Maternal Haplotypes in DHFR Promoter and MTHFR Gene in Tuning Childhood Acute Lymphoblastic Leukemia Onset-Latency: Genetic/Epigenetic Mother/Child Dyad Study (GEMCDS)

 , , ,
, , , 

Abstract
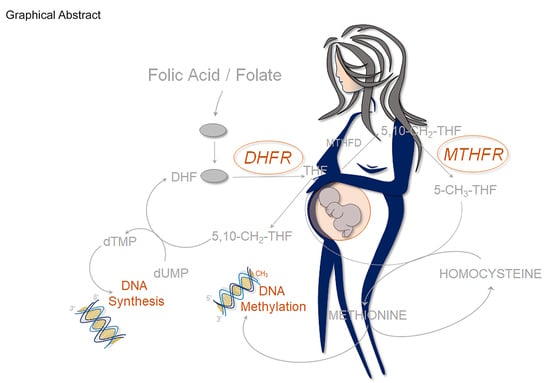
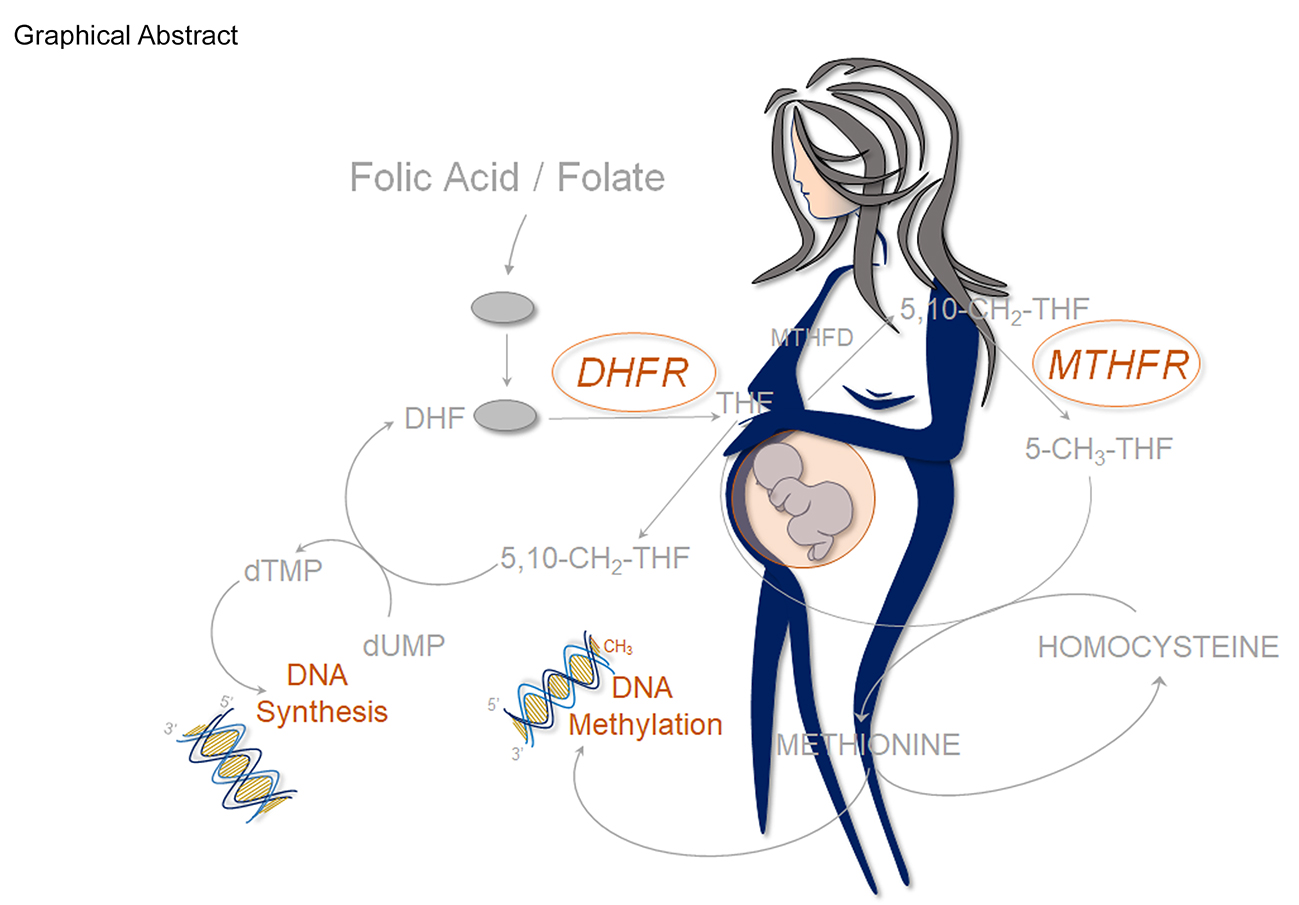
1. Introduction
2. Material and Methods
2.1. Study Population
2.2. Genotype Analysis
2.3. Statistical Analysis
3. Results
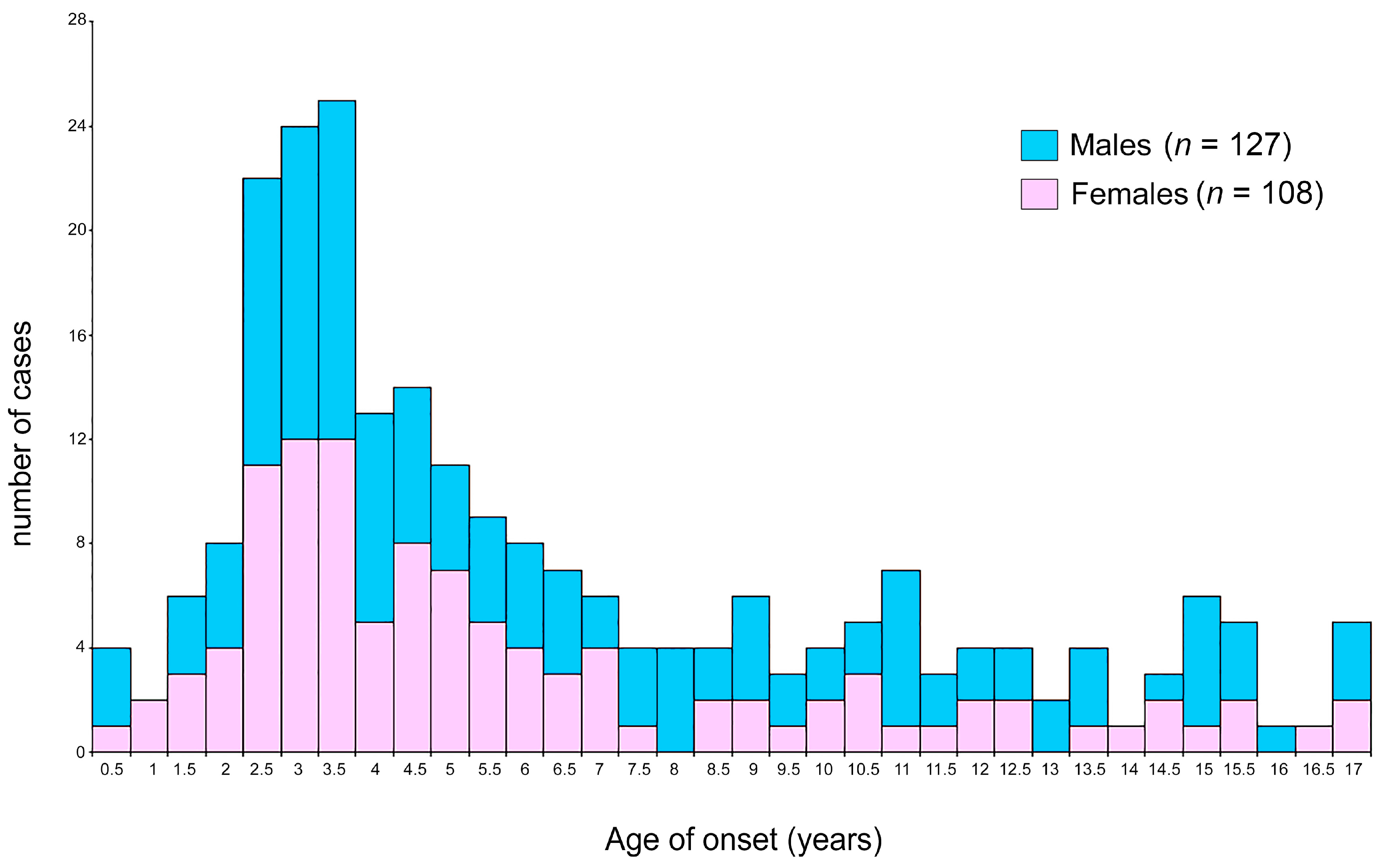
3.1. Clinical and Epidemiological Features of ALL Patients
3.2. DHFR and MTHFR Genes and ALL Age of Onset
3.2.1. DHFR and MTHFR Genotype Distribution in Children and Mothers (Single Analysis)
3.2.2. DHFR and MTHFR Genotype Distribution in Children (Combined Analysis)
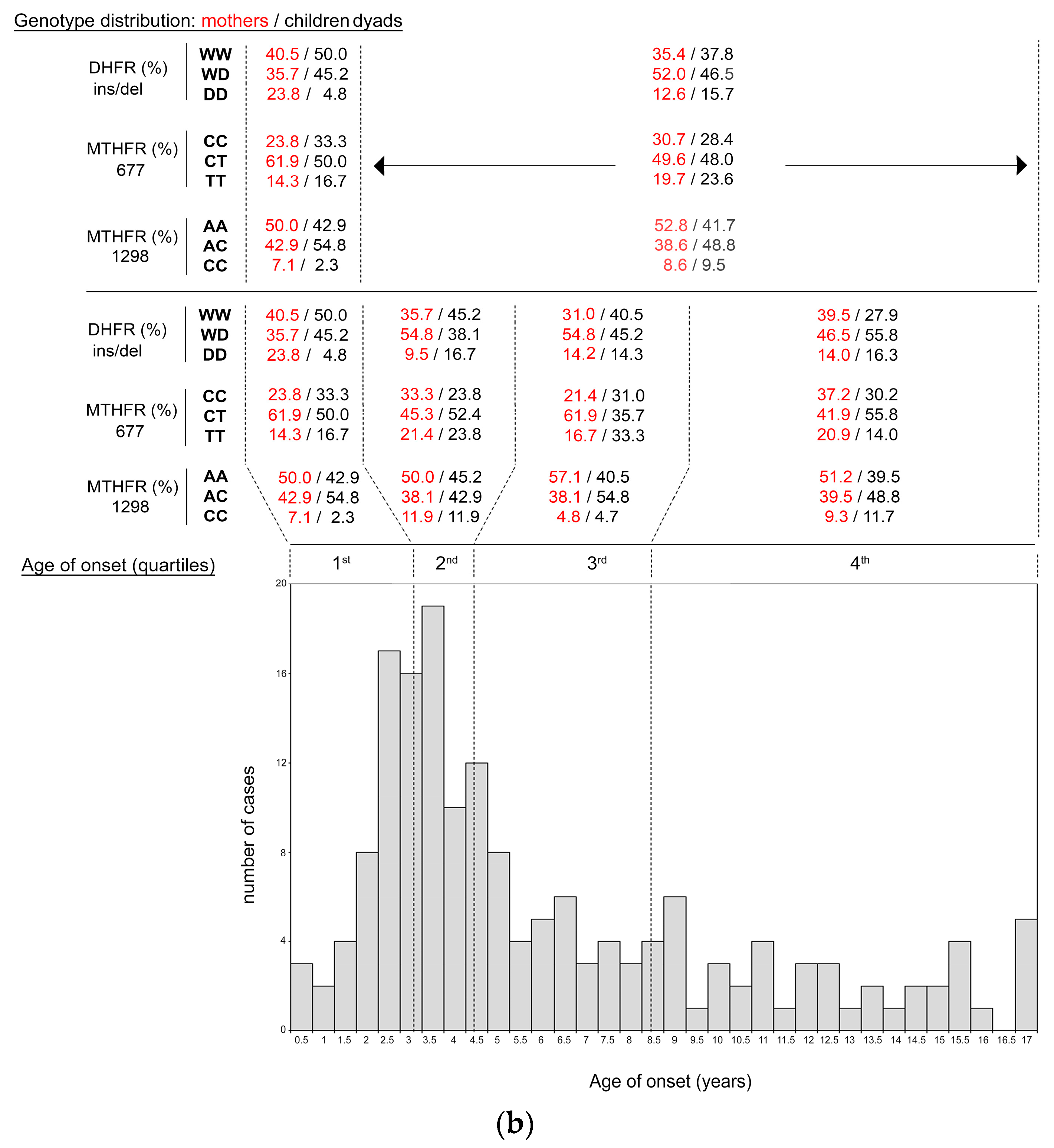
3.2.3. DHFR and MTHFR Genotype Distribution in Mother-child Dyads (Combined Analysis)
3.2.4. DHFR and MTHFR Genotype Distribution in Mother (Combined Analysis)
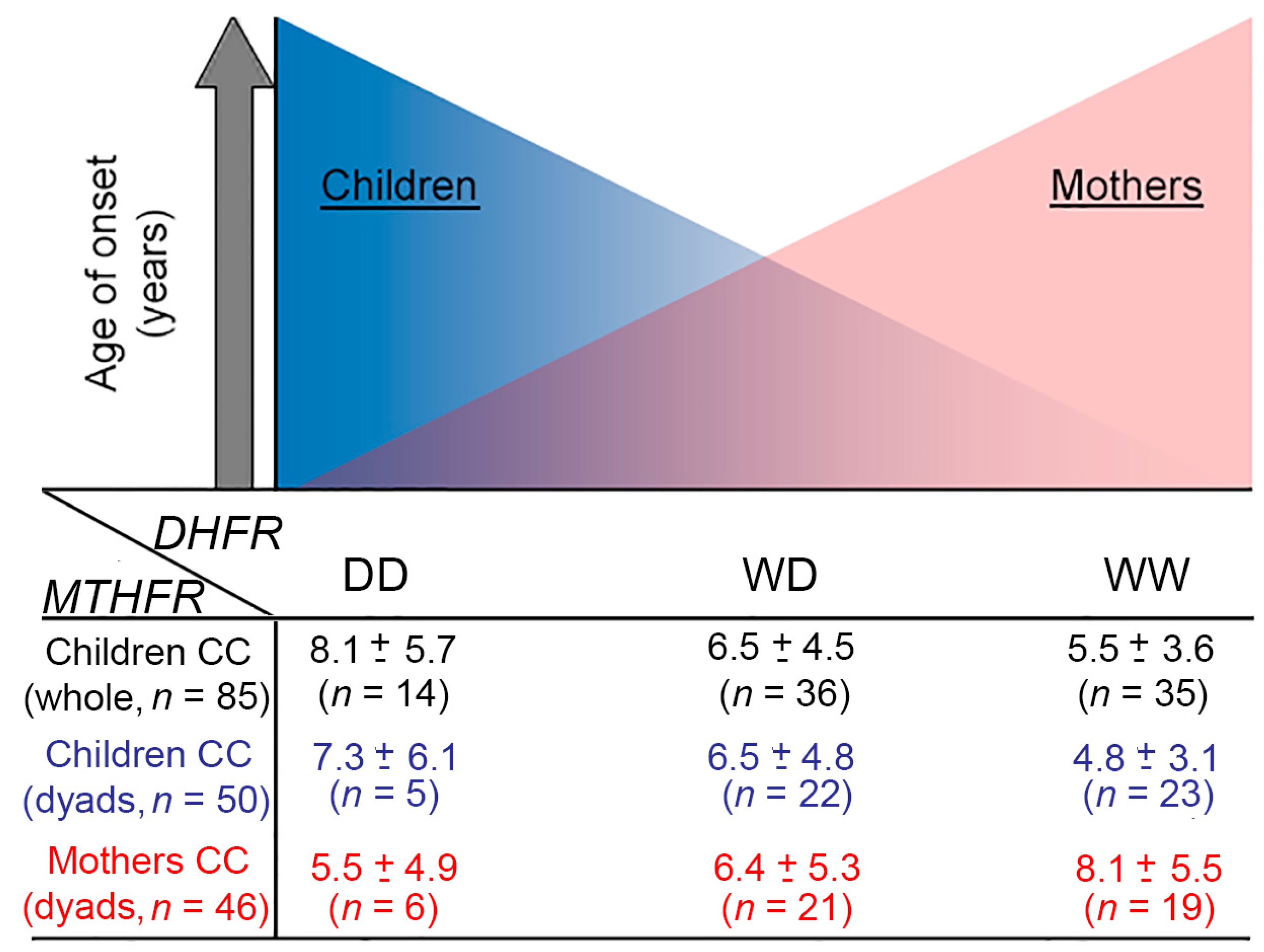
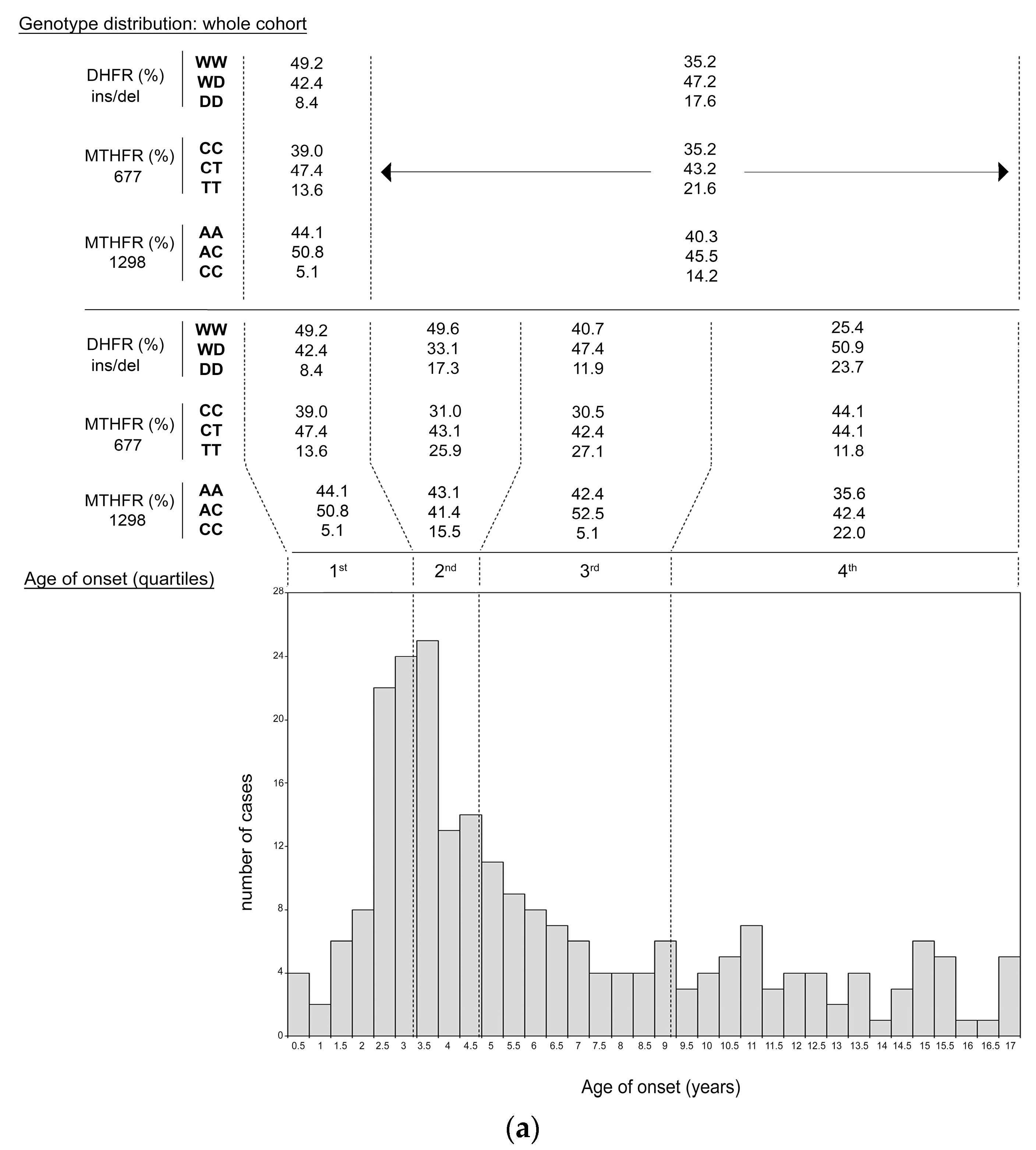
3.3. ALL Onset Age Distribution and DHFR/MTHFR Genotype Stratification
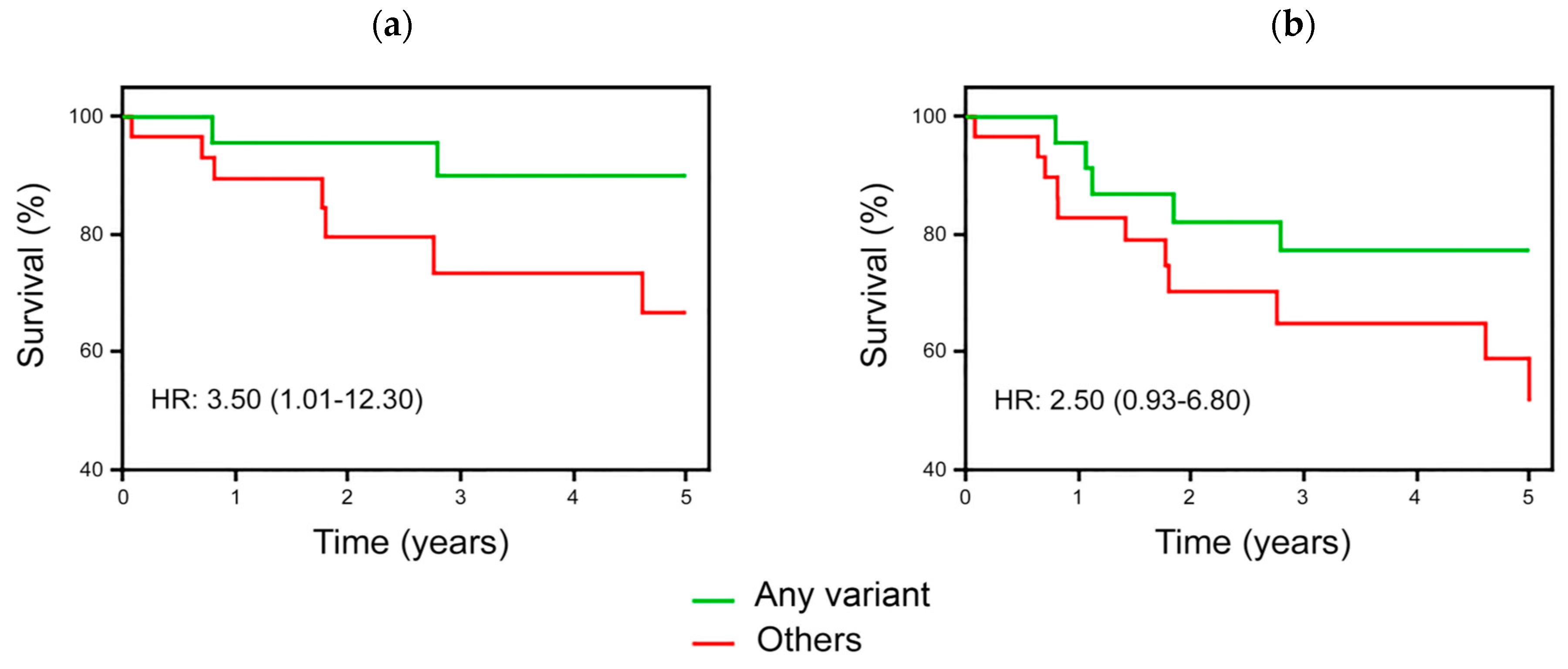
3.4. Survival Analyses in ALL Children According to DHFR/MTHFR Genotypes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Inaba, H.; Greaves, M.; Mullighan, C.G. Acute lymphoblastic leukaemia. Lancet 2013, 381, 1943–1955. [Google Scholar] [CrossRef]
- Hunger, S.P.; Loh, M.L.; Whitlock, J.A.; Winick, N.J.; Carroll, W.L.; Devidas, M.; Raetz, E.A.; Committee, C.O.G.A.L.L. Children’s Oncology Group’s 2013 blueprint for research: Acute lymphoblastic leukemia. Pediatr. Blood Cancer 2013, 60, 957–963. [Google Scholar] [CrossRef] [PubMed]
- WHO/Europe. Incidence of Childhood Leukaemia. Available online: http://www.euro.who.int/en/home (accessed on 3 May 2019).
- Hadley, L.G.; Rouma, B.S.; Saad-Eldin, Y. Challenge of pediatric oncology in Africa. Semin. Pediatr. Surg. 2012, 21, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Sandovici, I.; Hoelle, K.; Angiolini, E.; Constancia, M. Placental adaptations to the maternal-fetal environment: Implications for fetal growth and developmental programming. Reprod. Biomed. Online 2012, 25, 68–89. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M. In utero origins of childhood leukaemia. Early Hum. Dev. 2005, 81, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, T.; Metzger, M.L.; Wu, G.; Nishii, R.; Qian, M.; Devidas, M.; Yang, W.; Cheng, C.; Cao, X.; Quinn, E.; et al. Germline genetic variation in ETV6 and risk of childhood acute lymphoblastic leukaemia: A systematic genetic study. Lancet Oncol. 2015, 16, 1659–1666. [Google Scholar] [CrossRef]
- Knez, V.M.; Carstens, B.J.; Swisshelm, K.L.; McGranahan, A.N.; Liang, X. Heterogeneity of Abnormal RUNX1 Leading to Clinicopathologic Variations in Childhood B-Lymphoblastic Leukemia. Am. J. Clin. Pathol. 2015, 144, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Metayer, C.; Scelo, G.; Chokkalingam, A.P.; Barcellos, L.F.; Aldrich, M.C.; Chang, J.S.; Guha, N.; Urayama, K.Y.; Hansen, H.M.; Block, G.; et al. Genetic variants in the folate pathway and risk of childhood acute lymphoblastic leukemia. Cancer Causes Control 2011, 22, 1243–1258. [Google Scholar] [CrossRef]
