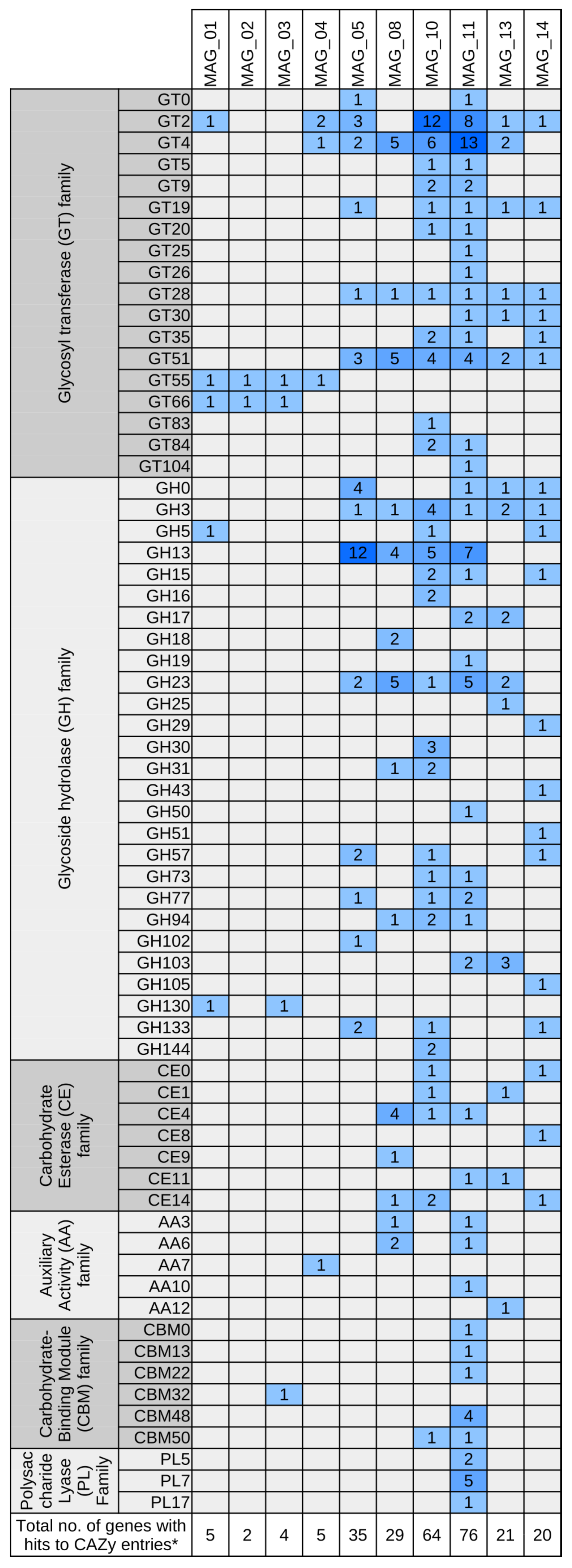
3.5. Functional Characterization of Bacterial MAGs
3.5.1. A Predicted Methylotrophic Bacterium of the Order Rhizobiales Is Represented by MAG_13
3.5.2. A Putative Plant-Growth-Promoting Pseudomonas Species Is Represented by MAG_11
3.5.3. A Member of the Candidate Order ‘Unclassified Deltaproteobacteria’ Predicted to Encode the DOXP/MEP Isoprenoid Biosynthesis Pathway Is Represented by MAG_05
3.5.4. A Putative Plant-Growth-Promoting Bacterium of the Family Bacillaceae Is Represented by MAG_08
3.5.5. A Member of the Flavobacteriaceae Predicted to Decompose Complex Carbohydrates Is Represented by MAG_10
3.5.6. A Member of the Phylum Acidobacteria Predicted to Be Versatile in Transport Activities and Carbohydrate Utilization Is Represented by MAG_14
3.6. Functional Characterization of Archaeal MAGs
A Putative Ammonia Oxidizer of the Phylum Thaumarchaeota Is Represented by MAG_01
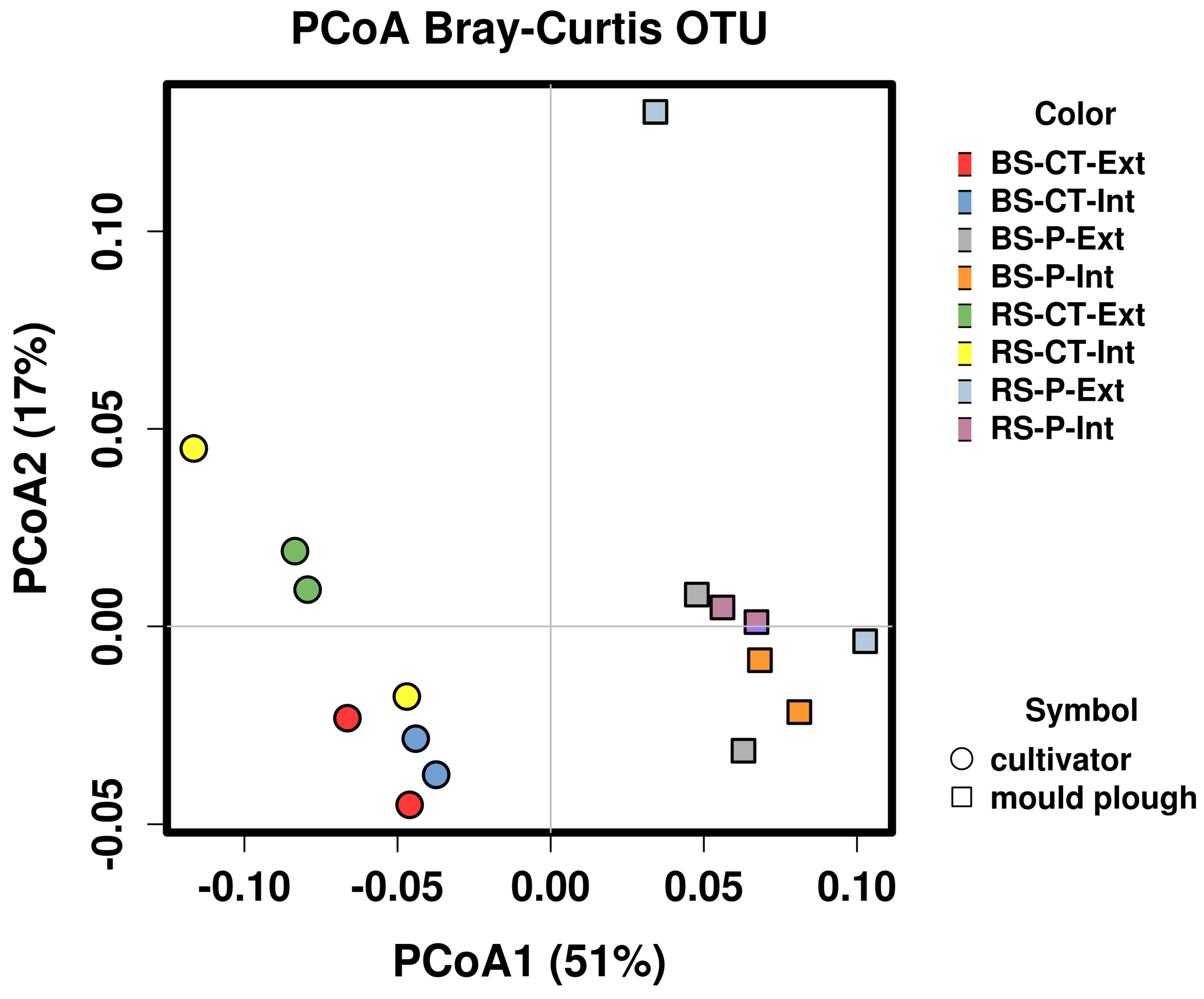
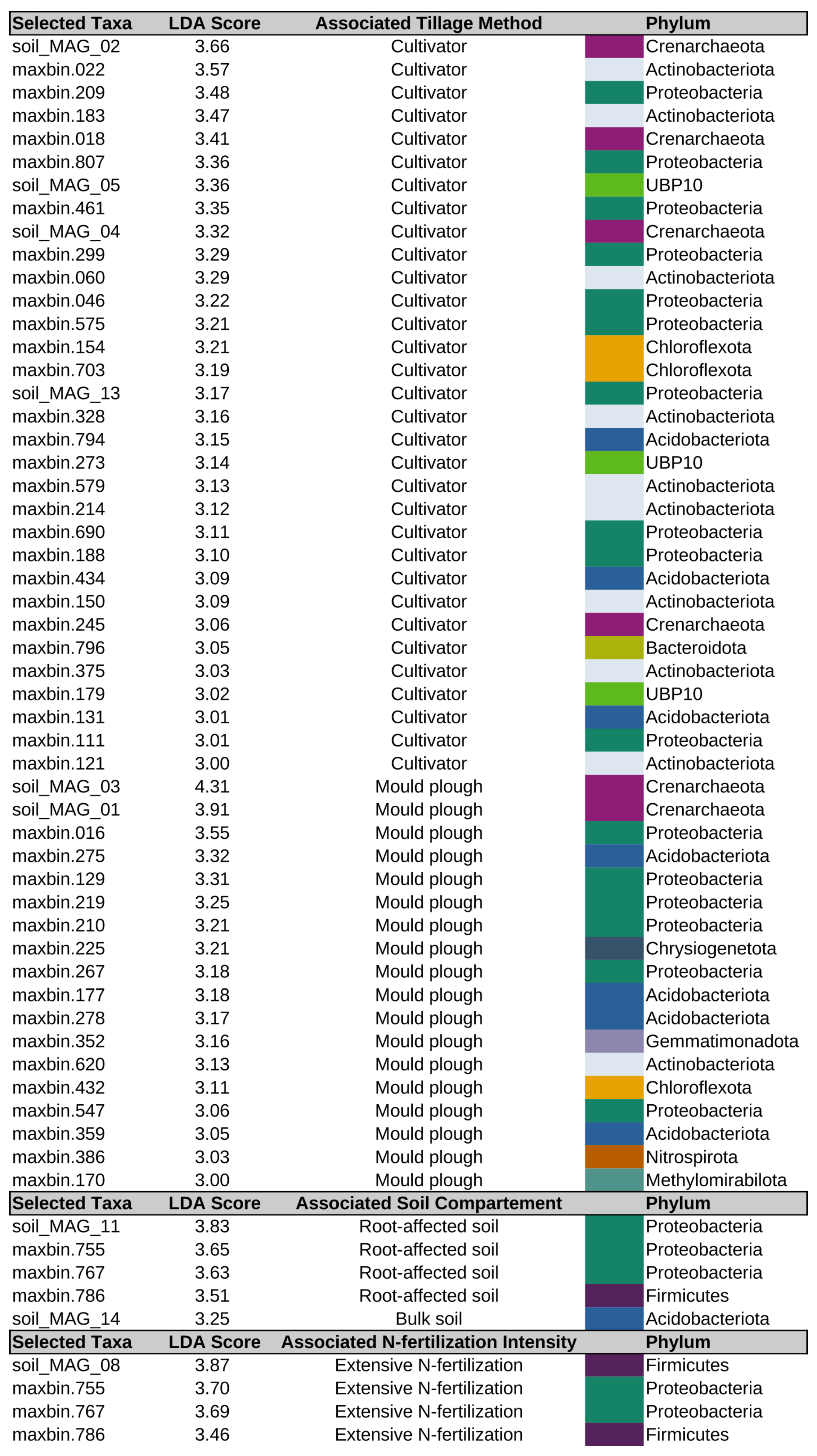
3.7. Differentially Abundant MAGs of the Soil Microbiome in Relation to Tillage and Fertilization Regimes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Orgiazzi, A.; Ballabio, C.; Panagos, P.; Jones, A.; Fernández-Ugalde, O. LUCAS Soil, the largest expandable soil dataset for Europe: A review. Eur. J. Soil. Sci. 2018, 69, 140–153. [Google Scholar] [CrossRef]
- Blum, W.E.H. Functions of Soil for Society and the Environment. Rev. Environ. Sci. Bio/Technol. 2005, 4, 75–79. [Google Scholar] [CrossRef]
- Matson, P.A.; Parton, W.J.; Power, A.G.; Swift, M.J. Agricultural intensification and ecosystem properties. Science 1997, 277, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Tscharntke, T.; Clough, Y.; Wanger, T.C.; Jackson, L.; Motzke, I.; Perfecto, I.; Vandermeer, J.; Whitbread, A. Global food security, biodiversity conservation and the future of agricultural intensification. Biol. Conserv. 2012, 151, 53–59. [Google Scholar] [CrossRef]
- Culman, S.W.; Young-Mathews, A.; Hollander, A.D.; Ferris, H.; Sánchez-Moreno, S.; O’Geen, A.T.; Jackson, L.E. Biodiversity is associated with indicators of soil ecosystem functions over a landscape gradient of agricultural intensification. Landsc. Ecol. 2010, 25, 1333–1348. [Google Scholar] [CrossRef]
- Garbeva, P.; van Veen, J.; van Elsas, J. MICROBIAL DIVERSITY IN SOIL: Selection of Microbial Populations by Plant and Soil Type and Implications for Disease Suppressiveness. Ann. Rev. Phytopathol. 2004, 42, 243–270. [Google Scholar] [CrossRef] [PubMed]
- Torsvik, V.; vreås, L. Microbial diversity and function in soil: From genes to ecosystems. Curr. Opin. Microbiol. 2002, 5, 240–245. [Google Scholar] [CrossRef]
- Dunbar, J.; Barns, S.M.; Ticknor, L.O.; Kuske, C.R. Empirical and theoretical bacterial diversity in four Arizona soils. Appl. Environ. Microbiol. 2002, 68, 3035–3045. [Google Scholar] [CrossRef]
- Tringe, S.G.; von Mering, C.; Kobayashi, A.; Salamov, A.A.; Chen, K.; Chang, H.W.; Podar, M.; Short, J.M.; Mathur, E.J.; Detter, J.C.; et al. Comparative Metagenomics of Microbial Communities. Science 2005, 308, 554–557. [Google Scholar] [CrossRef] [Green Version]
- Cretoiu, M.S.; Korthals, G.W.; Visser, J.H.M.; Van Elsas, J.D. Chitin amendment increases soil suppressiveness toward plant pathogens and modulates the actinobacterial and oxalobacteraceal communities in an experimental agricultural field. Appl. Environ. Microbiol. 2013, 79, 5291–5301. [Google Scholar] [CrossRef]
- Klein, E.; Katan, J.; Minz, D.; Gamliel, A. Soil Suppressiveness to Fusarium Disease: Shifts in Root Microbiome Associated with Reduction of Pathogen Root Colonization. Phytopathology 2012, 103, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Grantina-Ievina, L.; Nikolajeva, V.; Rostoks, N.; Skrabule, I.; Zarina, L.; Pogulis, A.; Ievinsh, G. Impact of Green Manure and Vermicompost on Soil Suppressiveness, Soil Microbial Populations, and Plant Growth in Conditions of Organic Agriculture of Northern Temperate Climate. In Organic Amendments and Soil Suppressiveness in Plant Disease Management; Springer: Cham, Switzerland, 2015; Volume 46, pp. 381–399. [Google Scholar]
