Joint RNA-Seq and miRNA Profiling Analyses to Reveal Molecular Mechanisms in Regulating Thickness of Pod Canopy in Brassica napus

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials, RNA Sequencing, and Small RNA Sequencing
2.2. Transcriptome Analysis
2.3. Small RNA Data Analysis
2.4. qRT-PCR Validation
3. Results
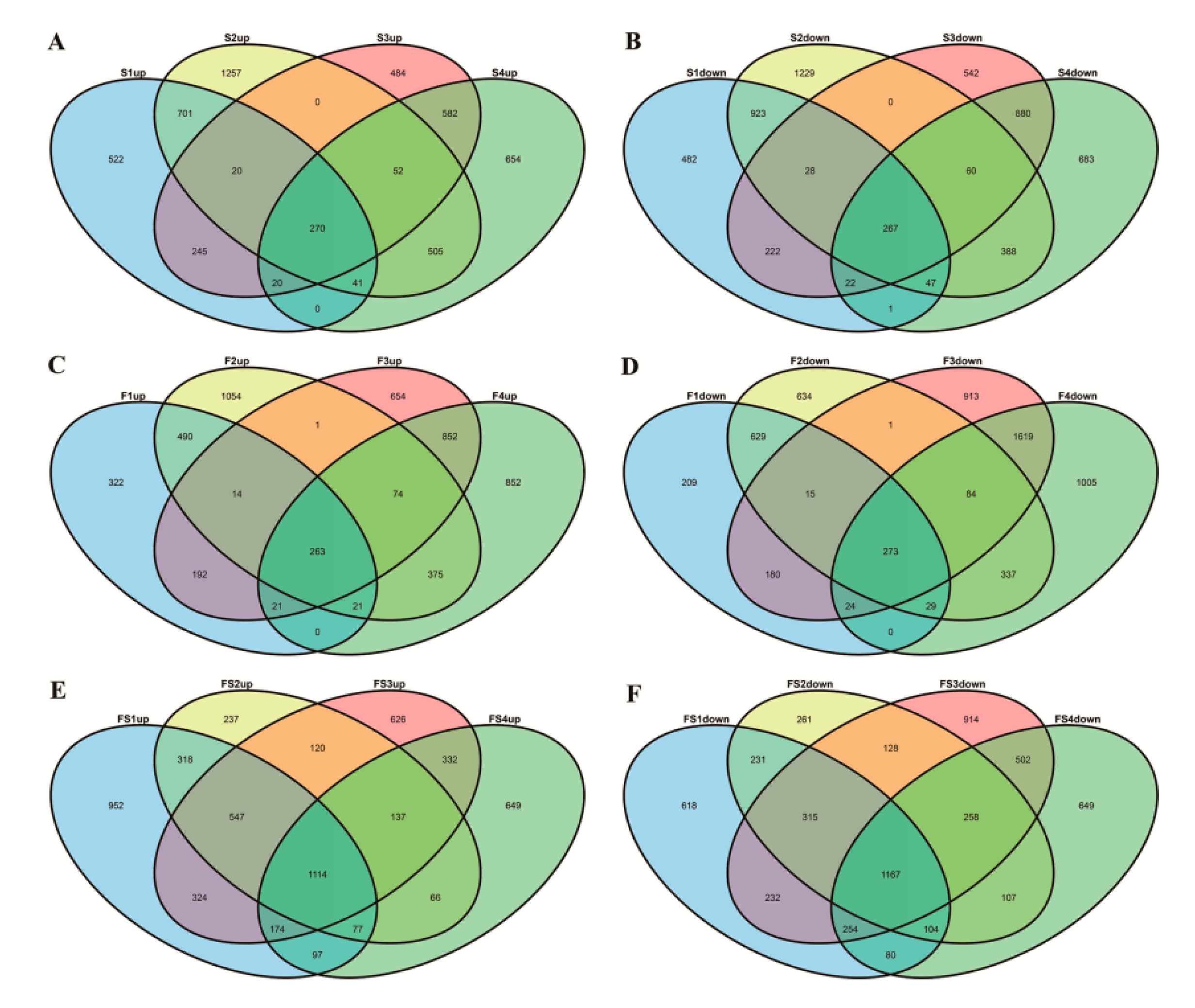
3.1. Identification of Differentially Expressed Genes by RNA-seq
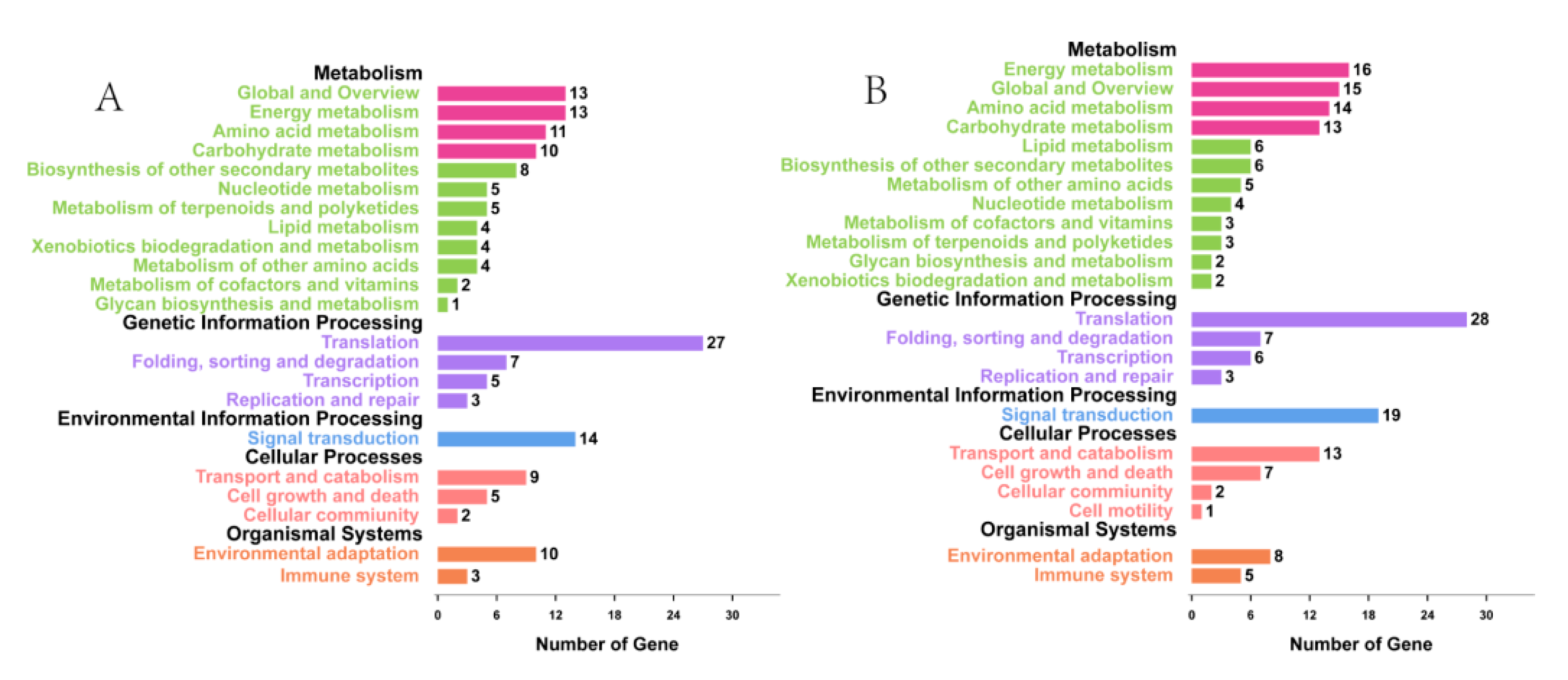
3.2. Functional Classification by Gene Ontology (GO) and Metabolic Pathway Analysis
3.3. MiRNAs Differentially Expressed in High- and Low-TPC Samples
3.4. Validation of Differential Gene and miRNA Expression by qRT-PCR
3.5. MiRNA and Gene Network Analysis in High- and Low-TPC Samples
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, S.; Xie, K.; Li, W.; Zou, T.; Ren, Y.; Wang, S.; Deng, Q.; Zheng, A.; Zhu, J.; Liu, H.; et al. Re-sequencing and genetic variation identification of a rice line with ideal plant architecture. Rice 2012, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Richards, R.A. Selectable traits to increase crop photosynthesis and yield of grain crops. J. Exp. Bot. 2000, 51, 447–458. [Google Scholar] [CrossRef]
- Peiffer, J.A.; Romay, M.C.; Gore, M.A.; Flint-Garcia, S.A.; Zhang, Z.; Millard, M.J.; Gardner, C.A.C.; McMullen, M.D.; Holland, J.B.; Bradbury, P.J.; et al. The Genetic Architecture Of Maize Height. Genetics 2014, 196, 1337–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, X.; Zhang, T. Quantitative trait loci controlling plant architectural traits in cotton. Plant Sci. 2009, 177, 317–323. [Google Scholar] [CrossRef]
- Peng, J.; Richards, D.E.; Hartley, N.M.; Murphy, G.P.; Devos, K.M.; Flintham, J.E.; Beales, J.; Fish, L.J.; Worland, A.J.; Pelica, F.; et al. ‘Green revolution’ genes encode mutant gibberellin response modulators. Nature 1999, 400, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Huyghe, C. Genetics and genetic modifications of plant architecture in grain legumes: A review. Agronomie 1998, 18, 383–411. [Google Scholar] [CrossRef]
- Busov, V.B.; Brunner, A.M.; Strauss, S.H. Genes for control of plant stature and form. New Phytol. 2008, 177, 589–607. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, J. Molecular basis of plant architecture. Annu. Rev. Plant Biol. 2008, 59, 253–279. [Google Scholar] [CrossRef] [PubMed]
- Monna, L.; Kitazawa, N.; Yoshino, R.; Suzuki, J.; Masuda, H.; Maehara, Y.; Tanji, M.; Sato, M.; Nasu, S.; Minobe, Y. Positional cloning of rice semidwarfing gene, sd-1: Rice “green revolution gene” encodes a mutant enzyme involved in gibberellin synthesis. DNA Res. 2002, 9, 11–17. [Google Scholar] [CrossRef]
