1. Introduction
In teleost, the pigment chromophores in the skin and scales could be involved in several important biological processes, such as camouflage, thermoregulation, mate choice, mimicry, warning of toxicity, and immunity [
1]. The pigments are produced by several pigment cells in fish, including melanophores, xanthophores, and iridophores [
2]. Pigmentation is a very complicated process, which is regulated by a series of cellular, genetic, endocrine, environmental, and physiological factors [
3]. Different pigment cells in fish display different colors due to their unique organelles. The melanin particles produced by melanocytes could absorb the incident light of a specific wavelength and make fish appear black or brown. The pigments produced by xanthophores are mainly pteridine, which can filter out certain wavelengths of light to make fish appear yellow or red [
4]. In addition, iridophores could produce silver pigments, in which guanine could combine with water to form purine-containing reflecting platelets [
5].
The transparent trait in fish is closely related to the development of pigment cells. Generally, most fish are transparent in their early lives, while the body in adult fish is no longer transparent due to the accumulation of pigment in skin and peritoneum. The cytoplasm of iridophores contains a large number of crystalline plates, which could reflect a certain wavelength of light and make them iridescent. The color presented by iridophores belongs to the configuration color, which is different from other kinds of pigment cells [
5]. The absence of silver pigments produced by iridophores could allow light to penetrate body and make fish transparent. Previous studies have reported that zebrafish
shady,
rose, and
barley transparent mutations are all associated with the lost of iridophores. Mutations in
shady could result in a lack of iridophores throughout the whole life [
6,
7]. Mutations in
rose, a gene encoding the endothelin receptor b1a (ednrb1a), could cause an adult-specific reduction of iridophores [
8,
9]. In addition, the transparent mutant not only shows the lost of iridophores, but also a significant decrease in the number of melanocytes.
To date, a series of genes have been reported to be involved in the determination of skin color, such as pro-opiomelanocortin (
POMC), melanocyte stimulating hormone (
MSH), microphthalmia-associated transcription factor (
MITF), kit oncogene (
KIT), tyrosinase (
TYR), tyrosine related protein-1 (
TYRP1), and tyrosine related protein-2 (
TYRP2) [
2,
10]. In melanocytes,
MITF could induce the biosynthesis of melanin through the up-regulation of melanogenic enzyme expression [
11]. Further studies found that endothelin receptor B (
ednrb) could induce the phosphorylation of
MITF and its mutation could reduce the pigmentation in melanocytes [
12,
13]. In addition, previous studies reported that purine nucleoside phosphorylase 4a (
pnp4a) was also involved in guanine synthesis and its mutation could cause the deficiency of iridophores [
14,
15]. However, most of the genetic resources involved in the color of fish remain to be discovered.
MicroRNAs (miRNAs) are a group of single-stranded, non-coding RNA molecules with an average size of approximately 22 nucleotides, which play crucial roles in numerous biological processes through promoting their target gene’s degradation or inhibiting mRNA translation. It is important for the crosstalk between miRNAs and mRNAs to steady the signal transduction and the transcriptional activities as well as maintain homeostasis in many organs [
16,
17,
18]. In mammals, it has been reported that the miR-137 could affect body color pattern in mice [
19]. In Drosophila, the loss of miR-8 could reduce pigmentation in dorsal abdomen [
20]. In addition, miR-429 silencing in common carp could increase the transcript level of
Foxd3, and influence the melanin production [
21]. These studies indicate that miRNAs could also play an important role in body color formation [
15]. However, little is known about the functional role of miRNA in the regulation of silver pigment synthesis.
