Abstract
The Irish national registry for inherited retinal degenerations (Target 5000) is a clinical and scientific program to identify individuals in Ireland with inherited retinal disorders and to attempt to ascertain the genetic cause underlying the disease pathology. Potential participants first undergo a clinical assessment, which includes clinical history and analysis with multimodal retinal imaging, electrophysiology, and visual field testing. If suitable for recruitment, a sample is taken and used for genetic analysis. Genetic analysis is conducted by use of a retinal gene panel target capture sequencing approach. With over 1000 participants from 710 pedigrees now screened, there is a positive candidate variant detection rate of approximately 70% (495/710). Where an autosomal recessive inheritance pattern is observed, an additional 9% (64/710) of probands have tested positive for a single candidate variant. Many novel variants have also been detected as part of this endeavor. The target capture approach is an economic and effective means of screening patients with inherited retinal disorders. Despite the advances in sequencing technology and the ever-decreasing associated processing costs, target capture remains an attractive option as the data produced is easily processed, analyzed, and stored compared to more comprehensive methods. However, with decreasing costs of whole genome and whole exome sequencing, the focus will likely move towards these methods for more comprehensive data generation.
1. Introduction
Inherited retinal degenerations (IRDs) are a broad set of clinically and genetically diverse conditions that represent the leading cause of visual dysfunction in those of working age. IRDs are typically caused by improper development or death of photoreceptor cells and have a substantial effect on both the quality of life of those affected and health economics. Inheritance patterns include autosomal recessive, autosomal dominant and X-linked, as well as rarer mitochondrial and digenic forms [1,2]. Since the discovery of the first IRD genes via linkage analysis [3,4,5], a comprehensive repertoire of disease-causing genes has been described.
To date, over 270 genes have been associated with a wide variety of IRDs across a phenotypic spectrum [6], which are routinely divided into large subcategories in order to readily distinguish the specific retinal regions and cell types affected; for example, retinitis pigmentosa (RP) versus macular dystrophy, which initially cause predominantly peripheral and central vision loss, respectively. In the majority of IRDs, ocular manifestations occur in isolation. However, many forms of syndromic IRD that result in systemic disease have also been described [7]; more common forms include Usher Syndrome and Bardet-Biedl syndrome. RP is the most frequent IRD presentation with an incidence of approximately 1 in 3000 [8]. Over 70 genes are implicated as causative of RP, making it one of the most diverse and genetically heterogeneous Mendelian conditions described to date [9]. Other IRDs are less heterogeneous—autosomal recessive Stargardt disease, for example, which is caused by pathogenic variations in ABCA4 in approximately 95% of cases [10,11,12,13]. Furthermore, IRDs such as X-linked retinoschisis are characterised by a single gene aetiology [14].
Using the incidence rates previously mentioned, it can be estimated that over 2.5 million individuals are affected by RP alone worldwide, and diagnoses are often based solely on clinical presentation [15]. The importance of identifying the genetic causes of IRDs is becoming increasingly clear, allowing for more accurate prognosis of disease progression and risk assessment for other family members. Importantly, a genetic diagnosis can also facilitate a better understanding of the disease for those affected. Furthermore, multiple clinical trials to treat these diseases are ongoing, with the first gene therapy for IRD, Luxturna, having been approved for use by both the US Food and Drug Administration and the European Medicines Agency [16]. Typically, such trials and treatments require that a causative variant has been identified in order to participate. Here, we present an update on Target 5000, a next-generation sequencing (NGS) study that aims to provide a large cohort of Irish patients with an accurate genetic diagnosis, whilst further elucidating the underlying genetic architecture of IRDs in Ireland.
Target 5000 is the Irish national registry for those affected by an IRD. It is both a research and clinical study that aims to elucidate the underlying genetic architecture of IRDs in Ireland and to provide an accurate genetic diagnosis for those affected. Target 5000 utilises target capture next generation sequencing, a time and cost-effective screening method that boasts more even coverage across captured regions, positively affecting diagnostic yield, in comparison with whole exome sequencing (WES) [17]. Exonic regions of over 250 IRD-associated genes as well as a number of intronic regions harbouring previously identified pathogenic variants were captured. Although whole genome sequencing (WGS) has become more accessible in recent years and facilitates the discovery of both deep intronic variants and novel genotype-phenotype correlations [18], significant challenges remain in terms of data handling, variant interpretation, and cost. Target 5000 significantly highlights the benefits of target capture NGS as a preliminary diagnostic measure, before employing more comprehensive sequencing methods such as WES or WGS.
Here we present novel findings from both newly recruited participants and retrospective analysis of 750 individuals previously screened, resulting over 1000 individuals across 710 pedigrees genetically screened to date. 458 individuals have been sequenced for variants in 210 genes, 689 individuals have been sequenced for variants in 254 genes, and confirmation sequencing is ongoing for other affected family members. The average age of recruited participants to Target 5000 is 55 (Figure S1). Target 5000 has identified positive candidate variants in almost 70% (495/710) of cases, including both previously reported pathogenic and novel potentially pathogenic variants. This includes novel structural variants, single nucleotide variants, and splice variants. Furthermore, 9% (64/710) of pedigrees were found to carry a pathogenic/likely pathogenic variant on one allele where the genotype and phenotype were associated with a recessive inheritance pattern.
2. Materials and Methods
2.1. Patient Identification and Recruitment
Probands and other family members are recruited nationwide and primarily assessed at the Research Foundation of the Royal Victoria Eye and Ear Hospital (Dublin, Ireland), the Mater Misericordiae University Hospital (Dublin, Ireland), and the Royal Victoria Hospital (Belfast, Northern Ireland, UK). This allows for the inclusion of participants from both The Republic of Ireland and Northern Ireland. Following informed consent, the applicable combination of tests were carried out to accurately assess the patients’ phenotype (Table 1).