- Petrásek, J.; Mravec, J.; Bouchard, R.; Blakeslee, J.J.; Abas, M.; Seifertová, D.; Wisniewska, J.; Tadele, Z.; Kubes, M.; Covanová, M.; et al. PIN proteins perform a rate-limiting function in cellular auxin efflux. Science 2006, 312, 914–918. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Maekawa, M.; Shimamoto, K.; Kyozuka, J. The LAX1 and FRIZZY PANICLE 2 genes determine the inflorescence architecture of rice by controlling rachis-branch and spikelet development. Dev. Biol. 2001, 231, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Ulmasov, T.; Hagen, G.; Guilfoyle, T.J. ARF1, a transcription factor that binds to auxin response elements. Science 1997, 276, 1865–1868. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Wang, R.; Qian, Q.; Yan, M.; Meng, X.; Fu, Z.; Yan, C.; Jiang, B.; Su, Z.; Li, J.; et al. DWARF27, an Iron-Containing Protein Required for the Biosynthesis of Strigolactones, Regulates Rice Tiller Bud Outgrowth. Plant Cell 2009, 21, 1512–1525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arite, T.; Umehara, M.; Ishikawa, S.; Hanada, A.; Maekawa, M.; Yamaguchi, S.; Kyozuka, J. d14, a strigolactone-insensitive mutant of rice, shows an accelerated outgrowth of tillers. Plant Cell Physiol. 2009, 50, 1416–1424. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wen, J.; Lease, K.A.; Doke, J.T.; Tax, F.E.; Walker, J.C. BAK1, an Arabidopsis LRR receptor-like protein kinase, interacts with BRI1 and modulates brassinosteroid signaling. Cell 2002, 110, 213–222. [Google Scholar] [CrossRef]
- Wang, X.; Chory, J. Brassinosteroids regulate dissociation of BKI1, a negative regulator of BRI1 signaling, from the plasma membrane. Science 2006, 313, 1118. [Google Scholar] [CrossRef] [PubMed]
- Rupp, H.-M.; Frank, M.; Werner, T.; Strnad, M.; Schmülling, T. Increased steady state mRNA levels of the STM and KNAT1 homeobox genes in cytokinin overproducing Arabidopsis thaliana indicate a role for cytokinins in the shoot apical meristem. Plant J. 1999, 18, 557–563. [Google Scholar] [CrossRef]
- Werner, T.; Motyka, V.; Strnad, M.; Schmülling, T. Regulation of plant growth by cytokinin. Proc. Natl. Acad. Sci. USA 2001, 98, 10487–10492. [Google Scholar] [CrossRef] [Green Version]