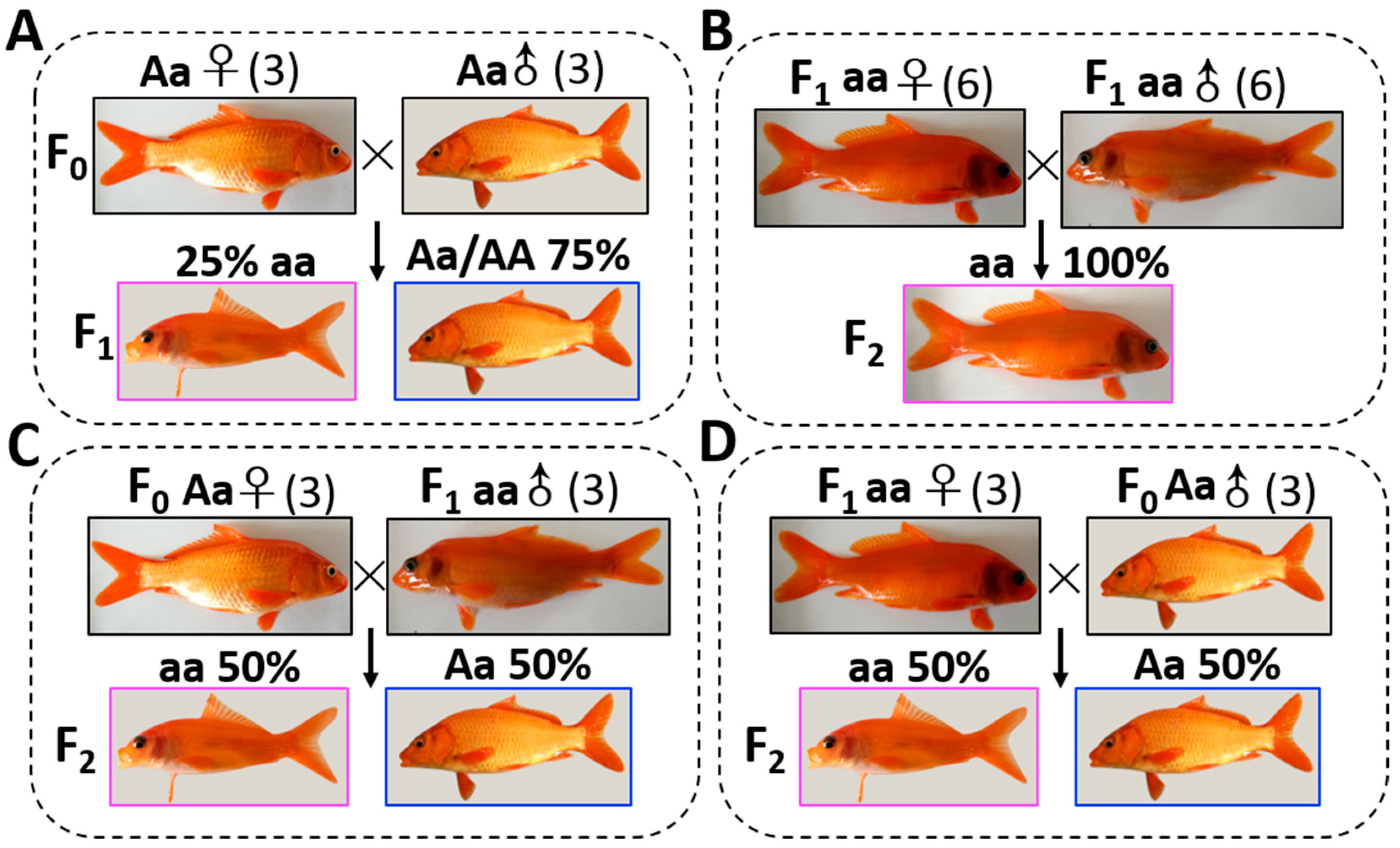
In our previous studies, some transparent mutant (TM) individuals were found in the wild type (WT) of the carp–goldfish nucleocytoplasmic hybrid (CyCa hybrid) population [
22]. In the WT, the silver pigments and melanin in the scales could block the light to pass through fish body, so they were opaque. However, there were no reflective substances in the skin and peritoneum of TM, so the internal organs of TM could be observed directly. In the present study, using the WT and TM as the model, (i) the hybridization experiment was used to further explore the genetic characteristics of the transparent trait in common carp; (ii) the RNA-Seq was used to examine the differentially expressed genes between the TM and WT; (iii) the miRNA-Seq was also recruited to detect the functional role of miRNA in the formation of pigment in common carp. This study will help us to understand the formation mechanism of the transparent trait in teleost.
4. Discussions
There are three main types of pigment cells in fish, namely melanophores, xanthophores, and iridophores [
2]. Among them, iridophores could produce silver pigments to reflect light in fish scales, so the loss of silver pigments could make the fish transparent. In goldfish, the transparent mutants have been reported in China, which were named as crystal goldfish. Further genetic studies on crystal goldfish indicated that the transparent mutants were homozygotes of guanophore (-/-) [
31]. Similarly, our present study demonstrated that the transparent trait in common carp was recessive over the reflective trait, and this phenomenon could completely conform to the Mendelian segregation rule of alleles. These results suggested that the transparent common carp could be a novel model to investigate the genetic basis of silver pigment synthesis.