Table 1.
Table of typical assessments, instruments, and manufacturers routinely used as part of Target 5000 recruitment.
2.2. DNA Acquisition and Next Generation Sequencing
DNA was isolated from either blood (DNA Blood Maxi Kit, Qiagen, Hilden, Germany) or saliva (Oragene-DNA, DNA Genotek, ON, Canada) samples from participants. Sample preparation was carried out using a hybridisation-based target capture sequencing method previously described [15]. The average read coverage achieved was 125× per captured region. All genomic locations refer to the Hg38 reference genome (Homo sapiens GRCh38).
2.3. Variant Confirmation Sequencing
To validate variants identified by NGS, relevant genomic loci containing the mutations were amplified by polymerase chain reaction (PCR) and investigated by direct sequencing. Primers were procured from Sigma-Aldrich (Gillingham, England, UK). DNA products were standardly amplified using Q5 High-Fidelity 2× Master Mix (New England Biolabs Inc., Ipswich, MA, USA). Segregation analyses for additional family members were amplified similarly or alternatively, directly from blood (Phusion Blood Direct PCR Kit, Thermo Scientific, MA, USA) or saliva (Phusion Human Specimen Direct PCR Kit, Thermo Scientific) where applicable. The annealing temperatures for reactions were optimised for each variant; all other details were executed as per the supplier’s recommendations. Sanger sequencing was performed by Eurofins Genomics (Ebersberg, Germany).
2.4. Choroideremia Deletion Confirmation
Primers were designed to probe for the exonic regions of CHM (MIM: 300390; ChrX: 85861180-86047558) and the neighbouring genes, POF1B (MIM: 300603; ChrX: 85277396-85379709) and DACH2 (MIM: 300608; ChrX: 86148451-86832602), as well as the intergenic space between. A full list of the primers utilised is available in Supplementary Materials (Table S1).
2.5. Sequencing of RPGR ORF15
A previously reported sequencing strategy of RPGR was employed in order to cover the highly repetitive sequences of ORF15, a major cause of X-linked RP [19]. Primers used to probe this region can be found in Figure S2.
2.6. Single-Molecule Molecular Inversion Probe (smMIP)-Based Sequencing of ABCA4
smMIPs-based whole gene sequencing of ABCA4 was carried out using a NextSeq 500 as part of a large-scale study by Khan et al. [13]. 3866 single-molecule molecular inversion probes (smMIPs) were designed as described previously [20] and employed to capture 110nt increments of both the sense and antisense strands of the ABCA4 gene, as well as 40 kb of flanking sequences. Sample processing, sequencing, data analysis, and variant interpretation was carried out, as described by Khan et al. [13].
2.7. Data Analysis and Variant Interpretation of Target Capture NGS Data
Raw sequencing data were demultiplexed and mapped to the IRD-relevant regions of the human genome (Hg38) as previously described [15]. The American College of Medical Genetics and Genomics (ACMG) criteria for classifying pathogenic variants was utilised to interpret variants [21]. Other proposed modifications to the ACMG guidelines to further quantitate certain lines of evidence were also employed during the study. These modifications effect three lines of evidence that can be applied. These adjustments consider using segregation data in a quantitative manner (code: PP1) [22], quantitating evidence that considers the rare incidence of variants (code: PS4) and the value of accurately phenotyping conditions with single gene aetiologies (code: PP4) [23]. To further implement these recommendations, REVEL and dbscSNV RF scores were added to the existing suite of ensemble predictors to more stringently detect agreement between predictors of pathogenicity [24,25]. Similarly, Manta was added to our structural variant pipeline to assist in the detection of genomic rearrangements [26].
2.8. Ethical Approval
Ethical approval for this study was awarded by the Research and Medical Ethics committee of the Royal Victoria Eye and Ear Hospital (13-06-2011: HRA-POR201097) and by the Institutional Review Board of the Mater Misericordiae University Hospital and Mater Private Hospital (MMUH IRB 1/378/1358), Dublin, Ireland, prior to commencement. All work was carried out in accordance with the approved guidelines. All patients have given informed consent before recruitment to the study.
3. Results
3.1. Clinical Presentation and Positive Candidate Detection Rates
Thus far, 710 pedigrees have been analysed as part of this study by target capture sequencing. 458 individuals have been sequenced for variants in 210 genes, 689 individuals have been sequenced for variants in 254 genes, and confirmation sequencing is ongoing for other affected family members. A spectrum of IRDs were observed in this study, as can be seen in Figure 1. The positive candidate dectection rate has improved from previous reports [15,27], now reaching approximately 70% (495/710). Furthermore, a single candidate variant only has been identified in an additional 9% (64/710) of pedigrees diagnosed with an IRD that typically exhibits a recessive inheritance pattern (Figure 2). In addition, a number of novel missense variants have been detected in the course of this study (Table 2).
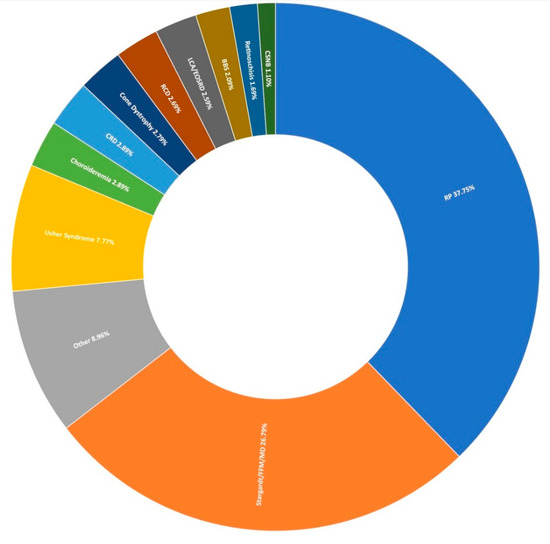
Figure 1.