- Feng, G.; Xie, T.; Wang, X.; Bai, J.; Tang, L.; Zhao, H.; Wei, W.; Wang, M.; Zhao, Y. Metagenomic analysis of microbial community and function involved in cd-contaminated soil. BMC Microbiol. 2018, 18, 11. [Google Scholar] [CrossRef] [PubMed]
- Mendes, R.; Kruijt, M.; de Bruijn, I.; Dekkers, E.; van der Voort, M.; Schneider, J.H.; Piceno, Y.M.; DeSantis, T.Z.; Andersen, G.L.; Bakker, P.A.; et al. Deciphering the Rhizosphere Microbiome for Disease-Suppressive Bacteria. Science 2011, 332, 1097–1100. [Google Scholar] [CrossRef] [PubMed]
- Hungria, M.; Franchini, J.C.; Brandão-Junior, O.; Kaschuk, G.; Souza, R.A. Soil microbial activity and crop sustainability in a long-term experiment with three soil-tillage and two crop-rotation systems. Appl. Soil Ecol. 2009, 42, 288–296. [Google Scholar] [CrossRef]
- Stirling, G.R.; Smith, M.K.; Smith, J.P.; Stirling, A.M.; Hamill, S.D. Organic inputs, tillage and rotation practices influence soil health and suppressiveness to soilborne pests and pathogens of ginger. Aust. Plant Pathol. 2012, 41, 99–112. [Google Scholar] [CrossRef]
- Kepler, R.M.; Ugine, T.A.; Maul, J.E.; Cavigelli, M.A.; Rehner, S.A. Community composition and population genetics of insect pathogenic fungi in the genus Metarhizium from soils of a long-term agricultural research system. Environ. Microbiol. 2015, 17, 2791–2804. [Google Scholar] [CrossRef]
- Cederlund, H.; Wessén, E.; Enwall, K.; Jones, C.M.; Juhanson, J.; Pell, M.; Philippot, L.; Hallin, S. Soil carbon quality and nitrogen fertilization structure bacterial communities with predictable responses of major bacterial phyla. Appl. Soil Ecol. 2014, 84, 62–68. [Google Scholar] [CrossRef]
- Gómez Expósito, R.; de Bruijn, I.; Postma, J.; Raaijmakers, J.M. Current Insights into the Role of Rhizosphere Bacteria in Disease Suppressive Soils. Front. Microbiol. 2017, 8, 2529. [Google Scholar] [CrossRef]
- Banik, J.J.; Brady, S.F. Recent application of metagenomic approaches toward the discovery of antimicrobials and other bioactive small molecules. Curr. Opin. Microbiol. 2010, 13, 603–609. [Google Scholar] [CrossRef] [Green Version]
- Thomas, T.; Gilbert, J.; Meyer, F. Metagenomics: A guide from sampling to data analysis. In The Role of Bioinformatics in Agriculture; BioMed Central: London, UK, 2014; Volume 2, pp. 357–383. [Google Scholar]
- Scholz, M.B.; Lo, C.C.; Chain, P.S.G. Next generation sequencing and bioinformatic bottlenecks: The current state of metagenomic data analysis. Curr. Opin. Biotechnol. 2012, 23, 9–15. [Google Scholar] [CrossRef]
- Culligan, E.P.; Sleator, R.D.; Marchesi, J.R.; Hill, C. Metagenomics and novel gene discovery: Promise and potential for novel therapeutics. Virulence 2014, 5, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Sommermann, L.; Geistlinger, J.; Wibberg, D.; Deubel, A.; Zwanzig, J.; Babin, D.; Schlüter, A.; Schellenberg, I. Fungal community profiles in agricultural soils of a long-term field trial under different tillage, fertilization and crop rotation conditions analyzed by high-throughput ITS-amplicon sequencing. PLoS ONE 2018, 13, e0195345. [Google Scholar] [CrossRef]
- Deubel, A.; Hofmann, B.; Orzessek, D. Long-term effects of tillage on stratification and plant availability of phosphate and potassium in a loess chernozem. Soil Tillage Res. 2011, 117, 85–92. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Jaenicke, S.; Albaum, S.P.; Blumenkamp, P.; Linke, B.; Stoye, J.; Goesmann, A. Flexible metagenome analysis using the MGX framework. Microbiome 2018, 6, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; LoCascio, P.F.; Hauser, L.J.; Uberbacher, E.C. Gene and translation initiation site prediction in metagenomic sequences. Bioinformatics 2012, 28, 2223–2230. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.J.; Tappu, R. MEGAN Community Edition - Interactive Exploration and Analysis of Large-Scale Microbiome Sequencing Data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef]
- Kang, D.D.; Froula, J.; Egan, R.; Wang, Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 2015, 3, e1165. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.W.; Simmons, B.A.; Singer, S.W. MaxBin 2.0: An automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 2015, 32, 605–607. [Google Scholar] [CrossRef]