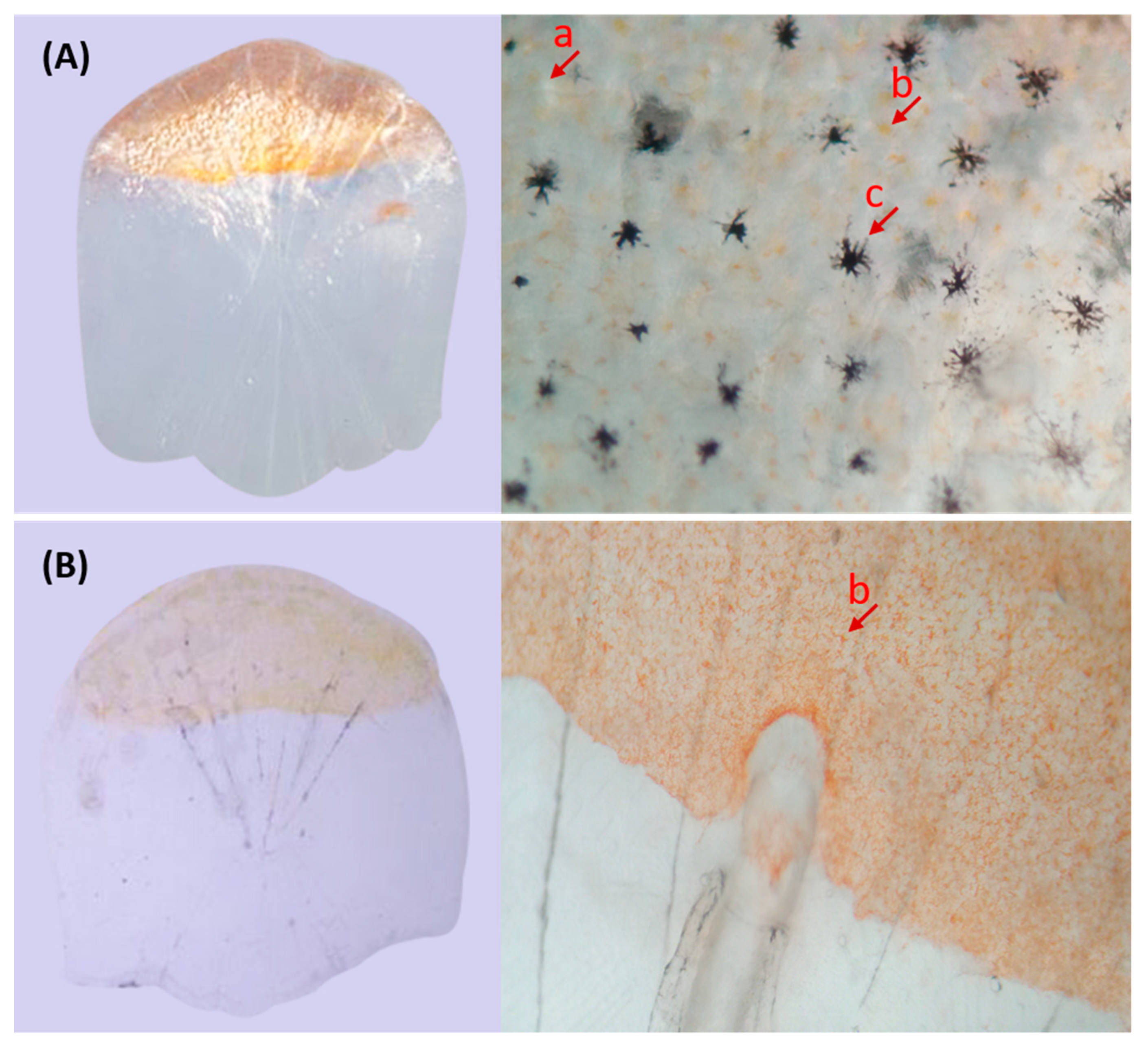
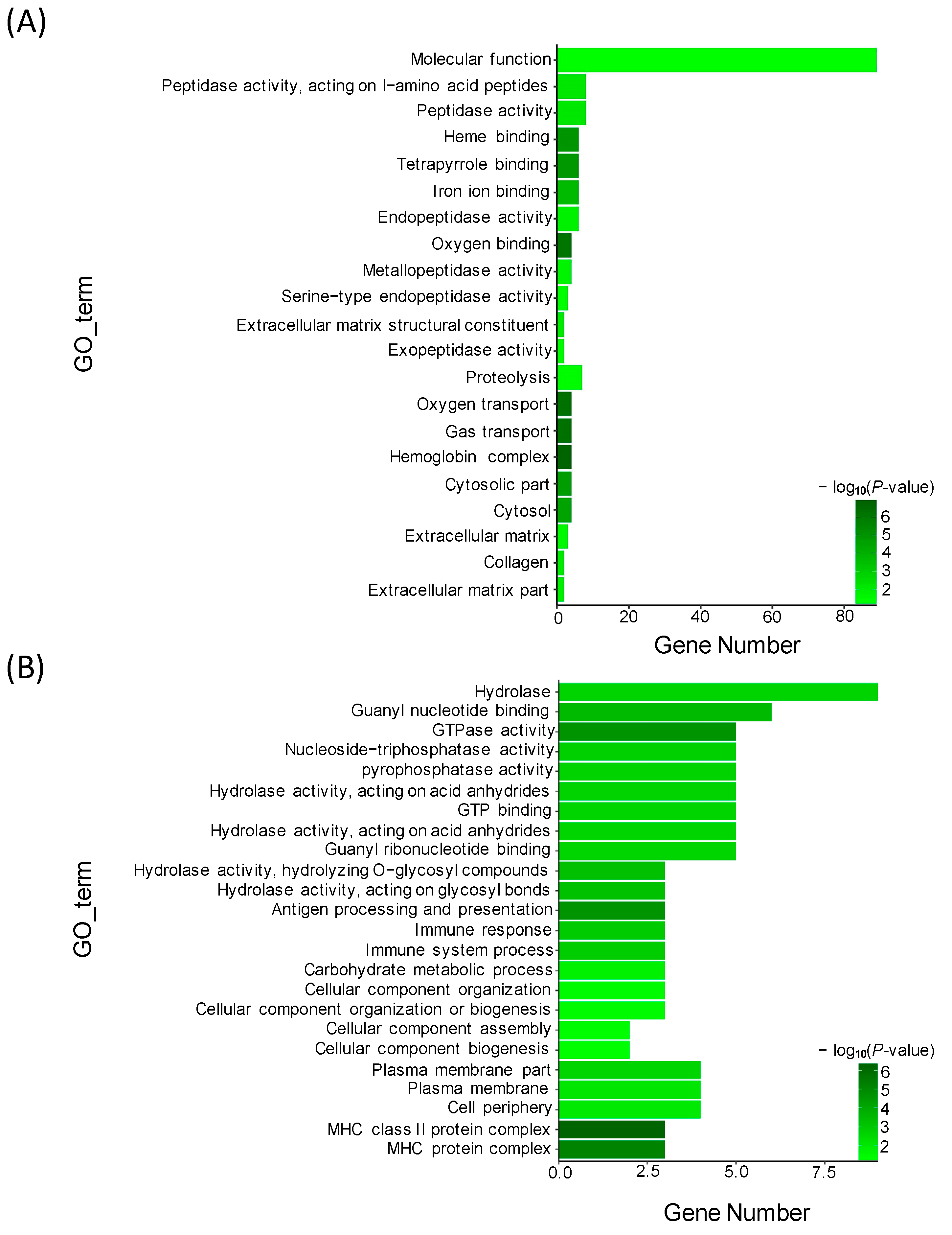
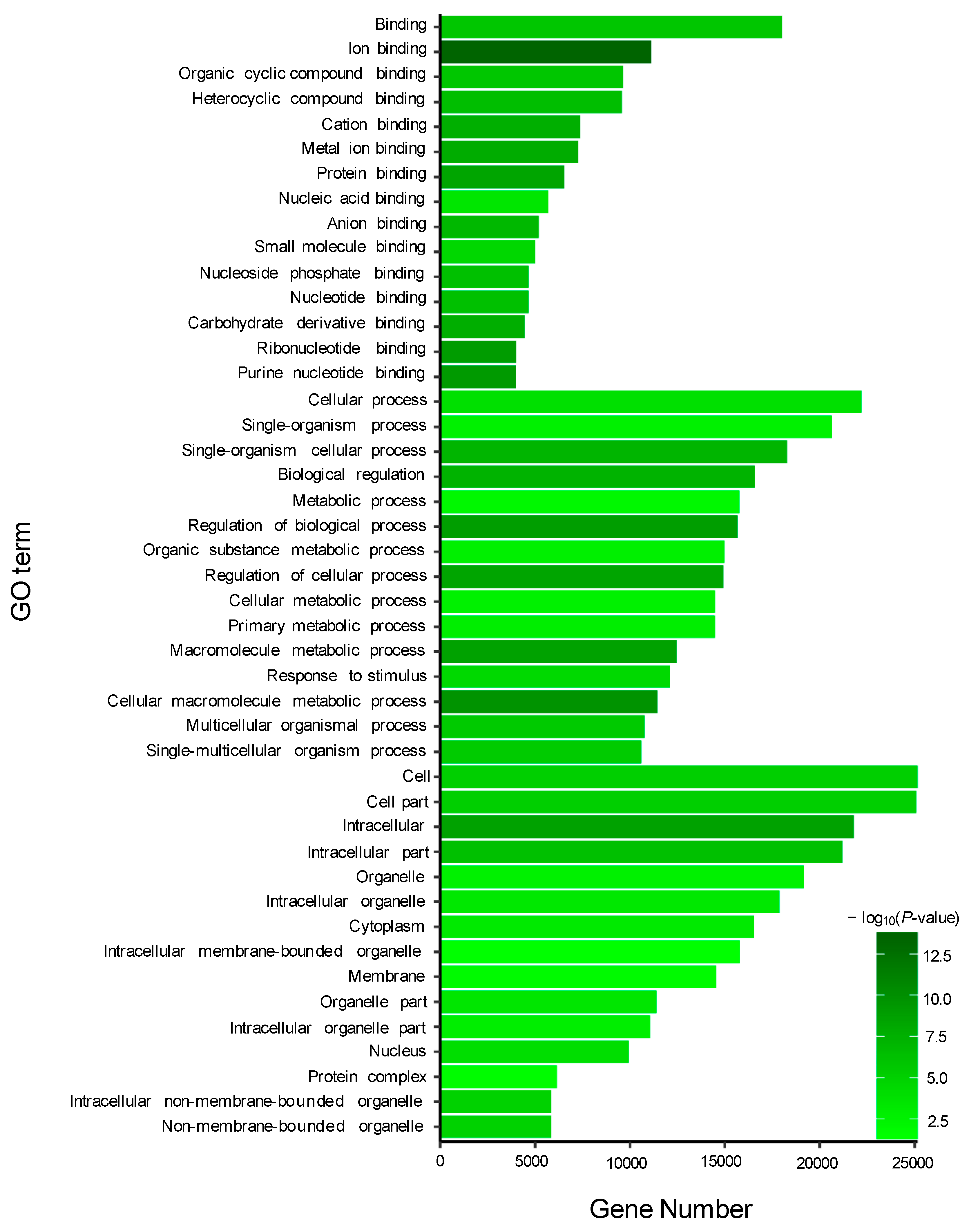
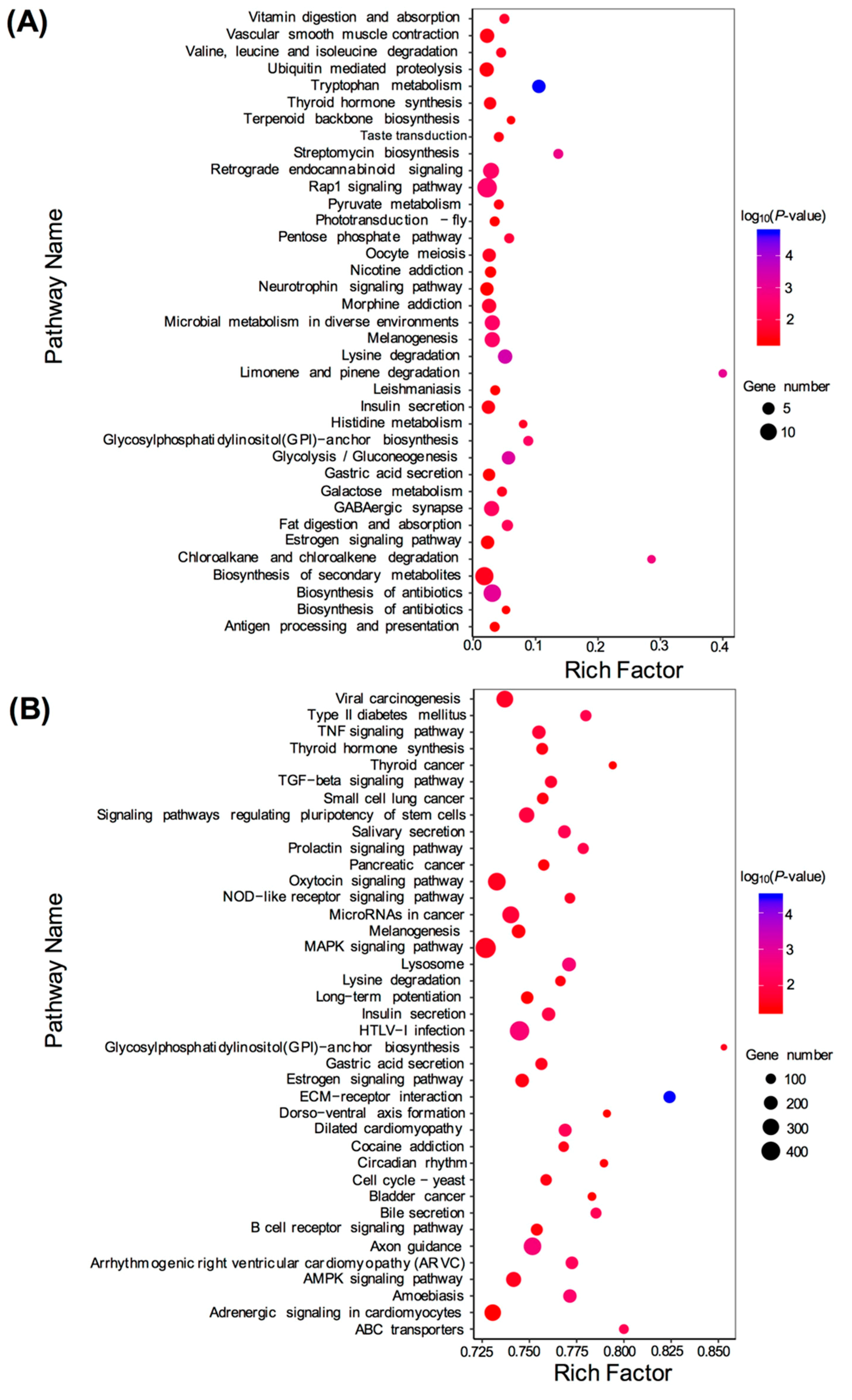
In the present study, microscopic observations showed that the silver pigments and melanin were both lost in the scales of transparent mutants compared to that in wild types. Furthermore, GO and KEGG analysis showed that the DEGs between wild type and transparent mutants were mainly enriched in the metabolism of guanine, such as hydrolase, guanyl nucleotide binding, guanyl ribonucleotide binding, and GTPase activity. Consistent with these, previous studies have shown that the guanine was the major component of silver pigments, and it could combine with water to form a crystal plate to make scales reflect light [
32]. The reduction in guanine deposition in iridophores could induce the establishment of the transparent medaka strains [