Clinical presentation of 1004 recruited Target 5000 pedigrees. RP: Retinitis Pigmentosa, FFM: Fundus Flavimaculatus, MD: Macular Dystrophy, CRD: Cone-Rod Dystrophy, RCD: Rod-Cone Dystrophy, LCA: Leber Congenital Amaurosis, EOSRD: Early-Onset Retinal Degeneration, BBS: Bardet-Biedl Syndrome, CSNB: Congenital Stationary Night Blindness (Table S2).
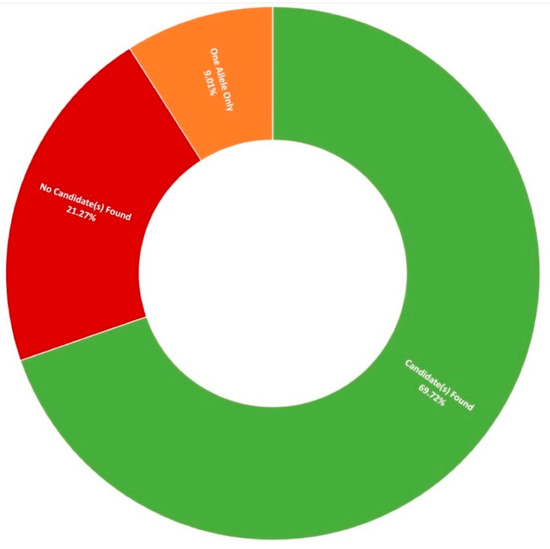
Figure 2.
Diagnostic yield rates for 710 Target 5000 pedigrees utilising target capture next-generation sequencing of the exonic regions of over 250 genes and previously identified pathogenic intronic variants (Table S3).
Table 2.
List of 19 novel missense variants discovered in Target 5000. This table does not include novel structural variants reported below. To the best of our knowledge, mutations listed here have not been previously associated with an IRD. American College of Medical Genetics and Genomics (ACMG) guidelines have been applied to all variants [21], with modifications made in accordance with recommendations by Romanet et al. [23]. (S): reclassified as strong evidence. VUS: Variant of unknown significance.
3.2. Retinitis Pigmentosa
The most prevalent clinical presentation was retinitis pigmentosa (RP; MIM: 268000), accounting for 37.75% (379/1004) of all recruited pedigrees (Figure 1). These figures incorporate various phenotypic presentations, including atypical, inverse, and paravenous RP. The total number of pedigrees analysed and clinically reported as having a family history of RP was 184. Of these, 131 have been genetically resolved. The total number of pedigrees analysed and clinically reported as having simplex RP was 79. 48 of these pedigrees have been genetically resolved. This gives a genetic diagnosis rate of 71% and 61%, respectively. Of these resolved cases, 81 exhibited an autosomal dominant inheritance pattern, 71 exhibited an autosomal recessive inheritance pattern, and 27 exhibited an X-linked inheritance pattern. Autosomal dominant was the most common inheritance pattern, accounting for over 45% (81/178) of all sequenced RP pedigrees in this cohort (Figure 3). This figure is larger than reported in other studies [28]. Variants in RHO (MIM: 180380) were the most common candidate variants for RP in this cohort, accounting for over 14% (26/178) of all sequenced RP pedigrees, and over 32% (26/81) of dominant pedigrees alone. It is important to note that there is likely an even higher prevalence of RHO-linked RP in the Irish IRD cohort, as many pedigrees involved in single-gene studies were excluded from this study due to the discovery of a causative variant previously [15,27]. Of these variants, namely c.533A>G (p.Tyr178Cys), c.541G>A (p.Glu181Lys), and c.620T>G (p.Met207Arg) collectively account for candidate RHO mutations across 13 different pedigrees to date.
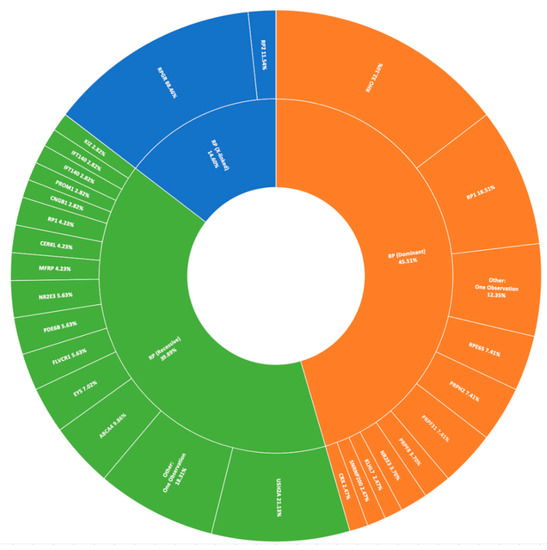
Figure 3.
A summary of the genetic architecture of retinitis pigmentosa (RP) in Target 5000 participants (Table S4).
The most commonly observed candidate gene for autosomal recessive RP was USH2A (MIM: 608400). Two variants in this gene were found in over 21% (15/71) of pedigrees with this phenotype (Figure 3). As variants in USH2A are also the primary cause of Type II Usher Syndrome, it is important to note that participants included in this section presented with recessive RP without syndromic disease manifestation. Non-syndromic USH2A-linked RP has been consistently reported in other IRD cohorts [29]. Interestingly, a proportion of recessive RP pedigrees (>9%) (7/71) carry variants in ABCA4, typically associated with Stargardt Disease, a form of macular dystrophy. In the Target 5000 patient cohort, it has become apparent that an atypical RP phenotype appears to be caused by variants in ABCA4, as previously described in other cohorts (Figure 4 and Figure 5) [10,30].
