The Conserved Non-Coding Sequence 2 (CNS2) Enhances CD69 Transcription through Cooperation between the Transcription Factors Oct1 and RUNX1

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
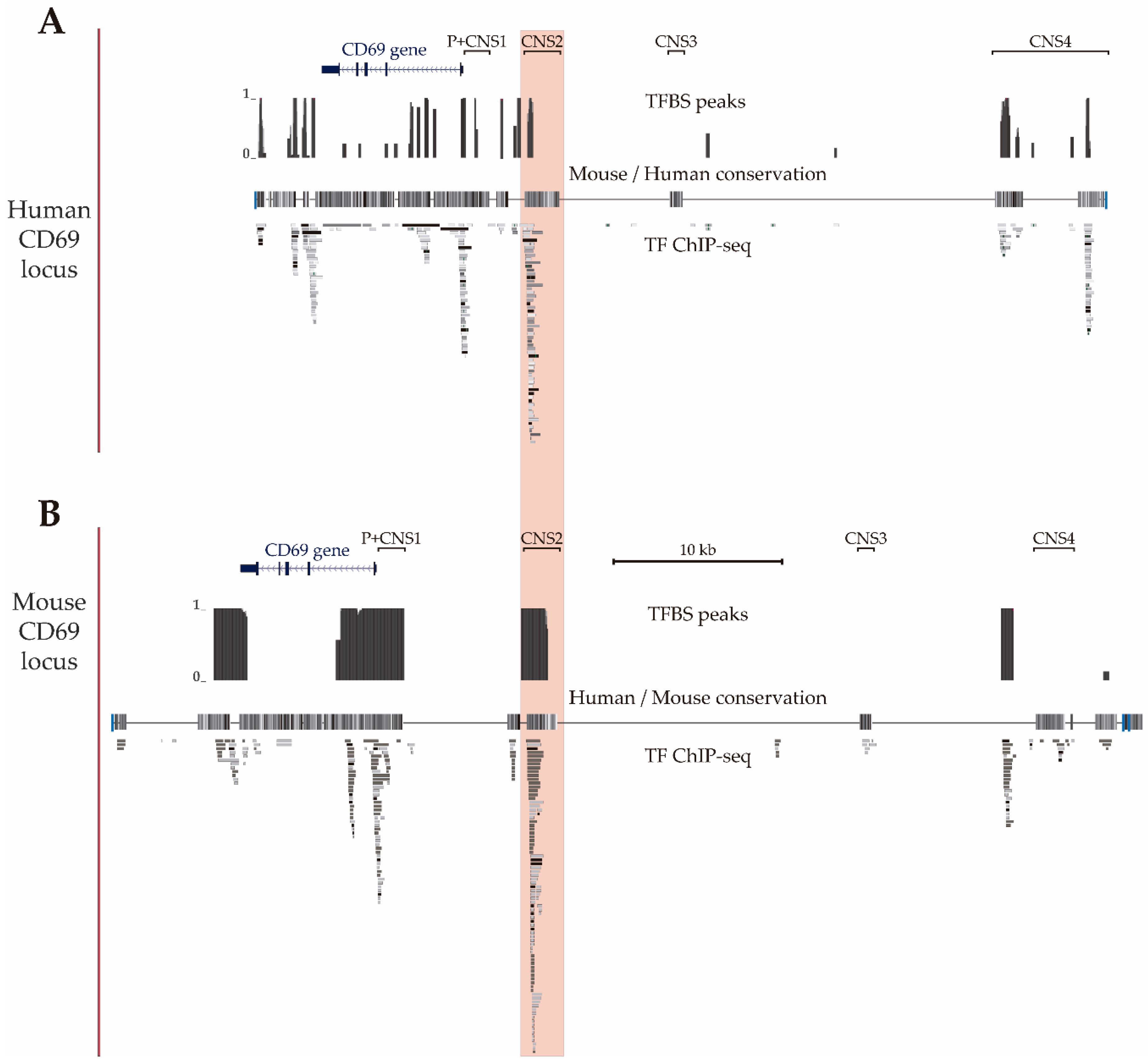
3.1. Mapping of Transcription Factor Binding Sites within Mouse CNS2 Enhancer
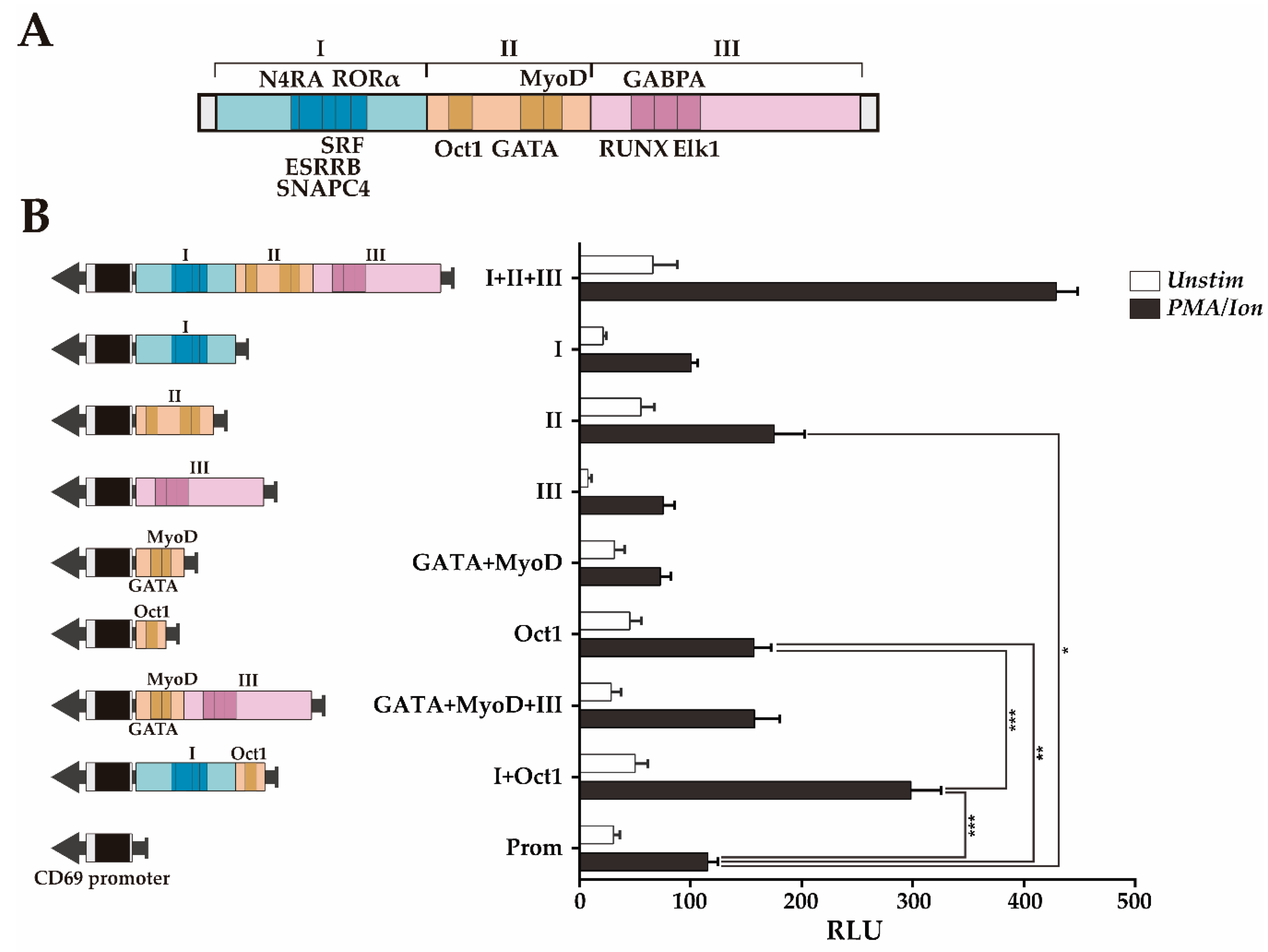
3.2. Transcriptional Enhancer Capacity of the CNS2 Region and Its Enhancer Core Activity
3.3. RUNX1 and Oct1 Silencing Affects the Transcriptional Regulation of CD69
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Stamatoyannopoulos, J.A. What does our genome encode? Genome Res. 2012, 22, 1602–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanno, Y.; Vahedi, G.; Hirahara, K.; Singleton, K.; O’Shea, J.J. Transcriptional and Epigenetic Control of T Cell Lineage Specification: Molecular Mechanisms Underlying Commitment and Plasticity. Annu. Rev. Immunol. 2012, 30, 707–731. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.R.; Kim, S.T.; Spilianakis, C.G.; Fields, P.E.; Flavell, R.A. T Helper Cell Differentiation: Regulation by cis Elements and Epigenetics. Immunity 2006, 24, 369–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montavon, T.; Duboule, D. Landscapes and archipelagos: Spatial organization of gene regulation in vertebrates. Trends Cell Biol. 2012, 22, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Esplugues, E.; Sancho, D.; Vega-Ramos, J.; Martínez-A, C.; Syrbe, U.; Hamann, A.; Engel, P.; Sánchez-Madrid, F.; Lauzurica, P. Enhanced Antitumor Immunity in Mice Deficient in CD69. J. Exp. Med. 2003, 197, 1093–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viedma, F.; De La Fuente, H.; Sancho, D.; Gomez, M.; Esplugues, E.; Gordon-Alonso, M.; García-López, M.A.; Martínez-A, C.; Lauzurica, P.; Sánchez-Madrid, F. CD69 downregulates autoimmune reactivity through active transforming growth factor-β production in collagen-induced arthritis. J. Clin. Investig. 2003, 112, 872–882. [Google Scholar]