33,
34] (Wakamatsu et al., 2001; Ohshima et al., 2013). In addition, the differentiation of melanophores is coupled closely with the onset of pteridine synthesis which starts from GTP and is regulated through the control of GTP cyclohydrolase I activity [
14].
Transcriptome analysis showed that several DEGs were detected between TMs and WTs, including purine nucleoside phosphorylase 4a (
pnp4a) and endothelin receptor B (
ednrb). As a member of
pnp gene family in vertebrates,
pnp4a was reported to have an important role in guanine synthesis in iridophores [
14]. The previously reported zebrafish
shady,
rosy, and
barley transparent mutations were all associated with the deficiency of iridophores caused by the mutant of
pnp4a [
7,
32,
35]. The CRISPR/CAS9 system has been used to further confirm that
pnp4a was a causal gene on the guanineless locus of zebrafish [
14,
15]. In the present study, the slivery pigments in TM were completely lost, and
pnp4a was less expressed in TM compared to WT. These results, taken together, suggested that
pnp4a could also be involved in the formation of silver pigments in common carp. Similar to
pnp4a, our results found that the
pnp5a was also differentially expressed between WT and TM, which indicated that the
pnp5a might be a guanine synthetic gene in teleost iridophores. Endothelin receptor b (
ednrb) and its ligand, endothelin (
edn), were vital in melanocyte development [
36]. The signaling pathway of
ednrb could promote the proliferation and differentiation of melanocyte stem cells and the regeneration of epidermal melanocytes, and
ednrb mutations could give rise to pigmentation defects [
12,
13]. Further studies found that the activation of
ednrb could induce the phosphorylation of microphthalmia-associated transcription factor (
MITF), which was the key transcription factor for catalyzing tyrosine synthesis into melanin in melanocytes [
15]. In our present study, the melanin in TM was completely lost and the expression level of
ednrb in TM was significantly lower than that in WT, which suggested that
ednrb might play an important role in the synthesis of melanin in common carp. Guanine nucleotide-binding protein was the subunit of the G protein-couple signaling pathway, and a previous study reported that it might be involved in the formation of coat color in mice [
37]. Interestingly, our present study also found that guanine nucleotide-binding protein subunit α-14 (
GNA14) was significantly differentially expressed between WT and TM, which suggested that
GNA14 might be involved in the formation of pigment in teleost.