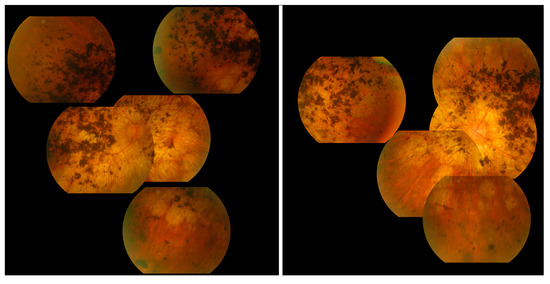
Figure 4.
Collage of fundus images of a patient initially presenting with a Stargardt phenotype that progressed to phenotype suggestive of retinitis pigmentosa. Extensive pigmentation can be seen at both maculae and also in the periphery of both eyes (left—right eye, right—left eye). There are also distinct punched-out areas in the pigment epithelium bilaterally.
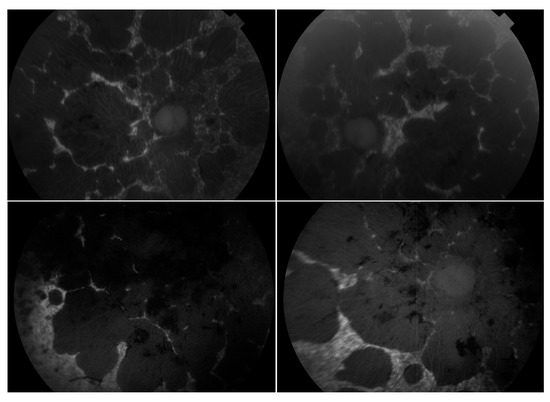
Figure 5.
Montage of autofluorescence fundus images of the patients initially presenting with a Stargardt phenotype that progressed to phenotype suggestive of retinitis pigmentosa. The right eyes are shown in the left column; the left eyes are shown in the right column. Patient example 1 is the first row and patient example 2 is the second row. All eyes demonstrate a dark choroid and large nummular regions of hypoautofluorescence throughout the retinae.
Pathogenic variants in RPGR (MIM: 312610) were the most frequent cause of X-Linked RP, explaining over 88% (23/26) of sequenced pedigrees (Figure 3). Employment of a bespoke sequencing strategy of ORF15 has enhanced the success of sequencing this region, which typically presents a diagnostic challenge due to low coverage and poor variant detection [19].
3.3. Stargardt Disease and Other Macular Dystrophies
Stargardt disease (STGD1) is the second largest phenotypic presentation in this study (Figure 1). As mentioned previously, variants in ABCA4 [31] were the most common cause, accounting for over 97% (109/112) of resolved STGD1 pedigrees in this cohort (Figure 6). Six potentially novel coding variants in ABCA4 were detected as part of this study, NM_000350.2: c.1865G>A (p.Ser622Asn), c.223T>C (p.Cys75Arg), c.4468T>C (p.Cys1490Arg), c.5329A>G (p.Met1777Val), c.5351T>C (p.Leu1784Pro), and c.6743T>C (Phe2248Ser). All of these variants, with the exception of c.5351T>C (p.Leu1784Pro), were observed in individual pedigrees where another known pathogenic ABCA4 variant was also observed. Variant c.5351T>C (p.Leu1784Pro) was observed in three individuals of two not knowingly related families also carrying a known pathogenic variant. The probands of these pedigrees do not appear to share any other rare variants in common in the regions captured.
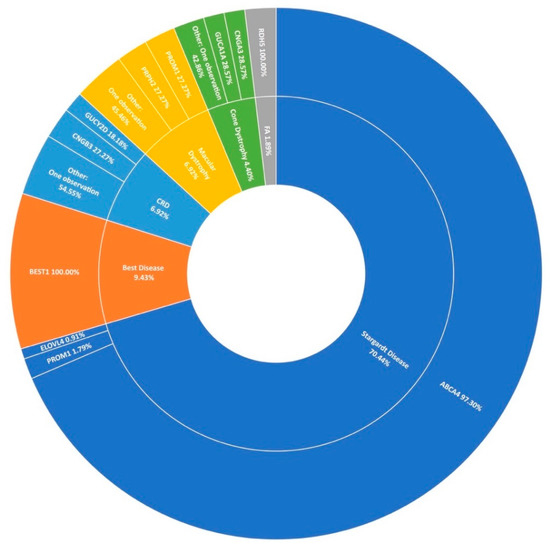
Figure 6.
A summary of the genetic architecture of macular dystrophies in Target 5000 participants. CRD: Cone-Rod Dystrophy FA: Fundus Albipunctatus (Table S5).
It is becoming increasingly clear that intronic variants play a major role in the causation of STGD1. Nine pathogenic intronic variants were included in the capture panel of this study and found in 26 cases in 20 pedigrees with another ABCA4 variant. In recent years, single-molecule molecular inversion probe (smMIP) based sequencing of the whole ABCA4 gene including intronic sequences has dramatically increased positive candidate detection rates for STGD1 [32,33]. Thirty-six unresolved probands from Target 5000 presenting as STGD1 or cone-rod dystophy included in this cohort were analysed as part of a landmark study on the genetic landscape of ABCA4 in over 1000 probands, with a positive candidate detection rate of 44%. (16/36) [13]. This has increased the rate at which ABCA4 is identified as the causal gene of STGD1 in this cohort (Figure 6). Of note from Irish participants in this study, 5 individuals were found to carry c.4539+2028C>T (p.[=,Arg1514Leufs*36]) in trans with another pathogenic ABCA4 variant. Upon retrospective analysis, 5 further incidences of this variant were detected in previously unresolved individuals, totaling 10 participants recruited by Target 5000 carrying this variant and another pathogenic ABCA4 variant, suggesting significant enrichment in the Irish STGD1 population.