- Sieber, C.M.K.; Probst, A.J.; Sharrar, A.; Thomas, B.C.; Hess, M.; Tringe, S.G.; Banfield, J.F. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 2018, 3, 836–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jünemann, S.; Kleinbölting, N.; Jaenicke, S.; Henke, C.; Hassa, J.; Nelkner, J.; Stolze, Y.; Albaum, S.P.; Schlüter, A.; Goesmann, A.; et al. Bioinformatics for NGS-based metagenomics and the application to biogas research. J. Biotechnol. 2017, 261, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Chuvochina, M.; Waite, D.W.; Rinke, C.; Skarshewski, A.; Chaumeil, P.A.; Hugenholtz, P. A proposal for a standardized bacterial taxonomy based on genome phylogeny. bioRxiv 2018, 256800. [Google Scholar] [CrossRef] [Green Version]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid Annotations using Subsystems Technology. BMC Genomics 2008, 9, 75. [Google Scholar] [CrossRef]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef]
- Weber, T.; Blin, K.; Duddela, S.; Krug, D.; Kim, H. antiSMASH 3.0—A comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids 2015, 43, W237–W243. [Google Scholar] [CrossRef]
- Blin, K.; Kim, H.U.; Medema, M.H.; Weber, T. Recent development of antiSMASH and other computational approaches to mine secondary metabolite biosynthetic gene clusters. Brief. Bioinform. 2017. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [Green Version]
- Zakrzewski, M.; Proietti, C.; Ellis, J.J.; Hasan, S.; Brion, M.J.; Berger, B.; Krause, L. Calypso: A user-friendly web-server for mining and visualizing microbiome–environment interactions. Bioinformatics 2016, 33, btw725. [Google Scholar] [CrossRef]
- Segata, N.; Huttenhower, C. Toward an efficient method of identifying core genes for evolutionary and functional microbial phylogenies. PLoS ONE 2011, 6, e24704. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef]
- Zhalnina, K.; de Quadros, P.D.; Gano, K.A.; Davis-Richardson, A.; Fagen, J.R.; Brown, C.T.; Giongo, A.; Drew, J.C.; Sayavedra-Soto, L.A.; Arp, D.J.; et al. Ca. Nitrososphaera and Bradyrhizobium are inversely correlated and related to agricultural practices in long-term field experiments. Front. Microbiol. 2013, 4, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganzert, L.; Lipski, A.; Hubberten, H.W.; Wagner, D. The impact of different soil parameters on the community structure of dominant bacteria from nine different soils located on Livingston Island, South Shetland Archipelago, Antarctica. FEMS Microbiol. Ecol. 2011, 76, 476–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fierer, N. Embracing the unknown: Disentangling the complexities of the soil microbiome. Nat. Rev. Microbiol. 2017, 15, 579–590. [Google Scholar] [CrossRef]
- Beneduzi, A.; Ambrosini, A.; Passaglia, L.M.P. Plant growth-promoting rhizobacteria (PGPR): Their potential as antagonists and biocontrol agents. Genet. Mol. Biol. 2012, 35, 1044–1051. [Google Scholar] [CrossRef]
- Berg, G. Plant-microbe interactions promoting plant growth and health: Perspectives for controlled use of microorganisms in agriculture. Appl. Microbiol. Biotechnol. 2009, 84, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Majeed, A.; Muhammad, Z.; Ahmad, H. Plant growth promoting bacteria: role in soil improvement, abiotic and biotic stress management of crops. Plant Cell Rep. 2018, 37, 1599–1609. [Google Scholar] [CrossRef]
- de Souza, R.; Ambrosini, A.; Passaglia, L.M. Plant growth-promoting bacteria as inoculants in agricultural soils. Genet. Mol. Biol. 2015, 38, 401–419. [Google Scholar] [CrossRef]
- Sahu, P.K.; Singh, D.P.; Prabha, R.; Meena, K.K.; Abhilash, P. Connecting microbial capabilities with the soil and plant health: Options for agricultural sustainability. Ecol. Indic. 2018. [Google Scholar] [CrossRef]
- Puga-Freitas, R.; Blouin, M. A review of the effects of soil organisms on plant hormone signalling pathways. Environ. Exp. Bot. 2015, 114, 104–116. [Google Scholar] [CrossRef]
- Vysloužilová, B.; Ertlen, D.; Schwartz, D.; Šefrna, L. Chernozem. From concept to classification: A review. AUC Geogr. 2016, 51, 85–95. [Google Scholar] [CrossRef]