- Notario, L.; Alari-Pahissa, E.; De Molina, A.; Lauzurica, P. CD69 Deficiency Enhances the Host Response to Vaccinia Virus Infection through Altered NK Cell Homeostasis. J. Virol. 2016, 90, 6464–6474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vega-Ramos, J.; Alari-Pahissa, E.; Valle, J.D.; Carrasco-Marin, E.; Esplugues, E.; Borras, M.; Martinez, A.C.; Lauzurica, P. CD69 limits early inflammatory diseases associated with immune response to Listeria monocytogenes infection. Immunol. Cell Biol. 2010, 88, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Alari-Pahissa, E.; Vega-Ramos, J.; Zhang, J.-G.; Castaño, A.R.; Turley, S.J.; Villadangos, J.A.; Lauzurica, P. Differential effect of CD69 targeting on bystander and antigen-specific T cell proliferation. J. Leukoc. Biol. 2012, 92, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Lanier, L.L. Interleukin 2 activation of natural killer cells rapidly induces the expression and phosphorylation of the Leu-23 activation antigen. J. Exp. Med. 1988, 167, 1572–1585. [Google Scholar] [CrossRef] [PubMed]
- Hara, T. Human T cell activation. III. Rapid induction of a phosphorylated 28 kD/32 kD disulfide-linked early activation antigen (EA 1) by 12-o-tetradecanoyl phorbol-13-acetate, mitogens, and antigens. J. Exp. Med. 1986, 164, 1988–2005. [Google Scholar] [CrossRef] [PubMed]
- Matloubian, M.; Lo, C.G.; Cinamon, G.; Lesneski, M.J.; Xu, Y.; Brinkmann, V.; Allende, M.L.; Proia, R.L.; Cyster, J.G. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 2004, 427, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Sancho, D.; Gómez, M.; Del Hoyo, G.M.; Lamana, A.; Esplugues, E.; Lauzurica, P.; Martinez-A, C.; Sánchez-Madrid, F.; Sancho-Madrid, D. CD69 targeting differentially affects the course of collagen-induced arthritis. J. Leukoc. Biol. 2006, 80, 1233–1241. [Google Scholar] [CrossRef]
- Esplugues, E.; Vega-Ramos, J.; Cartoixà, D.; Vazquez, B.N.; Salaet, I.; Engel, P.; Lauzurica, P. Induction of tumor NK-cell immunity by anti-CD69 antibody therapy. Blood 2005, 105, 4399–4406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, P.; Gómez, M.; Lamana, A.; Marín, A.M.; Cortés, J.R.; Ramirez-Huesca, M.; Barreiro, O.; López-Romero, P.; Gutiérrez-Vázquez, C.; De La Fuente, H.; et al. The leukocyte activation antigen CD69 limits allergic asthma and skin contact hypersensitivity. J. Allergy Clin. Immunol. 