In agreement with previous reports on Target 5000 [15,27], ABCA4 c.5603A>T (p.Asn1868Ile) is a significant causal variant of a milder form of STGD1 in this IRD cohort. Of 110 STGD1 pedigrees with 2 positive candidate variants identified, this variant was observed 14 times. In addition, 6 STGD1 patients were found to carry this variant homozygously. ELOVL4 and PROM1 were determined to be the cause of STGD1 disease in 2.7% (2/112) of sequenced pedigrees. Other maculopathies that presented prominantly include best disease accounting for 9.4% (15/159) of macular dystrophies in this study, cone-rod dystrophy, and a general macular dystrophy phenotype, each accounting for almost 7% (11/159) of all maculopathy pedigrees (Figure 6). However, it is clear from Figure 6 that STGD1 is the predominant cause of macular dystrophy in Target 5000 participants.
3.4. Usher Syndrome
Usher syndrome is the most common manifestation of syndromic-IRD in this cohort, accounting for over 7% (78/1004) of all clinical presentations in Target 5000 (Figure 1). Usher syndrome is a form of ciliopathy characterised by RP and sensorineural hearing loss. It is generally divided into sub-groups based on severity of symptoms, ranging from most severe in Type 1 to least severe in Type 3. In total 57 pedigrees were genetically diagnosed with Usher syndrome, where 2 candidate variants were detected, with Type 2 being the most common presentation. Seventy-eight pedigrees were clinically diagnosed with Usher syndrome, yielding a positive candidate detection rate of 73% (57/78). Usher type 2 is by far the predominant sub-group in the Target 5000 cohort (41/57), most frequently caused by variants in USH2A (28/41) (Figure 7). Variants in MYO7A were the most frequent cause of Usher type 1 (10/14). Variants in CLRN1 and MTTS2 (1/2) contributed equally to cases of Usher Type 3 in this study, with one individual carrying a novel candidate variant in CLRN1 (Table 2).
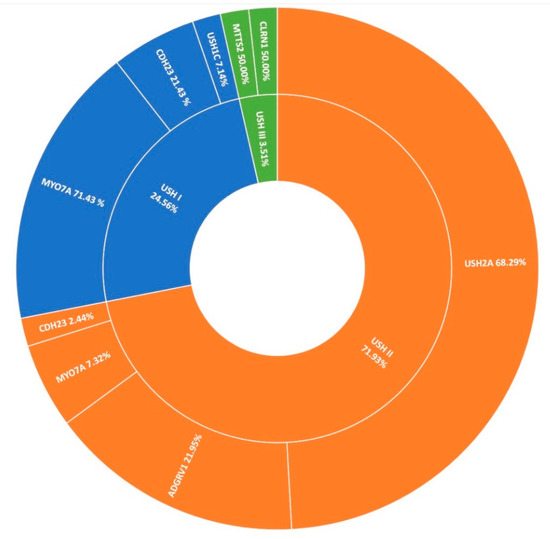
Figure 7.
A summary of the genetic architecture of Usher Syndrome (USH) in Target 5000 participants (Table S6).
3.5. Other IRDs Encompassed by Target 5000
The second most frequent syndromic IRD clinically diagnosed in this study was Bardet-Biedl syndrome (BBS), typically characterised by polydactyly, intellectual disability, and obesity. Over 2% of pedigrees (21/1004) in the entire cohort present with BBS at the clinic (Figure 1); however, this is thought to be an under-representation, as some of those diagnosed with simplex RP are later found to have had polydactyly removed as children. These individuals are re-diagnosed with BBS following detection of two candidate variants in a BBS-associated gene and confirmation with Target 5000 affiliated ophthalmologists. In total, 14 pedigrees received a genetic diagnosis of BBS having been initially clinically diagnosed with RP. Variants in BBS1 were the most frequent cause of this syndrome accounting for over 71% (23/32) of sequenced pedigrees. As reported previously, the most common variant is c.1169G>T (p.Met390Arg) [15]. This variant has been detected 28 times homozygously in 22 different pedigrees in this cohort. The second most frequent cause of BBS were variants in BBS10 (4/32), one of which is believed to be a novel variant (Table 2). BBS causality is followed by SDCCAG8 (n = 2), BBS9, BBS4, and TTC8 (n = 1 each) in the Target 5000 cohort (Figure 8).
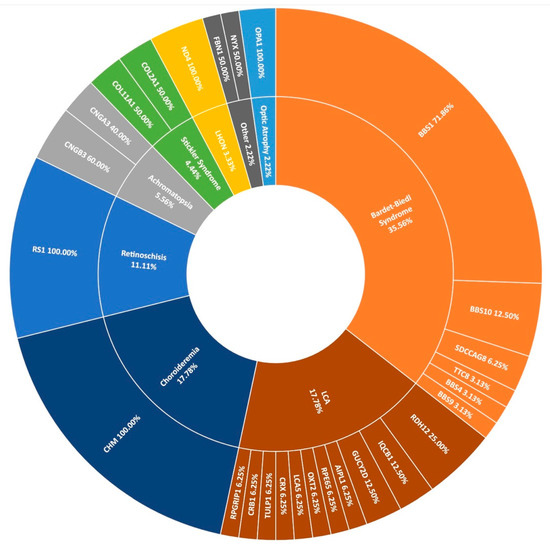
Figure 8.
A summary of the genetic architecture of less common phenotypes encountered in the Target 5000 study. LCA: Leber Congenital Amaurosis, LHON: Leber Hereditary Optic Neuropathy (Table S7).
Many other rare forms of IRD are also included in the Target 5000 study. Sixteen pedigrees with a diagnosis with Leber congenital amaurosis (LCA) have been genotyped with a spectrum of genes implicated as positive candidates (Figure 8). Other rare IRDs examined in this study include retinoschisis (n = 10), achromatopsia (n = 5), Stickler syndrome (n = 4), Leber hereditary optic neuropathy (n = 3), and optic atrophy (n = 2), among others. In total, 90 pedigrees with positive candidate variants are encompassed by Figure 8, illustrating the wide spectrum of disease presentation included in Target 5000.