- Thompson, J.R.; Gerald, P.F.; Willoughby, M.L.; Armstrong, B.K. Maternal folate supplementation in pregnancy and protection against acute lymphoblastic leukaemia in childhood: A case-control study. Lancet 2001, 358, 1935–1940. [Google Scholar] [CrossRef]
- Wiemels, J.L.; Smith, R.N.; Taylor, G.M.; Eden, O.B.; Alexander, F.E.; Greaves, M.F. United Kingdom Childhood Cancer Study Investigators. Methylenetetrahydrofolate reductase (MTHFR) polymorphisms and risk of molecularly defined subtypes of childhood acute leukemia. Proc. Natl. Acad. Sci. USA 2001, 98, 4004–4009. [Google Scholar] [CrossRef]
- Conter, V.; Bartram, C.R.; Valsecchi, M.G.; Schrauder, A.; Panzer-Grumayer, R.; Moricke, A.; Arico, M.; Zimmermann, M.; Mann, G.; De Rossi, G.; et al. Molecular response to treatment redefines all prognostic factors in children and adolescents with B-cell precursor acute lymphoblastic leukemia: Results in 3184 patients of the AIEOP-BFM ALL 2000 study. Blood 2010, 115, 3206–3214. [Google Scholar] [CrossRef]
- Larsen, E.C.; Devidas, M.; Chen, S.; Salzer, W.L.; Raetz, E.A.; Loh, M.L.; Mattano, L.A., Jr.; Cole, C.; Eicher, A.; Haugan, M.; et al. Dexamethasone and High-Dose Methotrexate Improve Outcome for Children and Young Adults With High-Risk B-Acute Lymphoblastic Leukemia: A Report From Children’s Oncology Group Study AALL0232. J. Clin. Oncol. 2016, 34, 2380–2388. [Google Scholar] [CrossRef]
- Marcoux, S.; Drouin, S.; Laverdiere, C.; Alos, N.; Andelfinger, G.U.; Bertout, L.; Curnier, D.; Friedrich, M.G.; Kritikou, E.A.; Lefebvre, G.; et al. The PETALE study: Late adverse effects and biomarkers in childhood acute lymphoblastic leukemia survivors. Pediatr. Blood Cancer 2017, 64. [Google Scholar] [CrossRef] [PubMed]
- Ongaro, A.; De Mattei, M.; Della Porta, M.G.; Rigolin, G.; Ambrosio, C.; Di Raimondo, F.; Pellati, A.; Masieri, F.F.; Caruso, A.; Catozzi, L.; et al. Gene polymorphisms in folate metabolizing enzymes in adult acute lymphoblastic leukemia: Effects on methotrexate-related toxicity and survival. Haematologica 2009, 94, 1391–1398. [Google Scholar] [CrossRef] [PubMed]
- Gemmati, D.; Ongaro, A.; Tognazzo, S.; Catozzi, L.; Federici, F.; Mauro, E.; Della Porta, M.; Campioni, D.; Bardi, A.; Gilli, G.; et al. Methylenetetrahydrofolate reductase C677T and A1298C gene variants in adult non-Hodgkin’s lymphoma patients: Association with toxicity and survival. Haematologica 2007, 92, 478–485. [Google Scholar] [CrossRef]
- Gemmati, D.; Ongaro, A.; Scapoli, G.L.; Della Porta, M.; Tognazzo, S.; Serino, M.L.; Di Bona, E.; Rodeghiero, F.; Gilli, G.; Reverberi, R.; et al. Common gene polymorphisms in the metabolic folate and methylation pathway and the risk of acute lymphoblastic leukemia and non-Hodgkin’s lymphoma in adults. Cancer Epidemiol. Biomark. Prev. 2004, 13, 787–794. [Google Scholar]
- Gemmati, D.; De Mattei, M.; Catozzi, L.; Della Porta, M.; Serino, M.L.; Ambrosio, C.; Cuneo, A.; Friso, S.; Krampera, M.; Orioli, E.; et al. DHFR 19-bp insertion/deletion polymorphism and MTHFR C677T in adult acute lymphoblastic leukaemia: Is the risk reduction due to intracellular folate unbalancing? Am. J. Hematol. 2009, 84, 526–529. [Google Scholar] [CrossRef]
- Gemmati, D. Folate-Pathway Gene Variants in Cancer: Haematological Malignancies; Gemmati, D., Ed.; India Publisher: Kerala, India, 2008. [Google Scholar]