2010, 126, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Gomez, M.; Sanz-Gonzalez, S.M.; Abu Nabah, Y.N.; Lamana, A.; Sanchez-Madrid, F.; Andres, V. Atherosclerosis development in apolipoprotein E-null mice deficient for CD69. Cardiovasc. Res. 2009, 81, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Adalia, A.; Jiménez-Borreguero, L.J.; Ramírez-Huesca, M.; Chico-Calero, I.; Barreiro, O.; López-Conesa, E.; Fresno, M.; Sánchez-Madrid, F.; Martín, P. CD69 Limits the Severity of Cardiomyopathy After Autoimmune Myocarditis. Circulation 2010, 122, 1396–1404. [Google Scholar] [CrossRef] [Green Version]
- Cibrián, D.; Sánchez-Madrid, F. CD69: From activation marker to metabolic gatekeeper. Eur. J. Immunol. 2017, 47, 946–953. [Google Scholar] [CrossRef]
- Rutella, S.; Rumi, C.; Lucia, M.B.; Barberi, T.; Puggioni, P.L.; Lai, M.; Romano, A.; Cauda, R.; Leone, G. Induction of CD69 antigen on normal CD4+ and CD8+ lymphocyte subsets and its relationship with the phenotype of responding T-cells. Cytometry 1999, 38, 95–101. [Google Scholar] [CrossRef]
- Testi, R.; Phillips, J.H.; Lanier, L.L. T cell activation via Leu-23 (CD69). J. Immunol. 1989, 143, 1123–1128. [Google Scholar]
- Shiow, L.R.; Rosen, D.B.; Brdičková, N.; Xu, Y.; An, J.; Lanier, L.L.; Cyster, J.G.; Matloubian, M. CD69 acts downstream of interferon-α/β to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature 2006, 440, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Zhang, D.; Palliser, D.; Zhu, P.; Cai, S.; Schlesinger, A.; Maliszewski, L.; Lieberman, J. Listeria-Infected Myeloid Dendritic Cells Produce IFN-β, Priming T Cell Activation. J. Immunol. 2005, 175, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-J.; Chao, H.-C.; Kuo, M.-L. The Effect of Interleukin-12 and Interleukin-15 on CD69 Expression of T-Lymphocytes and Natural Killer Cells from Umbilical Cord Blood. Neonatology 2000, 78, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Mateos, P.; Cebrián, M.; Acevedo, A.; López-Botet, M.; De Landázuri, M.O.; Sánchez-Madrid, F. Expression of a gp33/27,000 MW activation inducer molecule (AIM) on human lymphoid tissues. Induction of cell proliferation on thymocytes and B lymphocytes by anti-AIM antibodies. Immunology 1989, 68, 72–79. [Google Scholar] [PubMed]