3.6. Novel Variants
Here we present 19 novel missense variants that, to the best of our knowledge, have not yet been associated with an IRD (Table 2). In silico tools including MetaLR, M-CAP and REVEL were utilised to predict the pathogenicity of these variants. Segregation analysis was carried out when possible and was used as evidence according to the American College of Medical Genetics and Genomics (ACMG) guidelines where appropriate [21]. Two of the variants listed here, BBS10 c.155G>A (p.Gly52Asp) and PRPH2 c.464C>T (p.Thr155Ile), had been allocated dbSNP IDs; however, this is likely due to their detection in population sequencing studies, such as gnomAD [34], where they have not previously been associated with an IRD. Ten of these variants have been classified as variants of unknown significance (VUS) although having strong in silico predicted pathogenicities. This highlights the importance of functional analysis in vitro and in vivo in order to confidently call such variants likely pathogenic.
3.7. RPE65
In Ireland, the prevailing RPE65 phenotype has been that associated with the specific variant, (NM_000329.2) c.1430A>G (p.Asp477Gly). The disease phenotype is comparatively much milder than RPE65-LCA and closely resembles the clinical manifestations of choroideremia. This amino acid position has been shown to be highly conserved across multiple species [35]. This variant is one that continues to be detected in additional cases in the ongoing Target 5000 study. Initial reports and phenotype characterisation of this mutation were based on several pedigrees originating from Ireland [35,36]. The dominant c.1430A>G (p.Asp477Gly) variant remains to be probed for, yet is undetected, in large RPE65 screening studies (n > 2000) of non-Irish ethnicity [37]. Currently in the Target 5000 cohort, there are 23 genetically confirmed affected individuals, with many patients from these pedigrees that are pending recruitment. These 23 affected patients span 7 pedigrees although likely originate from a single source as observed in previous studies [38]. An example of segregation of the c.1430A>G (p.Asp477Gly) variant in one such pedigree is shown in Figure 9.
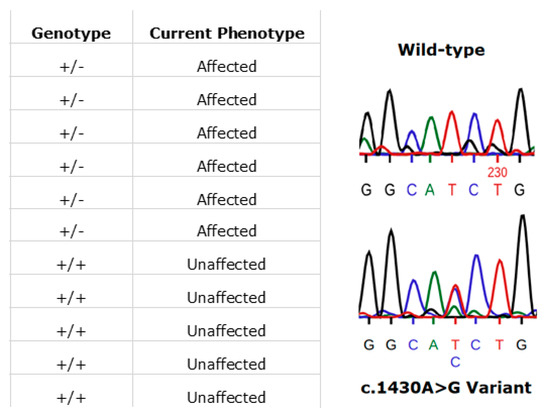
Figure 9.
An illustration of patients harbouring the RPE65 c.1430A>G (p.D477G) variant. In this pedigree, each individual’s phenotype corresponds to the expected genotype (left image). Also shown is a representative Sanger sequence trace from an unaffected (wild-type) family member (right image, top). The variant sequence shown below is that from an affected family member (right image, bottom). Sequences were read from the sense strand so the observed change above is T>G (GGCA[T>G]CTG).
3.8. FLVCR1
Typically, variants in FLVCR1 have been associated with a neurological syndrome, posterior column ataxia with retinitis pigmentosa (PCARP; MIM: 609033) [39,40,41], and more recently a specific splice variant (c.1092 + 5G>A) has been reported multiple times to be associated with non-syndromic RP [42,43,44]. Through Target 5000, substantial evidence has been obtained that suggests the first incidence of a protein coding FLVCR1 variant c.1022A>G (p.Tyr341Cys) implicated in non-syndromic RP [45]. RP is the most common clinical diagnosis for participants in the Target 5000 study, where the clinical diagnosis of RP accounts for nearly 40% (379/1004) of total pedigrees enrolled to date (Figure 1). Patients with this variant present with typical RP, without any extraocular features (Figure 10). Since its initial detection, this variant has been observed and deemed to segregate with the condition in three additional not knowingly related families in the Target 5000 cohort. Each of the genotyped affected individuals in these three pedigrees are homozygous for this variant.
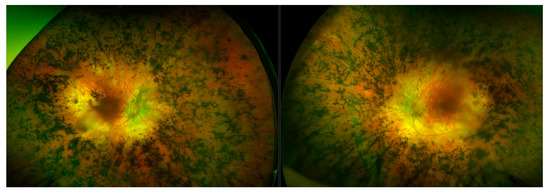
Figure 10.
Montage of wide-field fundus images from a retinitis pigmentosa patient with a FLVCR1 genotype. The FLVCR1 genotype results in a phenotype (left- right eye, right- left eye) that appears to bear a resemblance to classical retinitis pigmentosa, with features such as masses of bony spicules in the periphery, attenuated blood vessels, and waxy disc pallor. There is also a relative preservation of the maculae.
3.9. Choroideremia
Choroideremia is an X-Linked recessive chorioretinal degenerative condition with progressive atrophy of various retinal cell types and the surrounding blood retinal barrier. Here we describe a novel deletion in the CHM gene found in two Irish pedigrees. This nearly 500 kb deletion represents the largest as yet detected IRD-associated gene deletion in Ireland (Figure 11). Two members of a large X-linked Retinitis Pigmentosa pedigree (Figure 12) clinically presented with choroideremia and tested negative for the segregating RPGR variant found in other affected members of this pedigree. Both males were analysed with target capture sequencing and found to possess large deletions spanning the CHM gene, approximating 500 kb. The observation of two IRDs in this pedigree highlights the significant value of NGS-based diagnostics for IRDs (Figure 12).