- Johnson, W.G.; Stenroos, E.S.; Spychala, J.R.; Chatkupt, S.; Ming, S.X.; Buyske, S. New 19 bp deletion polymorphism in intron-1 of dihydrofolate reductase (DHFR): A risk factor for spina bifida acting in mothers during pregnancy? Am. J. Med. Genet. Part A 2004, 124, 339–345. [Google Scholar] [CrossRef]
- Orjuela, M.A.; Cabrera-Munoz, L.; Paul, L.; Ramirez-Ortiz, M.A.; Liu, X.; Chen, J.; Mejia-Rodriguez, F.; Medina-Sanson, A.; Diaz-Carreno, S.; Suen, I.H.; et al. Risk of retinoblastoma is associated with a maternal polymorphism in dihydrofolatereductase (DHFR) and prenatal folic acid intake. Cancer 2012, 118, 5912–5919. [Google Scholar] [CrossRef]
- Fisk Green, R.; Byrne, J.; Crider, K.S.; Gallagher, M.; Koontz, D.; Berry, R.J. Folate-related gene variants in Irish families affected by neural tube defects. Front. Genet. 2013, 4, 223. [Google Scholar] [CrossRef]
- Prasoona, K.R.; Sunitha, T.; Srinadh, B.; Muni Kumari, T.; Jyothy, A. Maternal association and influence of DHFR 19 bp deletion variant predisposes foetus to anencephaly susceptibility: A family-based triad study. Biomarkers 2018, 23, 640–646. [Google Scholar] [CrossRef]
- Sullivan, M.; Murray, T.; Assefa, H. Women with methylenetetrahydrofolate reductase gene polymorphism and the need for proper periconceptional folate supplementation. J. Pharm. Pharmacol. (DAVID PUBLISHING) 2015, 3, 204–222. [Google Scholar] [CrossRef][Green Version]
- Umerez, M.; Gutierrez-Camino, A.; Munoz-Maldonado, C.; Martin-Guerrero, I.; Garcia-Orad, A. MTHFR polymorphisms in childhood acute lymphoblastic leukemia: Influence on methotrexate therapy. Pharmgenom. Pers. Med. 2017, 10, 69–78. [Google Scholar] [CrossRef]
- Ceppi, F.; Gagne, V.; Douyon, L.; Quintin, C.J.; Colombini, A.; Parasole, R.; Buldini, B.; Basso, G.; Conter, V.; Cazzaniga, G.; et al. DNA variants in DHFR gene and response to treatment in children with childhood B ALL: revisited in AIEOP-BFM protocol. Pharmacogenomics 2018, 19, 105–112. [Google Scholar] [CrossRef]
- Al-Shakfa, F.; Dulucq, S.; Brukner, I.; Milacic, I.; Ansari, M.; Beaulieu, P.; Moghrabi, A.; Laverdiere, C.; Sallan, S.E.; Silverman, L.B.; et al. DNA variants in region for noncoding interfering transcript of dihydrofolate reductase gene and outcome in childhood acute lymphoblastic leukemia. Clin. Cancer Res. 2009, 15, 6931–6938. [Google Scholar] [CrossRef]
- Krajinovic, M.; Moghrabi, A. Pharmacogenetics of methotrexate. Pharmacogenomics 2004, 5, 819–834. [Google Scholar] [CrossRef]
- Moricke, A.; Zimmermann, M.; Valsecchi, M.G.; Stanulla, M.; Biondi, A.; Mann, G.; Locatelli, F.; Cazzaniga, G.; Niggli, F.; Arico, M.; et al. Dexamethasone vs. prednisone in induction treatment of pediatric ALL: Results of the randomized trial AIEOP-BFM ALL 2000. Blood 2016, 127, 2101–2112. [Google Scholar] [CrossRef]
- Tognazzo, S.; Gemmati, D.; Palazzo, A.; Catozzi, L.; Carandina, S.; Legnaro, A.; Tacconi, G.; Scapoli, G.L.; Zamboni, P. Prognostic role of factor XIII gene variants in nonhealing venous leg ulcers. J. Vasc. Surg. 2006, 44, 815–819. [Google Scholar] [CrossRef]