- Rogers, K.; Chen, X. Biogenesis, turnover, and mode of action of plant microRNAs. Plant Cell 2013, 25, 2383–2399. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Li, H.; Wang, Y.; Wu, M.; Li, L.; Jin, C.; Zhang, Q.; Chen, C.; Song, W.; Wang, C. Small RNA Sequencing Reveals Differential miRNA Expression in the Early Development of Broccoli (Brassica oleracea var. italica) Pollen. Front. Plant Sci. 2017, 8, 404. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Guo, Z.; Li, L. Evolutionary conservation of microRNA regulatory programs in plant flower development. Dev. Biol. 2013, 380, 133–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwab, R.; Ossowski, S.; Riester, M.; Warthmann, N.; Weigel, D. Highly specific gene silencing by artificial microRNAs in Arabidopsis. Plant Cell 2006, 18, 1121. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Song, Y.; Chen, Z.; Yu, D. Ectopic expression of miR396 suppresses GRF target gene expression and alters leaf growth in Arabidopsis. Physiol. Plant. 2009, 136, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Kutter, C.; Schöb, H.; Stadler, M.; Meins, F.; Si-Ammour, A. MicroRNA-mediated regulation of stomatal development in Arabidopsis. Plant Cell 2007, 19, 2417–2429. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Jung, J.-H.; Reyes, J.L.; Kim, Y.-S.; Kim, S.-Y.; Chung, K.-S.; Kim, J.A.; Lee, M.; Lee, Y.; Narry Kim, V.; et al. microRNA-directed cleavage of ATHB15 mRNA regulates vascular development in Arabidopsis inflorescence stems. Plant J. 2005, 42, 84–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.-W.; Wang, L.-J.; Mao, Y.-B.; Cai, W.-J.; Xue, H.-W.; Chen, X.-Y. Control of root cap formation by microRNA-targeted auxin response factors in Arabidopsis. Plant Cell 2005, 17, 2204. [Google Scholar] [CrossRef]
- Millar, A.A.; Gubler, F. The Arabidopsis GAMYB-Like genes, MYB33 and MYB65, are microRNA-regulated genes that redundantly facilitate anther development. Plant Cell 2005, 17, 705–721. [Google Scholar] [CrossRef]
- Alonso-Peral, M.M.; Sun, C.; Millar, A.A. MicroRNA159 can act as a switch or tuning microRNA independently of its abundance in Arabidopsis. PLoS ONE 2012, 7, e34751. [Google Scholar] [CrossRef]
- Wu, G.; Park, M.Y.; Conway, S.R.; Wang, J.-W.; Weigel, D.; Poethig, R.S. The sequential action of miR156 and miR172 regulates developmental timing in Arabidopsis. Cell 2009, 138, 750–759. [Google Scholar] [CrossRef]
- Aukerman, M.J.; Sakai, H. Regulation of flowering time and floral organ identity by a microRNA and its APETALA2-like target genes. Plant Cell 2003, 15, 2730–2741. [Google Scholar] [CrossRef] [PubMed]
- Chen, X. A MicroRNA as a Translational repressor of APETALA2 in Arabidopsis flower development. Science 2004, 303, 2022–2025. [Google Scholar] [CrossRef] [PubMed]