- Hassa, J.; Maus, I.; Off, S.; Pühler, A.; Scherer, P.; Klocke, M.; Schlüter, A. Metagenome, metatranscriptome, and metaproteome approaches unraveled compositions and functional relationships of microbial communities residing in biogas plants. Appl. Microbiol. Biotechnol. 2018, 102, 5045–5063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howe, A.C.; Jansson, J.K.; Malfatti, S.A.; Tringe, S.G.; Tiedje, J.M.; Brown, C.T. Tackling soil diversity with the assembly of large, complex metagenomes. Proc. Natl. Acad. Sci. USA 2014, 111, 4904–4909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.H.; Liao, Y.C. Accurate binning of metagenomic contigs via automated clustering sequences using information of genomic signatures and marker genes. Sci. Rep. 2016, 6, 24175. [Google Scholar] [CrossRef]
- Papudeshi, B.; Haggerty, J.M.; Doane, M.; Morris, M.M.; Walsh, K.; Beattie, D.T.; Pande, D.; Zaeri, P.; Silva, G.G.Z.; Thompson, F.; et al. Optimizing and evaluating the reconstruction of Metagenome-assembled microbial genomes. BMC Genomics 2017, 18, 915. [Google Scholar] [CrossRef]
- Sangwan, N.; Xia, F.; Gilbert, J.A. Recovering complete and draft population genomes from metagenome datasets. Microbiome 2016, 4, 8. [Google Scholar] [CrossRef] [PubMed]
- Sczyrba, A.; Hofmann, P.; Belmann, P.; Koslicki, D.; Janssen, S.; Dröge, J.; Gregor, I.; Majda, S.; Fiedler, J.; Dahms, E.; et al. Critical Assessment of Metagenome Interpretation—A benchmark of metagenomics software. Nat. Methods 2017, 14, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Furumichi, M.; Morishima, K.; Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019, 47, D590–D595. [Google Scholar] [CrossRef]
- Bulgarelli, D.; Schlaeppi, K.; Spaepen, S.; van Themaat, E.V.L.; Schulze-Lefert, P. Structure and Functions of the Bacterial Microbiota of Plants. Ann. Rev. Plant Biol. 2013, 64, 807–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamattina, L.; García-Mata, C.; Graziano, M.; Pagnussat, G. NITRIC OXIDE: The Versatility of an Extensive Signal Molecule. Ann. Rev. Plant Biol. 2003, 54, 109–136. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, H.; Fraga, R.; Gonzalez, T.; Bashan, Y. Genetics of phosphate solubilization and its potential applications for improving plant growth-promoting bacteria. Plant Soil 2006, 287, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Vassilev, N.; Martos, E.; Mendes, G.; Martos, V.; Vassileva, M. Biochar of animal origin: A sustainable solution to the global problem of high-grade rock phosphate scarcity? J. Sci. Food Agric. 2013, 93, 1799–1804. [Google Scholar] [CrossRef] [PubMed]
- Glick, B.R. Bacteria with ACC deaminase can promote plant growth and help to feed the world. Microbiol. Res. 2014, 169, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Leveau, J.; McSpadden Gardener, B.B.; Pierson, E.A.; Pierson, L.S.; Ryu, C.M. The multifactorial basis for plant health promotion by plant-associated bacteria. Appl. Environ. Microbiol. 2011, 77, 1548–1555. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.J.; O’Neill, D.P.; Wolbang, C.M.; Symons, G.M.; Reid, J.B. Auxin-Gibberellin Interactions and Their Role in Plant Growth. J. Plant Growth Regul. 2001, 20, 336–353. [Google Scholar] [CrossRef]
- Fincheira, P.; Quiroz, A. Microbial volatiles as plant growth inducers. Microbiol. Res. 2018, 208, 63–75. [Google Scholar] [CrossRef]
- Kai, M.; Effmert, U.; Piechulla, B. Bacterial-Plant-Interactions: Approaches to Unravel the Biological Function of Bacterial Volatiles in the Rhizosphere. Front. Microbiol. 2016, 7, 108. [Google Scholar] [CrossRef] [Green Version]
- Guttenberger, N.; Blankenfeldt, W.; Breinbauer, R. Recent developments in the isolation, biological function, biosynthesis, and synthesis of phenazine natural products. Bioorganic Med. Chem. 2017, 25, 6149–6166. [Google Scholar] [CrossRef]
- Vekeman, B.; Kerckhof, F.M.; Cremers, G.; de Vos, P.; Vandamme, P.; Boon, N.; Op den Camp, H.J.; Heylen, K. New Methyloceanibacter diversity from North Sea sediments includes methanotroph containing solely the soluble methane monooxygenase. Environ. Microbiol. 2016, 18, 4523–4536. [Google Scholar] [CrossRef] [PubMed]