- Gemmati, D.; Tognazzo, S.; Serino, M.L.; Fogato, L.; Carandina, S.; De Palma, M.; Izzo, M.; De Mattei, M.; Ongaro, A.; Scapoli, G.L.; et al. Factor XIII V34L polymorphism modulates the risk of chronic venous leg ulcer progression and extension. Wound Repair Regen. 2004, 12, 512–517. [Google Scholar] [CrossRef]
- Gemmati, D.; Occhionorelli, S.; Tisato, V.; Vigliano, M.; Longo, G.; Gonelli, A.; Sibilla, M.G.; Serino, M.L.; Zamboni, P. Inherited genetic predispositions in F13A1 and F13B genes predict abdominal adhesion formation: Identification of gender prognostic indicators. Sci. Rep. 2018, 8, 16916. [Google Scholar] [CrossRef]
- Ansani, L.; Marchesini, J.; Pestelli, G.; Luisi, G.A.; Scillitani, G.; Longo, G.; Milani, D.; Serino, M.L.; Tisato, V.; Gemmati, D. F13A1 Gene Variant (V34L) and Residual Circulating FXIIIA Levels Predict Short- and Long-Term Mortality in Acute Myocardial Infarction after Coronary Angioplasty. Int. J. Mol. Sci. 2018, 19, 2766. [Google Scholar] [CrossRef] [PubMed]
- Tisato, V.; Zuliani, G.; Vigliano, M.; Longo, G.; Franchini, E.; Secchiero, P.; Zauli, G.; Paraboschi, E.M.; Vikram Singh, A.; Serino, M.L.; et al. Gene-gene interactions among coding genes of iron-homeostasis proteins and APOE-alleles in cognitive impairment diseases. PLoS ONE 2018, 13, e0193867. [Google Scholar] [CrossRef] [PubMed]
- Gemmati, D.; Burini, F.; Talarico, A.; Fabbri, M.; Bertocco, C.; Vigliano, M.; Moratelli, S.; Cuneo, A.; Serino, M.L.; Avato, F.M.; et al. The Active Metabolite of Warfarin (3′-Hydroxywarfarin) and Correlation with INR, Warfarin and Drug Weekly Dosage in Patients under Oral Anticoagulant Therapy: A Pharmacogenetics Study. PLoS ONE 2016, 11, e0162084. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.; Krajinovic, M. Pharmacogenomics of acute leukemia. Pharmacogenomics 2007, 8, 817–834. [Google Scholar] [CrossRef] [PubMed]
- Dulucq, S.; Laverdiere, C.; Sinnett, D.; Krajinovic, M. Pharmacogenetic considerations for acute lymphoblastic leukemia therapies. Expert Opin. Drug Metab. Toxicol. 2014, 10, 699–719. [Google Scholar] [CrossRef] [PubMed]
- Krajinovic, M.; Labuda, D.; Sinnett, D. Childhood acute lymphoblastic leukemia: Genetic determinants of susceptibility and disease outcome. Rev. Environ. Health 2001, 16, 263–279. [Google Scholar] [CrossRef]
- Greenop, K.R.; Scott, R.J.; Attia, J.; Bower, C.; de Klerk, N.H.; Norris, M.D.; Haber, M.; Jamieson, S.E.; van Bockxmeer, F.M.; Gottardo, N.G.; et al. Folate pathway gene polymorphisms and risk of childhood brain tumors: Results from an Australian case-control study. Cancer Epidemiol. Biomark. Prev. 2015, 24, 931–937. [Google Scholar] [CrossRef]
- Lupo, P.J.; Nousome, D.; Kamdar, K.Y.; Okcu, M.F.; Scheurer, M.E. A case-parent triad assessment of folate metabolic genes and the risk of childhood acute lymphoblastic leukemia. Cancer Causes Control 2012, 23, 1797–1803. [Google Scholar] [CrossRef][Green Version]
- Lupo, P.J.; Dietz, D.J.; Kamdar, K.Y.; Scheurer, M.E. Gene-environment interactions and the risk of childhood acute lymphoblastic leukemia: Exploring the role of maternal folate genes and folic Acid fortification. Pediatr. Hematol. Oncol. 2014, 31, 160–168. [Google Scholar] [CrossRef]