- Cebrián, M.; Redondo, J.M.; De Landázuri, M.O.; Rodríguez-Tarduchy, G.; López-Rivas, A.; Rodriguez-Tarduchy, G.; Sánchez-Madrid, F.; López-Rivas, A.; Rodríguez-Tarduchy, G.; Sánchez-Madrid, F. Expression and function of AIM, an activation inducer molecule of human lymphocytes, is dependent on the activation of protein kinase C. Eur. J. Immunol. 1989, 19, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Bjorndahl, J.M.; Nakamura, S.; Hara, T.; Jung, L.K.; Fu, S.M. The 28-kDa/32-kDa activation antigen EA 1. Further characterization and signal requirements for its expression. J. Immunol. 1988, 141, 4094–4100. [Google Scholar] [PubMed]
- Risso, A.; Smilovich, D.; Capra, M.C.; Baldissarro, I.; Yan, G.; Bargellesi, A.; E Cosulich, M. CD69 in resting and activated T lymphocytes. Its association with a GTP binding protein and biochemical requirements for its expression. J. Immunol. 1991, 146, 4105–4114. [Google Scholar] [PubMed]
- Ziegler, S.F.; Levin, S.D.; Johnson, L.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Baker, E.; Sutherland, G.R.; Feldhaus, A.L.; Ramsdell, F. The mouse CD69 gene. Structure, expression, and mapping to the NK gene complex. J. Immunol. 1994, 152, 1228–1236. [Google Scholar] [PubMed]
- López-Cabrera, M.; Muñoz, E.; Blázquez, M.V.; Ursa, M.A.; Santis, A.G.; Sánchez-Madrid, F. Transcriptional regulation of the gene encoding the human C-type lectin leukocyte receptor AIM/CD69 and functional characterization of its tumor necrosis factor-alpha-responsive elements. J. Biol. Chem. 1995, 270, 21545–21551. [Google Scholar] [CrossRef]
- Castellanos, M.D.C.; López-Giral, S.; López-Cabrera, M.; De Landázuri, M.O. Multiple cis-acting elements regulate the expression of the early T cell activation antigen CD69. Eur. J. Immunol. 2002, 32, 3108–3117. [Google Scholar] [CrossRef]
- Castellanos, M.C.; Muñoz, C.; Montoya, M.C.; Lara-Pezzi, E.; López-Cabrera, M.; De Landázuri, M.O. Expression of the leukocyte early activation antigen CD69 is regulated by the transcription factor AP-1. J. Immunol. 1997, 159, 5463–5473. [Google Scholar] [PubMed]
- Vazquez, B.N.; Laguna, T.; Notario, L.; Lauzurica, P. Evidence for an intronic cis-regulatory element within CD69 gene. Genes Immun. 2012, 13, 356–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez, B.N.; Laguna, T.; Carabana, J.; Krangel, M.S.; Lauzurica, P. CD69 gene is differentially regulated in T and B cells by evolutionarily conserved promoter-distal elements1. J. Immunol. 2009, 183, 6513–6521. [Google Scholar] [CrossRef] [PubMed]