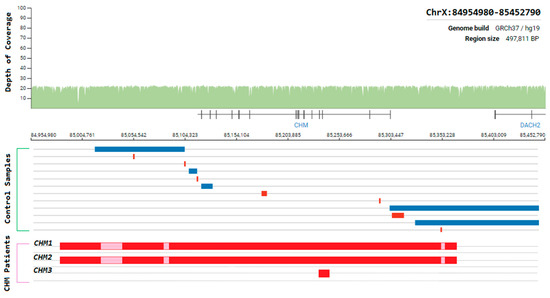
Figure 11.
Schematic representation of the genomic region surrounding the CHM gene. Whole-genome coverage data from a control population database (https://gnomad.broadinstitute.org/). Structural variants detected from controls samples and Target 5000 patients are aligned to this region to illustrate the instability of this genomic region. Blue = duplication, Red = deletion, Pink = template positive results likely due to highly similar sequence elsewhere in the genome.
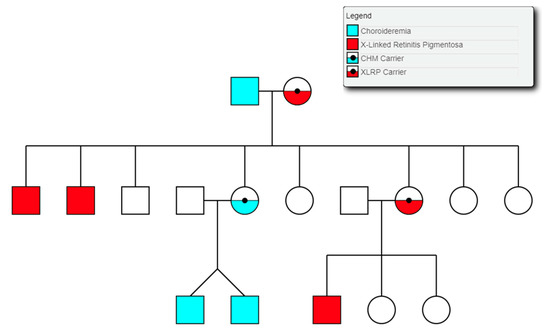
Figure 12.
Partial pedigree tree illustrating the segregation of two X-linked retinal conditions within one family.
The same CHM deletion has also been detected in a second Irish pedigree since its initial discovery. Two additional males and two carrier females from this second pedigree were all found to be affected with progressive choroideremia. In this pedigree, the incidence of choroideremia could be traced back 5 generations, 4 of which have been assessed by the clinical team. A third Irish pedigree with a large CHM deletion was also detected previously as part of Target 5000 [27]. This mutation spanned approximately 6.5 kb and encompassed exons 3 and 4 of CHM (Figure 11). In each instance, target capture NGS detected the presence of the deletion and breakpoints were approximated by tiled PCR analyses. Interestingly, some probing PCRs in the intergenic regions surrounding CHM suggested that genomic DNA was present between deleted regions. Upon analysis, these PCR products were likely the result of large regions of homology that exist within this genomic area and other regions of the genome. Several structural variants in this CHM-proximal intergenic region have also been observed in control samples (Figure 11).
4. Discussion
Up to now, over 1000 individuals have been genotyped as part of Target 5000, accounting for over 20% of the estimated Irish IRD cohort. The results of this study thus far highlight the unique genetic architecture of IRDs in Ireland, cumulatively resulting in over 89 novel variants identified to date [15,27], 19 of which were missense mutations in addition to 1 novel structural variant identified in the most recent analysis (Table 2, Figure 11). With a positive candidate detection rate of almost 70% (495/710 pedigrees), Target 5000 highlights the value of sequencing the exons of 254 IRD-associated genes as well as some known pathogenic intronic regions. This is consistent with other sequencing studies of various IRD cohorts, which report a diagnostic yield of between 38–75% [17,46,47,48,49]. This range can be expected due a number of factors. Firstly, deep-phenotyping to accurately categorise IRDs greatly assists with genotype-phenotype assessment. Assessor variability will exist between clinics and studies, contributing greatly to the variability of diagnostic yield. Secondly, there are differences between the numbers of genes and regions sequenced in these cohorts, where smaller gene panels will not capture some of the known genes associated with IRDs [46]. Lastly, the employment of additional analyses such as WES or WGS in other studies has increased the detection of candidate variants in previously unresolved cases [18,49,50,51]. The candidate variant detection rate of Target 5000 demonstrates that target capture sequencing is a cost and time-effective first-tier approach for genetic screening of those affected by IRDs in Ireland.
This study also highlights the importance of an accurate genetic diagnosis in addition to a clinical diagnosis. Genetic diagnoses continue to become more relevant as gene-based medicines move towards the forefront of IRD treatment given a first approved gene therapy for an IRD and an array of gene therapies for other forms of IRD in clinical and preclinical development. Furthermore, genetic diagnoses facilitate a better understanding of disease progression and manifestation for both patients and clinicians.
Stargardt disease (STGD1) is an autosomal recessive disorder caused almost exclusively by variants in the ATP-binding cassette subfamily A member 4 (ABCA4) gene (MIM: 601691) [52]. It is the most frequent form of macular dystrophy with an estimated prevalence of approximately 1 in 10,000 individuals [53]. STGD1 is characterised by progressive bilateral central vision loss, colour vision defects, delayed dark adaptation, and flecks in the retinal pigmentary epithelium [54]. A spectrum of disease severity underlies STGD1, with age of onset and progression governed by the specific combination of variants an individual carries, highlighting again the value of a specific genetic diagnosis for disease progression and risk assesment. An extensive list of 5962 likely pathogenic ABCA4 variants in 3928 cases was published in 2017 [55], providing a valuable resource to aid in the interpretation of variants discovered as part of this study. In addition to coding variants captured, it has become increasingly clear that many of the unresolved one allele only (Figure 2) cases may likely carry a pathogenic intronic variant where the disease is typically associated with a recessive inheritance pattern. This has been elegantly demonstrated by deep intronic variants in ABCA4 causing STGD1, such as c.4539+2028C>T (p.[=,Arg1514Leufs*36]). This variant has been observed as a candidate in 10 Target 5000 individuals, of which 5 were disovered as part of a landmark study of ABCA4-linked STGD1 employing smMIPs based whole gene sequencing [13] and 5 of which were discovered through target capture sequencing runs. This highlights a significant enrichment of this variant in the Irish IRD patient cohort. c.4539+2028C>T (p.[=,Arg1514Leufs*36]) was previously functionally analysed in photoreceptor precursor cells, and was shown to result in a 345 nucleotide pseudoexon inclusion due to strengthening of exonic splice enhancers. It is notable that such variants are an attractive target for antisense oligonucleotide-based splice correction therapy [56].