- Milne, E.; Greenop, K.R.; Scott, R.J.; Haber, M.; Norris, M.D.; Attia, J.; Jamieson, S.E.; Miller, M.; Bower, C.; Bailey, H.D.; et al. Folate pathway gene polymorphisms, maternal folic acid use, and risk of childhood acute lymphoblastic leukemia. Cancer Epidemiol. Biomark. Prev. 2015, 24, 48–56. [Google Scholar] [CrossRef]
- Lubinsky, M. An epigenetic association of malformations, adverse reproductive outcomes, and fetal origins hypothesis related effects. J. Assist. Reprod. Genet. 2018, 35, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Gammon, M.D.; Wetmur, J.G.; Rao, M.; Gaudet, M.M.; Teitelbaum, S.L.; Britton, J.A.; Neugut, A.I.; Santella, R.M.; Chen, J. A functional 19-base pair deletion polymorphism of dihydrofolate reductase (DHFR) and risk of breast cancer in multivitamin users. Am. J. Clin. Nutr. 2007, 85, 1098–1102. [Google Scholar] [CrossRef] [PubMed]
- Selhub, J.; Rosenberg, I.H. Excessive folic acid intake and relation to adverse health outcome. Biochimie 2016, 126, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Plumptre, L.; Masih, S.P.; Ly, A.; Aufreiter, S.; Sohn, K.J.; Croxford, R.; Lausman, A.Y.; Berger, H.; O’Connor, D.L.; Kim, Y.I. High concentrations of folate and unmetabolized folic acid in a cohort of pregnant Canadian women and umbilical cord blood. Am. J. Clin. Nutr. 2015, 102, 848–857. [Google Scholar] [CrossRef] [PubMed]
- Montalvao-de-Azevedo, R.; Vasconcelos, G.M.; Vargas, F.R.; Thuler, L.C.; Pombo-de-Oliveira, M.S.; de Camargo, B.; Brazilian Embryonic Tumor, G. RFC-1 80G>A polymorphism in case-mother/control-mother dyads is associated with risk of nephroblastoma and neuroblastoma. Genet. Test. Mol. Biomark. 2015, 19, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Luebeck, E.G.; Moolgavkar, S.H.; Liu, A.Y.; Boynton, A.; Ulrich, C.M. Does folic acid supplementation prevent or promote colorectal cancer? Results from model-based predictions. Cancer Epidemiol. Biomark. Prev. 2008, 17, 1360–1367. [Google Scholar] [CrossRef] [PubMed]
- Emerenciano, M.; Barbosa, T.C.; Lopes, B.A.; Blunck, C.B.; Faro, A.; Andrade, C.; Meyer, C.; Marschalek, R.; Pombo-de-Oliveira, M.S. Brazilian Collaborative Study Group of Infant Acute Leukemia. ARID5B polymorphism confers an increased risk to acquire specific MLL rearrangements in early childhood leukemia. BMC Cancer 2014, 14, 127. [Google Scholar] [CrossRef]
- Buffler, P.A.; Kwan, M.L.; Reynolds, P.; Urayama, K.Y. Environmental and genetic risk factors for childhood leukemia: Appraising the evidence. Cancer Invest. 2005, 23, 60–75. [Google Scholar] [CrossRef]
- Steluti, J.; Selhub, J.; Paul, L.; Reginaldo, C.; Fisberg, R.M.; Marchioni, D.M.L. An overview of folate status in a population-based study from Sao Paulo, Brazil and the potential impact of 10 years of national folic acid fortification policy. Eur. J. Clin. Nutr. 2017, 71, 1173–1178. [Google Scholar] [CrossRef]
- Jhaveri, M.S.; Wagner, C.; Trepel, J.B. Impact of extracellular folate levels on global gene expression. Mol. Pharmacol. 2001, 60, 1288–1295. [Google Scholar] [CrossRef]
- Kim, K.C.; Friso, S.; Choi, S.W. DNA methylation, an epigenetic mechanism connecting folate to healthy embryonic development and aging. J. Nutr. Biochem. 2009, 20, 917–926. [Google Scholar] [CrossRef]