- Laguna, T.; Notario, L.; Pippa, R.; Fontela, M.G.; Vazquez, B.N.; Maicas, M.; Aguilera-Montilla, N.; Odero, M.D.; Lauzurica, P.; Corbí Ángel, L.; et al. New insights on the transcriptional regulation of CD69 gene through a potent enhancer located in the conserved non-coding sequence 2. Mol. Immunol. 2015, 66, 171–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Oki, S.; Ohta, T.; Shioi, G.; Hatanaka, H.; Ogasawara, O.; Okuda, Y.; Kawaji, H.; Nakaki, R.; Sese, J.; Meno, C. ChIP-Atlas: A Data-Mining Suite Powered by Full Integration of Public ChIP-Seq Data; Reports; EMBO: Heidelberg, Germany, December 2018. [Google Scholar]
- Zhou, K.R.; Liu, S.; Sun, W.J.; Zheng, L.L.; Zhou, H.; Yang, J.H.; Qu, L.H. ChIPBase v2.0: Decoding transcriptional regulatory networks of non-coding RNAs and protein-coding genes from ChIP-seq data. Nucleic Acids Res. 2017, 45, D43–D50. [Google Scholar] [CrossRef] [PubMed]
- Dogan, N.; Wu, W.; Morrissey, C.S.; Chen, K.-B.; Stonestrom, A.; Long, M.; Keller, C.A.; Cheng, Y.; Jain, D.; Visel, A.; et al. Occupancy by key transcription factors is a more accurate predictor of enhancer activity than histone modifications or chromatin accessibility. Epigenetics Chromatin 2015, 8, 16. [Google Scholar] [CrossRef]
- Tsytsykova, A.V.; Rajsbaum, R.; Falvo, J.V.; Ligeiro, F.; Neely, S.R.; Goldfeld, A.E. Activation-dependent intrachromosomal interactions formed by the TNF gene promoter and two distal enhancers. Proc. Natl. Acad. Sci. USA 2007, 104, 16850–16855. [Google Scholar] [CrossRef]
- Li, L.; Zhang, J.A.; Dose, M.; Kueh, H.Y.; Mosadeghi, R.; Gounari, F.; Rothenberg, E.V. A far downstream enhancer for murine Bcl11b controls its T-cell specific expression. Blood 2013, 122, 902–911. [Google Scholar] [CrossRef] [Green Version]
- Schönheit, J.; Kuhl, C.; Gebhardt, M.L.; Klett, F.F.; Riemke, P.; Scheller, M.; Huang, G.; Naumann, R.; Leutz, A.; Stocking, C.; et al. PU.1 Level-Directed Chromatin Structure Remodeling at the Irf8 Gene Drives Dendritic Cell Commitment. Cell Rep. 2013, 3, 1617–1628. [Google Scholar] [CrossRef] [Green Version]
- Herr, W.; Cleary, M.A. The POU domain: Versatility in transcriptional regulation by a flexible two-in-one DNA-binding domain. Genes Dev. 1995, 9, 1679–1693. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, M.G. POU-domain transcription factors: Pou-er-ful developmental regulators. Genes Dev. 1991, 5, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Duncliffe, K.N.; Bert, A.G.; Vadas, M.A.; Cockerill, P.N. A T Cell–Specific Enhancer in the Interleukin-3 Locus Is Activated Cooperatively by Oct and NFAT Elements within a DNase I–Hypersensitive Site. Immunity 1997, 6, 175–185. [Google Scholar] [CrossRef]