- Wollmann, H.; Mica, E.; Todesco, M.; Long, J.A.; Weigel, D. On reconciling the interactions between APETALA2, miR172 and AGAMOUS with the ABC model of flower development. Development 2010, 137, 3633–3642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.-T.; Chen, P.-W.; Chen, L.-C.; Yang, C.-C.; Chen, S.-Y.; Huang, G.; Lin, T.C.; Ku, H.-M.; Chen, J.J.W. Suppressive effect of microRNA319 expression on rice plant height. Theor. Appl. Genet. 2017, 130, 1507–1518. [Google Scholar] [CrossRef] [PubMed]
- An, W.; Gong, W.; He, S.; Pan, Z.; Sun, J.; Du, X. MicroRNA and mRNA expression profiling analysis revealed the regulation of plant height in Gossypium hirsutum. BMC Genom. 2015, 16, 886. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Mai, Y.-X.; Zhang, Y.-C.; Luo, Q.; Yang, H.-Q. MicroRNA171c-targeted SCL6-II, SCL6-III, and SCL6-IV genes regulate shoot branching in Arabidopsis. Mol. Plant 2010, 3, 794–806. [Google Scholar] [CrossRef]
- Wang, L.; Sun, S.; Jin, J.; Fu, D.; Yang, X.; Weng, X.; Xu, C.; Li, X.; Xiao, J.; Zhang, Q. Coordinated regulation of vegetative and reproductive branching in rice. Proc. Natl. Acad. Sci. USA 2015, 112, 15504–15509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.; Hao, M.; Wang, W.; Mei, D.; Wells, R.; Liu, J.; Wang, H.; Sang, S.; Tang, M.; Zhou, R.; et al. Integrative RNA- and miRNA-profile analysis reveals a likely role of br and auxin signaling in branch angle regulation of B. napus. Int. J. Mol. Sci. 2017, 18, 887. [Google Scholar] [CrossRef]
- Han, R.; Jian, C.; Lv, J.; Yan, Y.; Chi, Q.; Li, Z.; Wang, Q.; Zhang, J.; Liu, X.; Zhao, H. Identification and characterization of microRNAs in the flag leaf and developing seed of wheat (Triticum aestivum L.). BMC Genom. 2014, 15, 289. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Yu, C.; Li, Y.; Lam, T.-W.; Yiu, S.-M.; Kristiansen, K.; Wang, J. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 2009, 25, 1966–1967. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef]
- Lu, K.; Li, T.; He, J.; Chang, W.; Zhang, R.; Liu, M.; Yu, M.; Fan, Y.; Ma, J.; Sun, W.; et al. qPrimerDB: A thermodynamics-based gene-specific qPCR primer database for 147 organisms. Nucleic Acids Res. 2018, 46, D1229–D1236. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.P.; Tang, H.; Wang, X.; Chiquet, J.; Belcram, H.; Tong, C.; Samans, B.; et al. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. [Google Scholar] [CrossRef]
- Cai, G.; Yang, Q.; Chen, H.; Yang, Q.; Zhang, C.; Fan, C.; Zhou, Y. Genetic dissection of plant architecture and yield-related traits in Brassica napus. Sci. Rep. 2016, 6, 21625. [Google Scholar] [CrossRef]