- Wasson, G.R.; McGlynn, A.P.; McNulty, H.; O’Reilly, S.L.; McKelvey-Martin, V.J.; McKerr, G.; Strain, J.J.; Scott, J.; Downes, C.S. Global DNA and p53 region-specific hypomethylation in human colonic cells is induced by folate depletion and reversed by folate supplementation. J. Nutr. 2006, 136, 2748–2753. [Google Scholar] [CrossRef]
- Kim, Y.I. Folate and carcinogenesis: Evidence, mechanisms, and implications. J. Nutr. Biochem. 1999, 10, 66–88. [Google Scholar] [CrossRef]
- Kim, Y.I. Folate and cancer prevention: A new medical application of folate beyond hyperhomocysteinemia and neural tube defects. Nutr. Rev. 1999, 57, 314–321. [Google Scholar] [CrossRef]
- Kim, Y.I. Role of folate in colon cancer development and progression. J. Nutr. 2003, 133, 3731S–3739S. [Google Scholar] [CrossRef]
- Gemmati, D.; Previati, M.; Serino, M.L.; Moratelli, S.; Guerra, S.; Capitani, S.; Forini, E.; Ballerini, G.; Scapoli, G.L. Low folate levels and thermolabile methylenetetrahydrofolate reductase as primary determinant of mild hyperhomocystinemia in normal and thromboembolic subjects. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1761–1767. [Google Scholar] [CrossRef]
- Orjuela, M.A.; Titievsky, L.; Liu, X.; Ramirez-Ortiz, M.; Ponce-Castaneda, V.; Lecona, E.; Molina, E.; Beaverson, K.; Abramson, D.H.; Mueller, N.E. Fruit and vegetable intake during pregnancy and risk for development of sporadic retinoblastoma. Cancer Epidemiol. Biomark. Prev. 2005, 14, 1433–1440. [Google Scholar] [CrossRef]
- Obeid, R.; Kirsch, S.H.; Dilmann, S.; Klein, C.; Eckert, R.; Geisel, J.; Herrmann, W. Folic acid causes higher prevalence of detectable unmetabolized folic acid in serum than B-complex: A randomized trial. Eur. J. Nutr. 2016, 55, 1021–1028. [Google Scholar] [CrossRef]
- Institute of Medicine (US). A Report of the Standing Committee on the Scientific Evaluation of Dietary Reference Intakes and its Panel on Folate, Other B Vitamins, and Choline. In Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline; National Academies Press (US): Washington, DC, USA, 1998. [Google Scholar] [CrossRef]
- Stover, P.J. Polymorphisms in 1-carbon metabolism, epigenetics and folate-related pathologies. J. Nutrigenet. Nutrigenom. 2011, 4, 293–305. [Google Scholar] [CrossRef]
- Pinto, R.Q.; Soares, I.; Carvalho-Correia, E.; Mesquita, A.R. Gene-environment interactions in psychopathology throughout early childhood: A systematic review. Psychiatr. Genet. 2015, 25, 223–233. [Google Scholar] [CrossRef]
- Oberg, E.; Givant, C.; Fisk, B.; Parikh, C.; Bradley, R. Epigenetics in Clinical Practice: Characterizing Patient and Provider Experiences with MTHFR Polymorphisms and Methylfolate. J. Nutrigenet. Nutrigenom. 2015, 8, 137–150. [Google Scholar] [CrossRef] [PubMed]
- James, S.J.; Melnyk, S.; Jernigan, S.; Pavliv, O.; Trusty, T.; Lehman, S.; Seidel, L.; Gaylor, D.W.; Cleves, M.A. A functional polymorphism in the reduced folate carrier gene and DNA hypomethylation in mothers of children with autism. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2010, 153B, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Eissa, D.S.; Ahmed, T.M. C677T and A1298C polymorphisms of the methylenetetrahydrofolate reductase gene: Effect on methotrexate-related toxicity in adult acute lymphoblastic leukaemia. Blood Coagul. Fibrinolysis 2013, 24, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Dulucq, S.; St-Onge, G.; Gagne, V.; Ansari, M.; Sinnett, D.; Labuda, D.; Moghrabi, A.; Krajinovic, M. DNA variants in the dihydrofolate reductase gene and outcome in childhood ALL. Blood 2008, 111, 3692–3700. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Demographic and Clinical Features | Whole Cohort n (%) | Male n (%) | Female n (%) | P |