In mammals, miRNAs played pivotal roles in a variety of developmental processes, and their dysregulations were linked to several skin pigment diseases, such as melisma development [
38] and melanoma [
39]. In teleost, previous studies have reported that miR-429 was highly expressed in red skin of common carp, and inhibition of miR-429 could cause a substantial decrease in skin pigmentation [
21], which suggested that the miRNAs also played an important role in the pigmentation of teleost. In our present study, DEMs analysis found that miR-146a and miR-153b were both more highly detected in TM compared to those in WT. In mammals, recent studies have reported that miR-146a played an important role in melanoma [
39,
40,
41] and retinal pigment epithelial cells [
42]. Similarly, miR-153 could suppress cell proliferation and invasion by targeting snail family transcriptional repressor 1 (SNAI1) in melanoma [
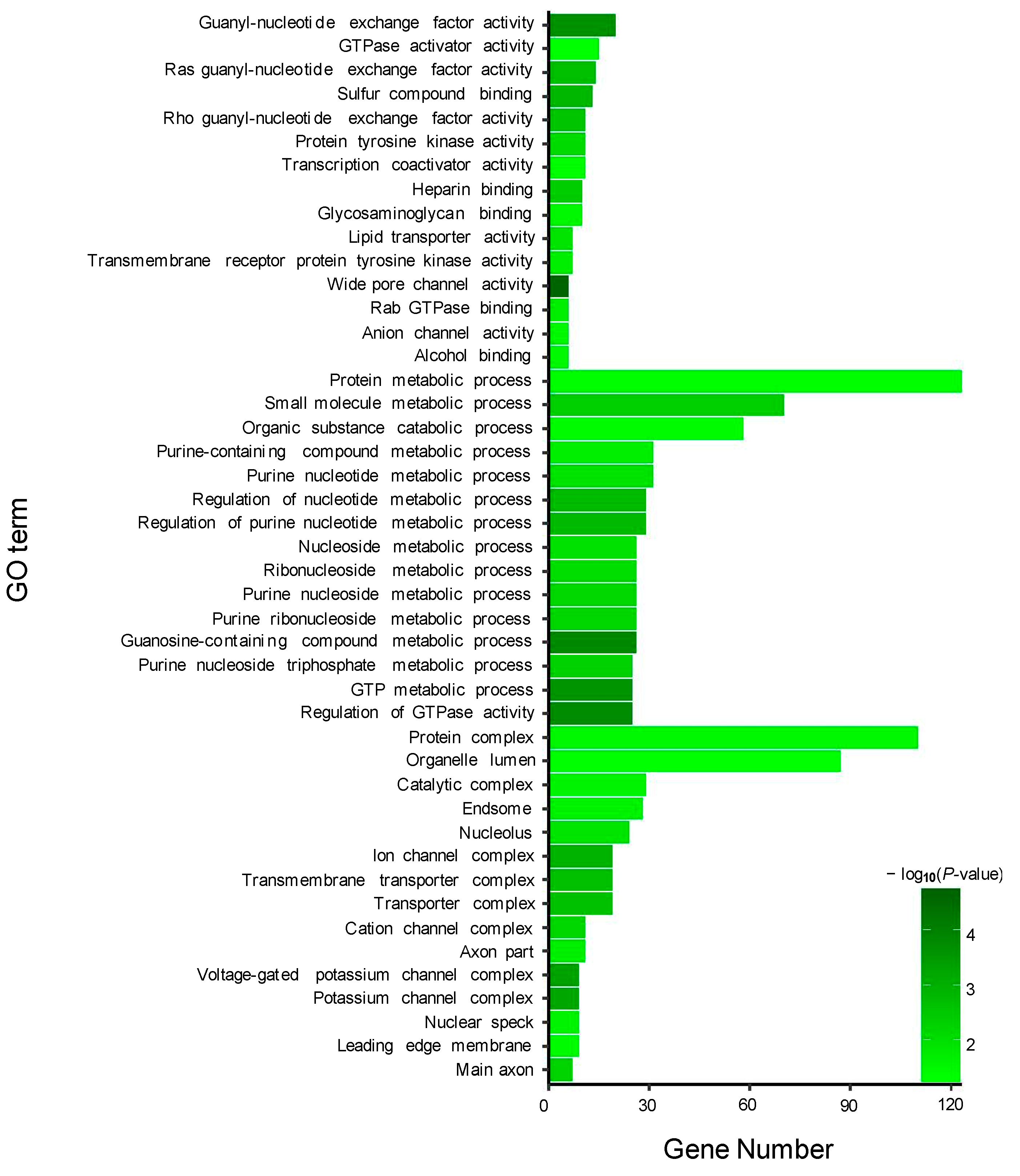
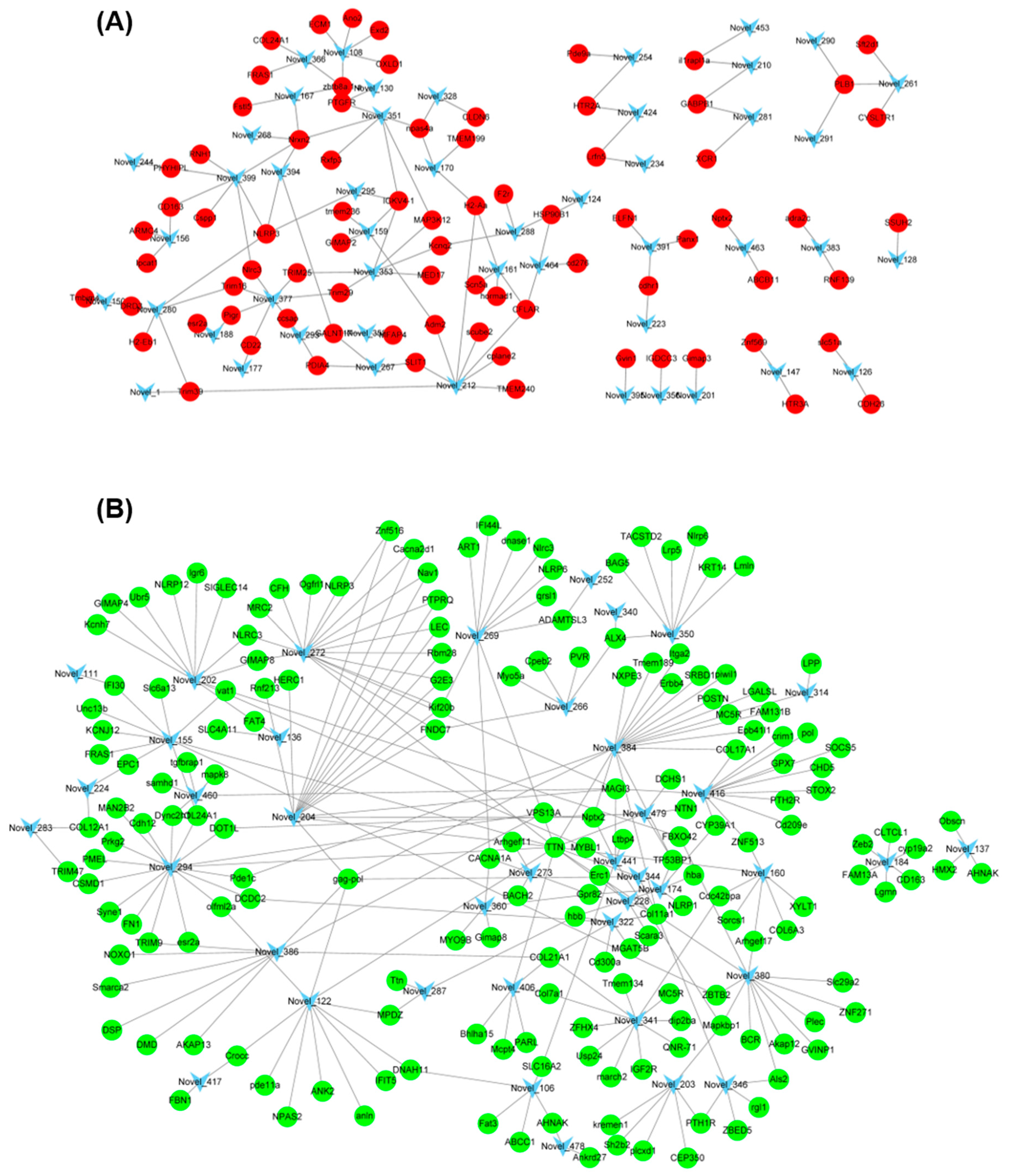
43]. In the present study, further GO and KEGG analysis showed that the target genes of miR-146a and miR-153b were mainly enriched in the guanyl-nucleotide exchange factor activity, GTPase activator activity, purine nucleotide metabolic process, and GTP metabolic process. These results suggested that miR-146a and miR-153b might also play a vital role in the regulation of pigment synthesis in common carp. In addition, we also found several novel DEMs between TMs and WTs, such as Novel_294, Novel_384, Novel_272, Novel_377, Novel_212, and Novel_399. Interaction analysis showed that these DEMs were associated with several DEGs between TMs and WTs. Further GO and KEGG analysis showed that these target DEGs were mainly enriched in purine nucleotide binding and nucleic acid binding. These results suggested that these novel DEMs might also play an important role in the regulation of pigmentation.
In the present study, the transparent mutants were detected in the CyCa hybrid carp. Microscopic examination showed that the scales of transparent mutants were completely devoid of silver pigments and melanin. Genetic analysis indicated that the transparent trait was recessive over the opaque trait in common carp. To further examine the molecular mechanism of the transparent trait, RNA-Seq and miRNA-Seq were both used to detect the DEGs and DEMs between TMs and WTs. RNA-Seq analysis showed that several DEGs were detected between TMs and WTs, such as pnp4a and ednrb. Interestingly, previous studies have reported that the pnp4a and ednrb played an important role in the synthesis of silver pigments and melanin, respectively. In addition, our GO and KEGG analysis showed that the DEGs between TM and WT were mainly enriched in the metabolism of guanine, which was consistent with the previous studies that the silver pigment was the metabolic product of guanine. By using miRNA-Seq analysis, several known and novel DEMs were also detected between TM and WT. In addition, interaction analysis showed that the DEMs were associated with several DEGs. These results suggested that the miRNAs were also involved in carp pigmentation. In general, the present study could provide some new ideas for investigating the genetic mechanism of the transparent trait and the formation mechanism of sliver pigment in common carp.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}