- Zhang, J.; Song, Q.; Cregan, P.B.; Nelson, R.L.; Wang, X.; Wu, J.; Jiang, G.-L. Genome-wide association study for flowering time, maturity dates and plant height in early maturing soybean (Glycine max) germplasm. BMC Genom. 2015, 16, 217. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lu, K.; Nie, H.; Zeng, Q.; Wu, B.; Qian, J.; Fang, Z. Rice nitrate transporter OsNPF7.2 positively regulates tiller number and grain yield. Rice 2018, 11, 12. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Anwar, S.; Kuai, J.; Ullah, S.; Fahad, S.; Zhou, G. Optimization of nitrogen rate and planting density for improving yield, nitrogen use efficiency, and lodging resistance in oilseed rape. Front. Plant Sci. 2017, 8, 532. [Google Scholar] [CrossRef] [PubMed]
- Kruger, N.J.; von Schaewen, A. The oxidative pentose phosphate pathway: Structure and organisation. Curr. Opin. Plant Biol. 2003, 6, 236–246. [Google Scholar] [CrossRef]
- Sahrawy, M.; Ávila, C.; Chueca, A.; Cánovas, F.M.; López-Gorgé, J. Increased sucrose level and altered nitrogen metabolism in Arabidopsis thaliana transgenic plants expressing antisense chloroplastic fructose-1,6-bisphosphatase. J. Exp. Bot. 2004, 55, 2495–2503. [Google Scholar] [CrossRef]
- Nunes-Nesi, A.; Fernie, A.R.; Stitt, M. Metabolic and signaling aspects underpinning the regulation of plant carbon nitrogen interactions. Mol. Plant 2010, 3, 973–996. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Xin, Z.; Zheng, Z.-L. The OSU1/QUA2/TSD2-encoded putative methyltransferase is a critical modulator of carbon and nitrogen nutrient balance response in Arabidopsis. PLoS ONE 2008, 3, e1387. [Google Scholar] [CrossRef]
- Yoneyama, K.; Xie, X.; Kisugi, T.; Nomura, T.; Yoneyama, K. Nitrogen and phosphorus fertilization negatively affects strigolactone production and exudation in sorghum. Planta 2013, 238, 885–894. [Google Scholar] [CrossRef]
- Razaq, M.; Zhang, P.; Shen, H.-L. Influence of nitrogen and phosphorous on the growth and root morphology of Acer mono. PLoS ONE 2017, 12, e0171321. [Google Scholar] [CrossRef]
- Enquist, B.J.; Niklas, K.J. Global allocation rules for patterns of biomass partitioning in seed plants. Science 2002, 295, 1517–1520. [Google Scholar] [CrossRef]
- Reinhardt, D.; Kuhlemeier, C. Plant architecture. EMBO Rep. 2002, 3, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Forde, B.G. local and long-range signaling pathways regulating plant responses to nitrate. Annu. Rev. Plant Biol. 2002, 53, 203–224. [Google Scholar] [CrossRef] [PubMed]