|---|---|---|---|---|
| n (%) | 235 (100) | 127 (54) | 108 (46) | ns |
| Age at onset (years) | ||||
| Mean ± SD | 6.13 ± 4.30 | 6.53 ± 4.45 | 5.66 ± 4.03 | 0.06 |
| Range | 0.003–16.9 | 0.003–16.9 | 0.068–16.81 | |
| Peak | 2.9 | 3.0–3.5 | 2.5–3.5 | |
| <10 years | 184 (78.3) | 95 (51.6) | 89 (48.4) | ns |
| ≥10 years | 51 (21.7) | 32 (62.7) | 19 (37.3) | |
| ALL phenotype | ||||
| B-phenotype | 204 (86.8) | 106 (52) | 98 (48) | 0.03 |
| T-phenotype | 27 (11.5) | 20 (74) | 7 (26) | |
| Other* | 4 (1.7) | 1 (25) | 3 (75) | - |
| DNA index (n = 192) | ||||
| ≥1.16 | 41 (21.3) | 22 (53.6) | 19 (46.4) | ns |
| <1.16 | 151 (78.6) | 84 (55.6) | 67 (44.4) | |
| not available | 43 | 21 | 22 | - |
| Translocations | ||||
| Bcr-Abl1/t(9;22) (y/n; n = 209) | 3/206 (1.43) | 3/112 (2.6) | 0/94 (0) | ns |
| t(4;11) (y/n; n = 207) | 3/204 (1.45) | 0/115 (0) | 3/89 (3.26) | 0.08 |
| Any translocation: t(9;22); t(4;11); t(12;21); t(1;19); t(9:11) (y/n; n = 209) | 49/160 (23.4) | 28/86 (24.6) | 21/74 (22.1) | ns |
| Response to prednisone | ||||
| Prednisone good responders | 217 (92.3) | 114 (52.5) | 103 (47.5) | 0.06 |
| Prednisone poor responders | 16 (6.8) | 12 (75) | 4 (25) | |
| Unknown | 2 (0.85) | 1 (50) | 1 (50) | - |
| Final risk stratification (n = 235) | ||||
| High-risk (HR) | 53 (22.5) | 31 (58.6) | 22 (41.4) | ns |
| Intermediate-risk (IR) | 125 (53.2) | 71 (56.8) | 54 (43.2) | |
| Standard risk (SR) | 57 (24.3) | 32 (56.1) | 25 (43.9) | |
| Relapses (n = 228) | ||||
| Yes | 20 (8.8) | 12 (60) | 8 (40) | ns |
| No | 208 (91.2) | 111 (53.4) | 97 (46.6) | |
| Death in continuous complete remission | 9 (3.8) | 6 (66.7) | 3 (33.3) | - |
| After chemotherapy | 3 (1.3) | 3 (100) | 0 (0) | ns |
| After stem cell transplantation | 6 (2.5) | 3 (50) | 3 (50) | |
| Continuous complete remission | 226 (96.2) | 121 (53.5) | 105 (46.5) | |
| Genotype Groups | Onset Age in Children with That Genotype (Whole-Cohort) Mean ± SD; Median (n) [Range] | P | Onset Age in Children with That Genotype (Dyad-group) Mean ± SD; Median (n) [Range] | P | Onset Age in Children with Mother Carrying That Genotype Mean ± SD; Median (n) [Range] | P | |
|---|---|---|---|---|---|---|---|
| DHFR ins/del | WW | 5.2 ± 3.7; 4.07 (91) [0.003–16.6] | 0.002 a 0.0034 b 0.03 c | 4.8 ± 3.6; 3.8 (69) [0.003–16.6] | 0.007 a 0.0037 b 0.033 c | 5.8 ± 4.5; 3.92 (62) [0.7–16.6] | ns a ns b ns c |
| WD | 6.7 ± 4.4; 5.04 (108) [0.07–16.8] | 6.4 ± 4.5; 4.8 (78) [0.07–16.8] | 5.9 ± 4.1; 4.38 (81) [0.003–16.9] | ||||
| DD | 7.5 ± 4.8; 5.77 (36) [0.5–16.9] | 7.2 ± 4.7; 5.4 (22) [2.05–16.9] | 5.8 ± 4.3; 3.72 (26) [0.8–15.1] | ||||
| MTHFR 677 | CC | 6.4 ± 4.4; 4.66 (85) [0.5–16.7] | ns a ns b ns | 5.82 ± 4.3; 4.1 (50) [0.67–16.7] | ns a ns b ns c | 7.0 ± 5.3; 4.27 (46) [0.2–16.9] | 0.057 a 0.017 b ns c |