- Iguchi, H.; Yurimoto, H.; Sakai, Y. Interactions of Methylotrophs with Plants and Other Heterotrophic Bacteria. Microorganisms 2015, 3, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Chistoserdova, L.; Kalyuzhnaya, M.G. Current Trends in Methylotrophy. Trends Microbiol. 2018, 26, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Tomar, R.S.; Lade, H.; Paul, D. Methylotrophic bacteria in sustainable agriculture. World J. Microbiol. Biotechnol. 2016, 32, 120. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Oter, R.; Nakano, R.T.; Dombrowski, N.; Ma, K.W.; McHardy, A.C.; Schulze-Lefert, P. Modular Traits of the Rhizobiales Root Microbiota and Their Evolutionary Relationship with Symbiotic Rhizobia. Cell Host Microbe 2018, 24, 155–167.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, R.; Salles, J.F.; Berg, G.; Smalla, K. Cultivation-independent analysis of Pseudomonas species in soil and in the rhizosphere of field-grown Verticillium dahliae host plants. Environ. Microbiol. 2006, 8, 2136–2149. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.L.; Waqas, M.; Kang, S.M.; Al-Harrasi, A.; Hussain, J.; Al-Rawahi, A.; Al-Khiziri, S.; Ullah, I.; Ali, L.; Jung, H.Y.; et al. Bacterial endophyte Sphingomonas sp. LK11 produces gibberellins and IAA and promotes tomato plant growth. J. Microbiol. 2014, 52, 689–695. [Google Scholar] [CrossRef]
- Verlinden, R.; Hill, D.; Kenward, M.; Williams, C.; Radecka, I. Bacterial synthesis of biodegradable polyhydroxyalkanoates. J. Appl. Microbiol. 2007, 102, 1437–1449. [Google Scholar] [CrossRef]
- Pankievicz, V.C.S.; Camilios-Neto, D.; Bonato, P.; Balsanelli, E.; Tadra-Sfeir, M.Z.; Faoro, H.; Chubatsu, L.S.; Donatti, L.; Wajnberg, G.; Passetti, F.; et al. RNA-seq transcriptional profiling of Herbaspirillum seropedicae colonizing wheat (Triticum aestivum) roots. Plant Mol. Biol. 2016, 90, 589–603. [Google Scholar] [CrossRef]
- Reuben, S.; Bhinu, V.S.; Swarup, S. Rhizosphere Metabolomics: Methods and Applications; Springer: Berlin/Heidelberg, Germany, 2008; pp. 37–68. [Google Scholar]
- Behnsen, J.; Raffatellu, M. Siderophores: More than stealing iron. mBio 2016. [Google Scholar] [CrossRef]
- Cimermancic, P.; Medema, M.; Claesen, J.; Kurita, K.; Wieland Brown, L.; Mavrommatis, K.; Pati, A.; Godfrey, P.; Koehrsen, M.; Clardy, J.; et al. Insights into Secondary Metabolism from a Global Analysis of Prokaryotic Biosynthetic Gene Clusters. Cell 2014, 158, 412–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schöner, T.A.; Gassel, S.; Osawa, A.; Tobias, N.J.; Okuno, Y.; Sakakibara, Y.; Shindo, K.; Sandmann, G.; Bode, H.B. Aryl Polyenes, a Highly Abundant Class of Bacterial Natural Products, Are Functionally Related to Antioxidative Carotenoids. ChemBioChem 2016, 17, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Bruce, J.B.; West, S.A.; Griffin, A.S. Bacteriocins and the assembly of natural Pseudomonas fluorescens populations. J. Evol. Biol. 2017, 30, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.; Cordovez, V.; de Boer, W.; Raaijmakers, J.; Garbeva, P. Volatile affairs in microbial interactions. ISME J. 2015, 9, 2329–2335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John, J.; Saranathan, R.; Adigopula, L.N.; Thamodharan, V.; Singh, S.P.; Lakshmi, T.P.; CharanTej, M.A.; Rao, R.S.; Krishna, R.; Rao, H.S.P.; et al. The quorum sensing molecule N -acyl homoserine lactone produced by Acinetobacter baumannii displays antibacterial and anticancer properties. Biofouling 2016, 32, 1029–1047. [Google Scholar] [CrossRef]
- Biggins, J.B.; Ternei, M.A.; Brady, S.F. Malleilactone, a Polyketide Synthase-Derived Virulence Factor Encoded by the Cryptic Secondary Metabolome of Burkholderia pseudomallei Group Pathogens. J. Am. Chem. Soc. 2012, 134, 13192–13195. [Google Scholar] [CrossRef] [Green Version]
- Helfrich, E.J.; Reiter, S.; Piel, J. Recent advances in genome-based polyketide discovery. Curr. Opin. Biotechnol. 2014, 29, 107–115. [Google Scholar] [CrossRef]
- Wanke, M.; Skorupinska-Tudek, K.; Swiezewska, E. Isoprenoid biosynthesis via 1-deoxy-D-xylulose 5-phosphate/2-C-methyl-D-erythritol 4-phosphate (DOXP/MEP) pathway. Acta Biochim. Pol. 2001, 48, 663–672. [Google Scholar]