- De Smet, I.; Signora, L.; Beeckman, T.; Inzé, D.; Foyer, C.H.; Zhang, H. An abscisic acid-sensitive checkpoint in lateral root development of Arabidopsis. Plant J. 2003, 33, 543–555. [Google Scholar] [CrossRef] [PubMed]
- Signora, L.; De Smet, I.; Foyer, C.H.; Zhang, H. ABA plays a central role in mediating the regulatory effects of nitrate on root branching in Arabidopsis. Plant J. 2001, 28, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Vlieghe, K.; Vuylsteke, M.; Florquin, K.; Rombauts, S.; Maes, S.; Ormenese, S.; Van Hummelen, P.; Van de Peer, Y.; Inzé, D.; De Veylder, L. Microarray analysis of E2Fa-DPa-overexpressing plants uncovers a cross-talking genetic network between DNA replication and nitrogen assimilation. J. Cell Sci. 2003, 116, 4249–4259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Q.; Wang, Y.; Li, X.; Wu, S.; Wang, Y.; Luo, J.; Mattson, N.; Xu, Y. Interactions between ethylene, gibberellin and abscisic acid in regulating submergence induced petiole elongation in Nelumbo nucifera. Aquat. Bot. 2017, 137, 9–15. [Google Scholar] [CrossRef]
- Pellny, T.K.; Van Aken, O.; Dutilleul, C.; Wolff, T.; Groten, K.; Bor, M.; De Paepe, R.; Reyss, A.; Van Breusegem, F.; Noctor, G.; et al. Mitochondrial respiratory pathways modulate nitrate sensing and nitrogen-dependent regulation of plant architecture in Nicotiana sylvestris. Plant J. 2008, 54, 976–992. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Pu, H.; Gao, J.; Long, W.; Chen, F.; Zhang, W.; Zhou, X.; Peng, Q.; Chen, S.; Zhang, J. Comparative analysis of miRNAs of two rapeseed genotypes in response to acetohydroxyacid synthase-inhibiting herbicides by high-throughput sequencing. PLoS ONE 2017, 12, e0184917. [Google Scholar] [CrossRef]
- Guoxing, H.A.U. Wuhan Chen. Study of Overexpressing miRNA 167a to Regulate the Architecture in Oryza sativa. Mol. Plant Breed. 2011, 9, 390–396. [Google Scholar]
- Zhao, Y.-T.; Wang, M.; Fu, S.-X.; Yang, W.-C.; Qi, C.-K.; Wang, X.-J. Small RNA Profiling in Two Brassica napus Cultivars Identifies MicroRNAs with Oil Production- and Development-Correlated Expression and New Small RNA Classes. Plant Physiol. 2012, 158, 813–823. [Google Scholar] [CrossRef]
- Wang, J.; Jian, H.; Wang, T.; Wei, L.; Li, J.; Li, C.; Liu, L. Identification of microRNAs actively involved in fatty acid biosynthesis in developing Brassica napus seeds using high-throughput sequencing. Front. Plant Sci. 2016, 7, 1570. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Jiang, J.; Hu, Z.; Lyu, T.; Yang, Y.; Jiang, J.; Cao, J. Over-expression of miR158 causes pollen abortion in Brassica campestris ssp. chinensis. Plant Mol. Biol. 2017, 93, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wen, H.; Teotia, S.; Du, Y.; Zhang, J.; Li, J.; Sun, H.; Tang, G.; Peng, T.; Zhao, Q. Suppression of microRNA159 impacts multiple agronomic traits in rice (Oryza sativa L.). BMC Plant Biol. 2017, 17, 215. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Liu, Y.; Liu, Z.; Kong, D.; Duan, M.; Luo, L. Genome-wide identification and analysis of drought-responsive microRNAs in Oryza sativa. J. Exp. Bot. 2010, 61, 4157–4168. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Shu, J.; Jin, W. Identification and validation of miRNAs associated with the resistance of maize (Zea mays L.) to Exserohilum turcicum. PLoS ONE 2014, 9, e87251. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.N.; Rothstein, S.J.; Spangenberg, G.; Kant, S. Role of microRNAs involved in plant response to nitrogen and phosphorous limiting conditions. Front. Plant Sci. 2015, 6, 629. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Gong, S.; Wang, Y.; Wang, F.; Bao, H.; Sun, J.; Cai, C.; Yi, K.; Chen, Z.; Zhu, C. MicroRNA166 modulates cadmium tolerance and accumulation in rice. Plant Physiol. 2018, 177, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Dragomir, M.; Mafra, A.C.P.; Dias, S.M.G.; Vasilescu, C.; Calin, G.A. Using microRNA Networks to Understand Cancer. Int. J. Mol. Sci. 2018, 19, 1871. [Google Scholar] [CrossRef]
- Roy, A.; Sahoo, D.; Tripathy, B.C. Involvement of phytochrome A in suppression of photomorphogenesis in rice seedling grown in red light. Plant Cell Environ. 2013, 36, 2120–2134. [Google Scholar] [CrossRef]
- Liu, J.; Wang, X.; Huang, H.; Wang, J.; Li, Z.; Wu, L.; Zhang, G.; Ma, Z. Efficiency of phosphorus utilization in phyA-expressing cotton lines. Indian J. Biochem. Biophys. 2012, 49, 250–256. [Google Scholar] [CrossRef]
- Donohue, K.; Heschel, M.S.; Butler, C.M.; Barua, D.; Sharrock, R.A.; Whitelam, G.C.; Chiang, G.C. Diversification of phytochrome contributions to germination as a function of seed-maturation environment. New Phytol. 2008, 177, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Shah, K.; Sharma, N.; Kaur, R.; Singh, J.; Vinson, C.; Rishi, V. A-ZIP53, a dominant negative reveals the molecular mechanism of heterodimerization between bZIP53, bZIP10 and bZIP25 involved in Arabidopsis seed maturation. Sci. Rep. 2017, 7, 14343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, J.H.; Namasivayam, P.; Abdullah, M.P. The PAL2 promoter activities in relation to structural development and adaptation in Arabidopsis thaliana. Planta 2012, 235, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Simon, N.M.L.; Sawkins, E.; Dodd, A.N. Involvement of the SnRK1 subunit KIN10 in sucrose-induced hypocotyl elongation. Plant Signal. Behav. 2018, e1457913. [Google Scholar] [CrossRef] [PubMed]