| CT | 6.2 ± 4.6; 5.23 (104) [0.003–16.9] | 6.21 ± 4.8; 4.04 (82) [0.003–16.9] | 5.5 ± 3.9; 4.09 (89) [0.068–16.8] | ||||
| TT | 5.5 ± 3.2; 4.5 (46) [0.77–15] | 5.19 ± 2.8; 4.4 (37) [0.77–11.6] | 5.4 ± 3.3; 4.07 (34) [0.003–11.7] | ||||
| MTHFR 1298 | AA | 5.78 ± 4.15; 4.07 (97) [0.48–16.9] | 0.006 a 0.005 b 0.05 c | 5.79 ± 4.21; 4.07 (71) [0.77–16.92] | 0.05 a 0.04 b ns c | 5.9 ± 4.1; 4.3 (88) [0.003–16.3] | ns a ns b ns c |
| AC | 5.96 ± 4.22; 4.49 (110) [0.003–16.8] | 5.76 ± 4.25; 4.21 (85) [0.0027–16.82] | 5.85 ± 4.4; 4.1 (67) [0.07–16.8] | ||||
| CC | 8.03 ± 4.82; 7.60 (28) [1.85–16.21] | 7.05 ± 4.84; 4.09 (13) [1.85–15.13] | 6.0 ± 4.7; 3.5 (14) [1.3–15.3] | ||||
| Genotype/Groups (n = 235) | DHFR | ||||||
|---|---|---|---|---|---|---|---|
| WW | WD | DD | Pa | Pb | |||
| The onset age of children with that genotype | Mean ± SD median (n) | 5.2 ± 3.7 4.07 (91) | 6.7 ± 4.4 5.04 (108) | 7.5 ± 4.8 5.77 (36) | 0.002 | 0.0034 | |
| MTHFR 677 | CC | 6.4 ± 4.4 4.66 (85) | 5.5 ± 3.6 4.21 (35) | 6.5 ± 4.5 5.16 (36) | 8.1 ± 5.7 7.14 (14) | 0.034 | 0.069 |
| CT | 6.2 ± 4.6 4.23 (104) | 5.1 ± 4.3 3.78 (39) | 6.6 ± 4.6 5.10 (52) | 7.8 ± 4.8 5.86 (13) | 0.038 | 0.026 | |
| TT | 5.5 ± 3.2 4.5 (46) | 4.7 ± 2.1 4.4 (17) | 5.9 ± 3.9 4.34 (20) | 6.2 ± 3.4 5.68 (9) | 0.21 | 0.09 | |
| MTHFR 1298 | AA | 5.78 ± 4.15 4.07 (97) | 4.64 ± 3.46 3.74 (37) | 6.24 ± 4.18 4.60 (43) | 7.07 ± 5 5.27 (17) | 0.0217 | 0.016 |
| AC | 5.96 ± 4.22 4.49 (110) | 5.35 ± 3.71 4.25 (44) | 5.90 ± 4.4 4.51 (51) | 7.97 ± 4.67 6.19 (15) | 0.0156 | 0.10 | |
| CC | 8.03 ± 4.82 7.60 (28) | 6.43 ± 4.48 4.06 (10) | 9.28 ± 4.58 10.25 (14) | 7.66 ± 6.43 6.08 (4) | 0.34 | 0.09 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tisato, V.; Muggeo, P.; Lupiano, T.; Longo, G.; Serino, M.L.; Grassi, M.; Arcamone, E.; Secchiero, P.; Zauli, G.; Santoro, N.; et al. Maternal Haplotypes in DHFR Promoter and MTHFR Gene in Tuning Childhood Acute Lymphoblastic Leukemia Onset-Latency: Genetic/Epigenetic Mother/Child Dyad Study (GEMCDS). Genes 2019, 10, 634. https://doi.org/10.3390/genes10090634
Tisato V, Muggeo P, Lupiano T, Longo G, Serino ML, Grassi M, Arcamone E, Secchiero P, Zauli G, Santoro N, et al. Maternal Haplotypes in DHFR Promoter and MTHFR Gene in Tuning Childhood Acute Lymphoblastic Leukemia Onset-Latency: Genetic/Epigenetic Mother/Child Dyad Study (GEMCDS). Genes. 2019; 10(9):634. https://doi.org/10.3390/genes10090634
Chicago/Turabian StyleTisato, Veronica, Paola Muggeo, Tracy Lupiano, Giovanna Longo, Maria Luisa Serino, Massimo Grassi, Ermanno Arcamone, Paola Secchiero, Giorgio Zauli, Nicola Santoro, and et al. 2019. "Maternal Haplotypes in DHFR Promoter and MTHFR Gene in Tuning Childhood Acute Lymphoblastic Leukemia Onset-Latency: Genetic/Epigenetic Mother/Child Dyad Study (GEMCDS)" Genes 10, no. 9: 634. https://doi.org/10.3390/genes10090634
APA StyleTisato, V., Muggeo, P., Lupiano, T., Longo, G., Serino, M. L., Grassi, M., Arcamone, E., Secchiero, P., Zauli, G., Santoro, N., & Gemmati, D. (2019). Maternal Haplotypes in DHFR Promoter and MTHFR Gene in Tuning Childhood Acute Lymphoblastic Leukemia Onset-Latency: Genetic/Epigenetic Mother/Child Dyad Study (GEMCDS). Genes, 10(9), 634. https://doi.org/10.3390/genes10090634