- Simontacchi, M.; García-Mata, C.; Bartoli, C.G.; Santa-María, G.E.; Lamattina, L. Nitric oxide as a key component in hormone-regulated processes. Plant Cell Rep. 2013, 32, 853–866. [Google Scholar] [CrossRef]
- Brodhagen, M.; Peyron, M.; Miles, C.; Inglis, D.A. Biodegradable plastic agricultural mulches and key features of microbial degradation. Appl. Microbiol. Biotechnol. 2015, 99, 1039–1056. [Google Scholar] [CrossRef]
- Mandic-Mulec, I.; Stefanic, P.; van Elsas, J.D. Ecology of Bacillaceae. In The Bacterial Spore: From Molecules to Systems; ASM Press: Washington, DC, USA, 2015; Volume 3, pp. 59–85. [Google Scholar]
- Mirouze, N.; Dubnau, D. Chance and Necessity in Bacillus subtilis Development. In The Bacterial Spore: From Molecules to Systems; American Society of Microbiology: Washington, DC, USA, 2016; pp. 105–127. [Google Scholar]
- Ransom-Jones, E.; McCarthy, A.J.; Haldenby, S.; Doonan, J.; McDonald, J.E. Lignocellulose-Degrading Microbial Communities in Landfill Sites Represent a Repository of Unexplored Biomass-Degrading Diversity. mSphere 2017, 2, e00300-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Mondéjar, R.; Zühlke, D.; Becher, D.; Riedel, K.; Baldrian, P. Cellulose and hemicellulose decomposition by forest soil bacteria proceeds by the action of structurally variable enzymatic systems. Sci. Rep. 2016, 6, 25279. [Google Scholar] [CrossRef]
- Abe, K.; Nakajima, M.; Yamashita, T.; Matsunaga, H.; Kamisuki, S.; Nihira, T.; Takahashi, Y.; Sugimoto, N.; Miyanaga, A.; Nakai, H.; et al. Biochemical and structural analyses of a bacterial endo-β-1,2-glucanase reveal a new glycoside hydrolase family. J. Biol. Chem. 2017, 292, 7487–7506. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Nakajima, M.; Miyanaga, A.; Takahashi, Y.; Tanaka, N.; Kobayashi, K.; Sugimoto, N.; Nakai, H.; Taguchi, H. Characterization and Structural Analysis of a Novel exo -Type Enzyme Acting on β-1,2-Glucooligosaccharides from Parabacteroides distasonis. Biochemistry 2018, 57, 3849–3860. [Google Scholar] [CrossRef]
- Kwak, M.J.; Kong, H.G.; Choi, K.; Kwon, S.K.; Song, J.Y.; Lee, J.; Lee, P.A.; Choi, S.Y.; Seo, M.; Lee, H.J.; et al. Rhizosphere microbiome structure alters to enable wilt resistance in tomato. Nat. Biotechnol. 2018, 36, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, P.; Bottini, R. Terpene Production by Bacteria and its Involvement in Plant Growth Promotion, Stress Alleviation, and Yield Increase. In Molecular Microbial Ecology of the Rhizosphere; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; pp. 335–343. [Google Scholar]
- Yu, D.; Xu, F.; Zeng, J.; Zhan, J. Type III polyketide synthases in natural product biosynthesis. IUBMB Life 2012, 64, 285–295. [Google Scholar] [CrossRef]
- Katsuyama, Y.; Ohnishi, Y. Type III Polyketide Synthases in Microorganisms. Methods Enzymol. 2012, 515, 359–377. [Google Scholar] [CrossRef]
- Kielak, A.M.; Barreto, C.C.; Kowalchuk, G.A.; van Veen, J.A.; Kuramae, E.E. The Ecology of Acidobacteria: Moving beyond Genes and Genomes. Front. Microbiol. 2016, 7, 744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichorst, S.A.; Trojan, D.; Roux, S.; Herbold, C.; Rattei, T.; Woebken, D. Genomic insights into the Acidobacteria reveal strategies for their success in terrestrial environments. Environ. Microbiol. 2018, 20, 1041–1063. [Google Scholar] [CrossRef]
- Kielak, A.M.; Cipriano, M.A.P.; Kuramae, E.E. Acidobacteria strains from subdivision 1 act as plant growth-promoting bacteria. Arch. Microbiol. 2016, 198, 987–993. [Google Scholar] [CrossRef] [Green Version]
- Pester, M.; Schleper, C.; Wagner, M. The Thaumarchaeota: An emerging view of their phylogeny and ecophysiology. Curr. Opin. Microbiol. 2011, 14, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Tourna, M.; Stieglmeier, M.; Spang, A.; Konneke, M.; Schintlmeister, A.; Urich, T.; Engel, M.; Schloter, M.; Wagner, M.; Richter, A.; et al. Nitrososphaera viennensis, an ammonia oxidizing archaeon from soil. Proc. Natl. Acad. Sci. USA 2011, 108, 8420–8425. [Google Scholar] [CrossRef] [PubMed]