- Fletcher, C.; Heintz, N.; Roeder, R.G. Purification and characterization of OTF-1, a transcription factor regulating cell cycle expression of a human histone H2b gene. Cell 1987, 51, 773–781. [Google Scholar] [CrossRef]
- Zhou, L.; Nazarian, A.A.; Xu, J.; Tantin, D.; Corcoran, L.M.; Smale, S.T. An Inducible Enhancer Required for Il12b Promoter Activity in an Insulated Chromatin Environment. Mol. Cell. Biol. 2007, 27, 2698–2712. [Google Scholar] [CrossRef] [PubMed]
- dela Paz, N.G.; Simeonidis, S.; Leo, C.; Rose, D.W.; Collins, T. Regulation of NF-kappaB-dependent gene expression by the POU domain transcription factor Oct-1. J. Biol. Chem. 2007, 282, 8424–8434. [Google Scholar] [CrossRef] [PubMed]
- Kakizawa, T.; Miyamoto, T.; Ichikawa, K.; Takeda, T.; Suzuki, S.; Mori, J.; Kumagai, M.; Yamashita, K.; Hashizume, K. Silencing mediator for retinoid and thyroid hormone receptors interacts with octamer transcription factor-1 and acts as a transcriptional repressor. J. Biol. Chem. 2001, 276, 9720–9725. [Google Scholar] [CrossRef] [PubMed]
- Schwachtgen, J.L.; Remacle, J.E.; Janel, N.; Brys, R.; Huylebroeck, D.; Meyer, D.; Kerbiriou-Nabias, D. Oct-1 is involved in the transcriptional repression of the von willebrand factor gene promoter. Blood 1998, 92, 1247–1258. [Google Scholar]
- Hwang, S.S.; Kim, L.K.; Lee, G.R.; Flavell, R.A. Role of OCT-1 and partner proteins in T cell differentiation. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2016, 1859, 825–831. [Google Scholar] [CrossRef]
- Shakya, A.; Goren, A.; Shalek, A.K.; German, C.N.; Snook, J.; Kuchroo, V.K.; Yosef, N.; Chan, R.C.; Regev, A.; Williams, M.A.; et al. Oct1 and OCA-B are selectively required for CD4 memory T cell function. J. Exp. Med. 2015, 212, 2115–2131. [Google Scholar] [CrossRef]
- Shakya, A.; Kang, J.; Chumley, J.; Williams, M.A.; Tantin, D. Oct1 is a switchable, bipotential stabilizer of repressed and inducible transcriptional states. J. Biol. Chem. 2011, 286, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Ebihara, T.; Seo, W.; Taniuchi, I. Roles of RUNX Complexes in Immune Cell Development. Results Probl. Cell Differ. 2017, 962, 395–413. [Google Scholar]