- Vullo, D.; Lehneck, R.; Pöggeler, S.; Supuran, C.T. Sulfonamide inhibition studies of two β-carbonic anhydrases from the ascomycete fungus Sordaria macrospora, CAS1 and CAS2. J. Enzym. Inhib. Med. Chem. 2018, 33, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Truernit, E.; Haseloff, J. A Role for KNAT class II genes in root development. Plant Signal. Behav. 2007, 2, 10–12. [Google Scholar] [CrossRef] [PubMed]
- Blanvillain, R.; Boavida, L.C.; McCormick, S.; Ow, D.W. EXPORTIN1 genes are essential for development and function of the gametophytes in Arabidopsis thaliana. Genetics 2008, 180, 1493–1500. [Google Scholar] [CrossRef]
- Lange, P.R.; Geserick, C.; Tischendorf, G.; Zrenner, R. Functions of chloroplastic adenylate kinases in Arabidopsis. Plant Physiol. 2008, 146, 492–504. [Google Scholar] [CrossRef]
- Song, H.X.; Ping, A.M.; Sun, M.X.; Qi, X.H.; Gao, M.Y.; Xu, X.Y.; Zhu, Z.J.; Li, M.L.; Hou, L.P. Identification of genes related to floral organ development in pak choi by expression profiling. Genet. Mol. Res. 2017, 16, 1–14. [Google Scholar] [CrossRef]
- Chen, Z.; Gao, X.; Zhang, J. Alteration of osa-miR156e expression affects rice plant architecture and strigolactones (SLs) pathway. Plant Cell Rep. 2015, 34, 767–781. [Google Scholar] [CrossRef]
- Shen, Y.; Sun, S.; Hua, S.; Shen, E.; Ye, C.-Y.; Cai, D.; Timko, M.P.; Zhu, Q.-H.; Fan, L. Analysis of transcriptional and epigenetic changes in hybrid vigor of allopolyploid Brassica napus uncovers key roles for small RNAs. Plant J. 2017, 91, 874–893. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Z.; Huo, Q.; Yang, H.; Jian, H.; Qu, C.; Lu, K.; Li, J. Joint RNA-Seq and miRNA Profiling Analyses to Reveal Molecular Mechanisms in Regulating Thickness of Pod Canopy in Brassica napus. Genes 2019, 10, 591. https://doi.org/10.3390/genes10080591
Chen Z, Huo Q, Yang H, Jian H, Qu C, Lu K, Li J. Joint RNA-Seq and miRNA Profiling Analyses to Reveal Molecular Mechanisms in Regulating Thickness of Pod Canopy in Brassica napus. Genes. 2019; 10(8):591. https://doi.org/10.3390/genes10080591
Chicago/Turabian StyleChen, Zhiyou, Qiang Huo, Hong Yang, Hongju Jian, Cunmin Qu, Kun Lu, and Jiana Li. 2019. "Joint RNA-Seq and miRNA Profiling Analyses to Reveal Molecular Mechanisms in Regulating Thickness of Pod Canopy in Brassica napus" Genes 10, no. 8: 591. https://doi.org/10.3390/genes10080591