- Offre, P.; Nicol, G.W.; Prosser, J.I. Community profiling and quantification of putative autotrophic thaumarchaeal communities in environmental samples. Environ. Microbiol. Rep. 2011, 3, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Nicol, G.W.; Schleper, C. Ammonia-oxidising Crenarchaeota: Important players in the nitrogen cycle? Trends Microbiol. 2006, 14, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Könneke, M.; Schubert, D.M.; Brown, P.C.; Hügler, M.; Standfest, S.; Schwander, T.; Schada von Borzyskowski, L.; Erb, T.J.; Stahl, D.A.; Berg, I.A. Ammonia-oxidizing archaea use the most energy-efficient aerobic pathway for CO2 fixation. Proc. Natl. Acad. Sci. USA 2014, 111, 8239–8244. [Google Scholar] [CrossRef]
- Berg, I.A.; Kockelkorn, D.; Buckel, W.; Fuchs, G. A 3-hydroxypropionate/4-hydroxybutyrate autotrophic carbon dioxide assimilation pathway in archaea. Science 2007, 318, 1782–1786. [Google Scholar] [CrossRef]
- Cibis, K.G.; Gneipel, A.; König, H. Isolation of acetic, propionic and butyric acid-forming bacteria from biogas plants. J. Biotechnol. 2016, 220, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Müller, H.; Berg, C.; Landa, B.B.; Auerbach, A.; Moissl-Eichinger, C.; Berg, G. Plant genotype-specific archaeal and bacterial endophytes but similar Bacillus antagonists colonize Mediterranean olive trees. Front. Microbiol. 2015, 6, 138. [Google Scholar] [CrossRef] [Green Version]
- Hedden, P.; Thomas, S.G. Gibberellin biosynthesis and its regulation. Biochem. J. 2012, 444, 11–25. [Google Scholar] [CrossRef] [Green Version]
- Cuskin, F.; Baslé, A.; Ladevèze, S.; Day, A.M.; Gilbert, H.J.; Davies, G.J.; Potocki-Véronèse, G.; Lowe, E.C. The GH130 Family of Mannoside Phosphorylases Contains Glycoside Hydrolases That Target β-1,2-Mannosidic Linkages in Candida Mannan. J. Biol. Chem. 2015, 290, 25023–25033. [Google Scholar] [CrossRef]
- Hong, J.K.; Cho, J.C. Environmental Variables Shaping the Ecological Niche of Thaumarchaeota in Soil: Direct and Indirect Causal Effects. PLoS ONE 2015, 10, e0133763. [Google Scholar] [CrossRef] [PubMed]
- Kielak, A.; Pijl, A.S.; van Veen, J.A.; Kowalchuk, G.A. Phylogenetic diversity of Acidobacteria in a former agricultural soil. ISME J. 2009, 3, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Schreiter, S.; Ding, G.C.; Heuer, H.; Neumann, G.; Sandmann, M.; Grosch, R.; Kropf, S.; Smalla, K. Effect of the soil type on the microbiome in the rhizosphere of field-grown lettuce. Front. Microbiol. 2014, 5, 144. [Google Scholar] [CrossRef]
- Feng, L.; Chen, K.; Han, D.; Zhao, J.; Lu, Y.; Yang, G.; Mu, J.; Zhao, X. Comparison of nitrogen removal and microbial properties in solid-phase denitrification systems for water purification with various pretreated lignocellulosic carriers. Bioresour. Technol. 2017, 224, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.S.; Son, J.; Oren, A. A phylogenomic and molecular markers based taxonomic framework for members of the order Entomoplasmatales: proposal for an emended order Mycoplasmatales containing the family Spiroplasmataceae and emended family Mycoplasmataceae comprised of six genera. Antonie van Leeuwenhoek 2018. [Google Scholar] [CrossRef]
- Torres-Cortés, G.; Ghignone, S.; Bonfante, P.; Schüßler, A. Mosaic genome of endobacteria in arbuscular mycorrhizal fungi: Transkingdom gene transfer in an ancient mycoplasma-fungus association. Proc. Natl. Acad. Sci. USA 2015, 112, 7785–7790. [Google Scholar] [CrossRef] [Green Version]
- Treseder, K.K.; Allen, E.B.; Egerton-Warburton, L.M.; Hart, M.M.; Klironomos, J.N.; Maherali, H.; Tedersoo, L. Arbuscular mycorrhizal fungi as mediators of ecosystem responses to nitrogen deposition: A trait-based predictive framework. J. Ecol. 2018, 106, 480–489. [Google Scholar] [CrossRef]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | Bernburg-Strenzfeld (LTE-1 field site), |
| Saxony-Anhalt, Germany | |
| Latitude/Longitude | 51.82° N, 11.70° E |
| Altitude | 80 m (above sea level) |
| Environment | Agricultural farmland |
| Cultivated crop | Winter wheat cultivar ‘Pamir’ |
| Soil type | Loess chernozem over limestone |
| Soil texture | 22% clay, 70% silt, 8% sand |