- Tolhuis, B.; Palstra, R.J.; Splinter, E.; Grosveld, F.; de Laat, W. Looping and interaction between hypersensitive sites in the active beta-globin locus. Mol. Cell 2002, 10, 1453–1465. [Google Scholar] [CrossRef]
- Amano, T.; Sagai, T.; Tanabe, H.; Mizushina, Y.; Nakazawa, H.; Shiroishi, T. Chromosomal Dynamics at the Shh Locus: Limb Bud-Specific Differential Regulation of Competence and Active Transcription. Dev. Cell 2009, 16, 47–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoshi, M.; Fukaya, T. Dynamics of transcriptional enhancers and chromosome topology in gene regulation. Dev. Growth Differ. 2019, 61, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Kulaeva, O.I.; Nizovtseva, E.V.; Polikanov, Y.S.; Ulianov, S.V.; Studitsky, V.M. Distant Activation of Transcription: Mechanisms of Enhancer Action. Mol. Cell. Biol. 2012, 32, 4892–4897. [Google Scholar] [CrossRef] [Green Version]
- Levine, M.; Cattoglio, C.; Tjian, R. Looping Back to Leap Forward: Transcription Enters a New Era. Cell 2014, 157, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Bender, M.A.; Byron, R.; Ragoczy, T.; Telling, A.; Bulger, M.; Groudine, M. Flanking HS-62.5 and 3′ HS1, and regions upstream of the LCR, are not required for β-globin transcription. Blood 2006, 108, 1395–1401. [Google Scholar] [CrossRef]
- Vernimmen, D.; De Gobbi, M.; Sloane-Stanley, J.A.; Wood, W.G.; Higgs, D.R. Long-range chromosomal interactions regulate the timing of the transition between poised and active gene expression. EMBO J. 2007, 26, 2041–2051. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence 5′ → 3′ |
|---|---|
| I_Fw | GAGGAATTCTTTCAGAAACCTCCCTCCGG |
| I_Rv | AGACTCGAGCAGCATCTGTTGTGATTAGCAT |
| I+II+III_Fw | CCGAATTCCCAGTACCTCATTCACTCACTG |
| I+OCT_Fw | AAAAGAATTCAGCTGCCGTGATAAGGACTGTA |
| III_Fw | ATAAAGAATTCCCAGTACCTCATTCACTCACTG |
| I+OCT_Rv | AGTCTCGAGCACTAAAACCCAGTGCAGTTCT |
| III+OCT_Fw | AAAGAATTCCCAGTACCTCATTCACTCACTG |
| OCT_Fw | AAAAGAATTCAGCTGCCGTGATAAGGACTGTA |
| OCT_Rv | AAACTCGAGCTGAAAGGAAATGATGTAATGT |
| III_Rv | AAACTCGAGGCCTTAGTGGTATGTGTTCAAAG |
| I+II_Fw | AAAAGAATTCTGGTTCCTTTGAACACATAC |
| III-GATA_Rv | AAAACTCGAGAGGAAGGCAGGGTGT |
| MyoD+GATA_Rv | AAACTCGAGAGGAAGGCAGGGTGTACA |
| RV3_Fw * | CTAGCAAAATAGGCTGTCCC |
| Prom_Seq_Rv * | TGACATGGGAAAAGCACTGGA |
| Construct | Primers Used | Amplicon Length |
|---|---|---|
| I+II+III | I+II+III_Fw + I + Oct1_Rv | 297 bp |
| II+III | III+Oct1_Fw + Oct1_Rv | 227 bp |
| I+II | Oct1_Fw + I + Oct1_Rv | 177 bp |
| I | I_Fw + I+Oct1_Rv | 70 bp |
| II | I+II_Fw + Oct1_Rv | 107 bp |
| III | III_Fw + III_Rv | 120 bp |
| GATA+MyoD | I+II_Fw + MyoD + GATA_Rv | 65 bp |
| Oct1 | Oct1_Fw + Oct1_Rv | 60 bp |
| GATA+MyoD+III | III_Fw + III-GATA_Rv | 156 bp |
| I+Oct1 | I+Oct1_Fw + I+Oct1_Rv | 142 bp |
| DsiRNA | Sequences | Cross-Reacting Transcript |
|---|---|---|
| Runx1.13.1 | 5′-AAGAAAGAUAUCAAGUACUACAUtt-3′ 3′-AAUUCUUUCUAUAGUUCAUGAUGUAAA-5′ | NM_001111022 NM_001111021 NM_001111023 NM_009821 |
| Runx1.13.2 | 5′-AUGGCAGGCAACGAUGAAAACUAct-3′ 3′-AGUACCGUCCGUUGCUACUUUUGAUGA-5′ | NM_009821 NM_001111023 NM_001111021 NM_001111022 |
| Runx1.13.3 | 5′-GAAGAACCAGGUAGCGAGAUUCAac-3′ 3′-UACUUCUUGGUCCAUCGCUCUAAGUUG-5′ | NM_001111022 NM_001111021 NM_001111023 NM_009821 |
| Pou2f1.13.1 | 5′-UAAAUUUCAUGAAAGCUUUACUUgt-3′ 3′-CGAUUUAAAGUACUUUCGAAAUGAACA-5′ | NM_198932 NM_011137 NM_198934 NM_198933 |
| Pou2f1.13.2 | 5′-CAAGAAUGAAUAAUCCAUCAGAAac-3′ 3′-GAGUUCUUACUUAUUAGGUAGUCUUUG-5′ | NM_198933 NM_198934 NM_011137 NM_198932 |
| Pou2f1.13.3 | 5′-GCAGUUUGCCAAGACUUUCAAACaa-3′ 3′-CUCGUCAAACGGUUCUGAAAGUUUGUU-5′ | NM_198932 NM_011137 NM_198934 NM_198933 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fontela, M.G.; Notario, L.; Alari-Pahissa, E.; Lorente, E.; Lauzurica, P. The Conserved Non-Coding Sequence 2 (CNS2) Enhances CD69 Transcription through Cooperation between the Transcription Factors Oct1 and RUNX1. Genes 2019, 10, 651. https://doi.org/10.3390/genes10090651
Fontela MG, Notario L, Alari-Pahissa E, Lorente E, Lauzurica P. The Conserved Non-Coding Sequence 2 (CNS2) Enhances CD69 Transcription through Cooperation between the Transcription Factors Oct1 and RUNX1. Genes. 2019; 10(9):651. https://doi.org/10.3390/genes10090651
Chicago/Turabian StyleFontela, Miguel G., Laura Notario, Elisenda Alari-Pahissa, Elena Lorente, and Pilar Lauzurica. 2019. "The Conserved Non-Coding Sequence 2 (CNS2) Enhances CD69 Transcription through Cooperation between the Transcription Factors Oct1 and RUNX1" Genes 10, no. 9: 651. https://doi.org/10.3390/genes10090651