| Horizon (sampled) | Ap |
| pH | 7.0–7.4 |
| P supply | 45–70 mg per kg |
| K supply | 130–185 mg per kg |
| Average annual temperature (1980–2010) | 9.7 °C |
| Average annual precipitation | 511 mm |
| Crop rotation | Grain maize (Zea mays), winter wheat (Triticum aestivum), |
| winter barley (Hordeum vulgare), | |
| winter rapeseed (Brassica napus ssp. Napus), winter wheat | |
| Treatment: N-fertilization | 220 kg per ha as ammonium sulfate and calcium ammonium |
| nitrate + fungicides and growth regulators as described | |
| in [24] (intensive—Int) or 90 kg per ha as ammonium sulfate | |
| and calcium ammonium nitrate (extensive—Ext) | |
| Treatment: Tillage | Ploughed using a mould-board plough (P), |
| carrier board with combined alternating ploughshares, | |
| ploughing depth 30 cm, incl. soil inversion or treated with | |
| cultivator (CT), 10 cm flat non-inverting soil loosening | |
| History | Long-term field trial started in 1992 |
| Collection date | 30 July 2015 |
| Sampling depth | 0–25 cm (soil corer) |
| Physicochemical soil properties | [25] |
| Basic soil parameters | [24] Table S3 |
| Winter wheat yields of the LTE-1 field plots | Described by [24] |
| Comp- | Con- | Abun- | Maximal | Sample | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| lete- | tami- | Size | Dance | Abundance | with Highest | |||||
| MAG ID | LCA a | GTDB Taxonomy | ness | nation | [bp] | Contigs | Features b | Rank c | (RPKM) | Abundance |
| MAG_01 | Thaumarchaeota | Nitrososphaeraceae | 88.69 | 4.85 | 1,828,699 | 692 | 2570 | 1 | 7.614 | BS-P-Int_R2 |
| MAG_02 | Thaumarchaeota | Nitrososphaeraceae | 61.58 | 7.28 | 1,705,306 | 1021 | 2486 | 2 | 5.0159 | BS-CT-Int_R1 |
| MAG_03 | Thaumarchaeota | Nitrososphaeraceae | 81.37 | 0.97 | 1,526,325 | 556 | 2279 | 7 | 3.5153 | BS-P-Int_R2 |
| MAG_04 | Thaumarchaeota | Nitrososphaera | 70.8 | 11.04 | 1,219,884 | 718 | 1776 | 6 | 1.5651 | BS-CT-Int_R2 |
| MAG_05 | uc_Deltaproteobacteria | GR-WP33-30 | 75.42 | 12.44 | 4,002,969 | 2515 | 5905 | 12 | 1.1615 | BS-CT-Ext_R1 |
| MAG_08 | Bacillaceae | Virgibacillus_F | 81.28 | 6.4 | 2,457,004 | 1229 | 3488 | 15 | 3.8837 | RS-P-Ext_R1 |
| MAG_10 | Flavobacteriaceae | Gillisia | 90.07 | 4.75 | 3,058,208 | 1271 | 4033 | 72 | 3.3439 | RS-P-Ext_R1 |
| MAG_11 | Pseudomonas | Pseudomonas | 97.39 | 4.71 | 4,749,133 | 612 | 4886 | 55 | 3.3354 | RS-CT-Int_R1 |
| MAG_13 | Rhizobiales | Methyloceanibacter | 71.18 | 0.9 | 1,553,703 | 289 | 1757 | 11 | 1.1708 | BS-P-Int_R2 |
| MAG_14 | Acidobacteria | OLB17 | 56.83 | 1.71 | 1,904,854 | 464 | 2270 | 69 | 0.3698 | BS-CT-Int_R2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nelkner, J.; Henke, C.; Lin, T.W.; Pätzold, W.; Hassa, J.; Jaenicke, S.; Grosch, R.; Pühler, A.; Sczyrba, A.; Schlüter, A. Effect of Long-Term Farming Practices on Agricultural Soil Microbiome Members Represented by Metagenomically Assembled Genomes (MAGs) and Their Predicted Plant-Beneficial Genes. Genes 2019, 10, 424. https://doi.org/10.3390/genes10060424
Nelkner J, Henke C, Lin TW, Pätzold W, Hassa J, Jaenicke S, Grosch R, Pühler A, Sczyrba A, Schlüter A. Effect of Long-Term Farming Practices on Agricultural Soil Microbiome Members Represented by Metagenomically Assembled Genomes (MAGs) and Their Predicted Plant-Beneficial Genes. Genes. 2019; 10(6):424. https://doi.org/10.3390/genes10060424
Chicago/Turabian StyleNelkner, Johanna, Christian Henke, Timo Wentong Lin, Wiebke Pätzold, Julia Hassa, Sebastian Jaenicke, Rita Grosch, Alfred Pühler, Alexander Sczyrba, and Andreas Schlüter. 2019. "Effect of Long-Term Farming Practices on Agricultural Soil Microbiome Members Represented by Metagenomically Assembled Genomes (MAGs) and Their Predicted Plant-Beneficial Genes" Genes 10, no. 6: 424. https://doi.org/10.3390/genes10060424
APA StyleNelkner, J., Henke, C., Lin, T. W., Pätzold, W., Hassa, J., Jaenicke, S., Grosch, R., Pühler, A., Sczyrba, A., & Schlüter, A. (2019). Effect of Long-Term Farming Practices on Agricultural Soil Microbiome Members Represented by Metagenomically Assembled Genomes (MAGs) and Their Predicted Plant-Beneficial Genes. Genes, 10(6), 424. https://doi.org/10.3390/genes10060424