Identified variants proximal to the RHO p.Met207 region appear predominant in our sub-cohort of autosomal dominant RP patients, as reported in Section 3.2. Amino acids in this region of the protein form part of the fifth transmembrane domain of rhodopsin. This domain has been shown to be vital to retinal binding in bovine rhodopsin [57]. It is possible that undesirable changes in the protein folding of this region result in steric hindrance that prevents the apoprotein from successfully binding retinal. This impaired binding capacity may possibly underpin the reason for the prevalence of pathogenic variants found in the coding region of this protein domain.
Feline leukaemia virus subgroup C cellular receptor 1 (FLVCR1; MIM: 609144) is a transmembrane protein involved in erythropoiesis and heme transport [58]. FLVCR1 was first discovered for its role in aplastic anaemia in domestic cats and subsequently erythroblast destruction in vitro [59]. This was also the first study that theorised that FLVCR1 was a receptor for an organic anion and identified its component domains as strikingly similar to other ancient Major Facilitator Superfamily (MFS) members. In a later study it was shown this receptor protein provided the first description of a mammalian heme transporter [60]. The same group proceeded to investigate its effect in knockout mouse models, which resulted in embryonic lethality [58]. Candidate variant detection in FLVCR1 has significantly increased in recently recruited pedigrees. It is notable the c.1022A>G (p.Tyr341Cys) variant is only the second disease-associated variant to be found in FLVCR1 associated with RP without posterior column degeneration [43]. Moreover, it is the first protein coding variant found in this gene to be affiliated with non-syndromic RP [45]. There were no extraocular features associated with this variant (Figure 10). It is possible that the distinct families in this cohort with this variant share a common ancestry. However, they do not share any rare variants in the regions captured and originate from distinct geographical locations in Ireland. The age of onset for the symptoms of ataxia in PCARP has been reported as typically in the third decade of life. Of note in the Target 5000 cohort, all patients carrying this variant have reached this age, with the oldest patient currently in their seventh decade of life. It is unclear yet as to whether the disease pathology associated with this variant will remain completely non-syndromic throughout the entirety of a patient’s lifespan or will result in a milder, later onset of additional symptoms compared to other pathogenic variants found in this gene. Fortunately, the small size of this gene (2.6 kb) in principle makes it suitable for inclusion in AAV vectors for use as a gene therapy. Such a therapy may help to alleviate the toxic effects of intracellular free-heme [39]. The serotype of AAV could be chosen based on the presence or absence of systemic phenotypes. For example, if only the retina was to be targeted, an AAV 2/5 or 2/8 might be effective serotypes; on the other hand, if the whole central nervous system was to be targeted, then AAV9 or AAVB1 would be more valuable [61]. Many additional bespoke serotypes are now emerging as a result of site-directed evolution of AAV serotypes [62,63,64,65]. Given the recent results here on the role of FLVCR1 in this form of non-syndromic RP, FLVCR1 along with other IRD genes, becomes an increasingly interesting candidate for exploration of AAV-mediated gene therapies.
The RPE65 enzyme is active in the retinoid cycle that recycles retinoids in the RPE, which are utilised by photoreceptor cells. Mutations in the RPE65 gene were initially found to be causative of some cases of autosomal recessive LCA, a severe and clinically distinct IRD [66,67,68]. This was subsequently expanded to include a milder, dominant form of retinopathy and more recently, to include less severe autosomal recessive retinopathies [35,37]. Luxturna, the first ocular gene therapy approved by the FDA, is for biallelic RPE65 retinal disease [16]. RPE65 c.1430A>G (p.Asp477Gly) remains the predominant RPE65 genotype observed in the Irish population. This variant has since been detected in other population studies alongside the clinical presentation of similar phenotypes, largely described as a choroideremia phenocopy [36,38]. However, until recently the exact disease mechanism caused by this variant has evaded researchers. It was initially believed that this variant produced an abnormal protein structure that may affect enzymatic function [35]. More recently, it has been shown in a mouse model that although this variant is not located in a critical functional domain, the variant induces sufficient change to alter the physiochemical properties of the p.D477 loop to produce an aggregation-prone surface. This surface gains the aberrant function of enabling abnormal protein-protein interactions [69]. One hypothesised type of interaction is with ubiquitin ligases, which would likely result in proteasomal degradation as seen in several other RPE65 mutations [70]. This theory may support the variability observed phenotypically as severity might then become dependent on several other factors such as ubiquitination rates and proteasome response. It is of note that aberrant RNA splicing has been shown to have a major pathogenic effect in a knock-in mouse model with this variant [71]. Given the success of the gene therapy treatment of biallelic RPE65 retinopathies with Luxturna [16], an AAV-based therapy, it is interesting to speculate whether the therapy might have any utility for monoallelic dominant RPE65 cases.
The CHM gene hosts the largest collection of pathogenic structural variants detected in any IRD-related gene as part of the Target 5000 study to date. The presence of several structural variants in the general population may also indicate a natural instability in this genomic region. Additionally, female carriers of CHM mutations typically show mild stationary signs with no symptoms, while males are severely affected. In this instance, some females were more severely affected than expected with advanced signs of degeneration and progressive visual decline. One female carrier showed radial pigmentation at the retinal pigment epithelium level and scattered choroidal atrophy, whereas another showed more marked reactive pigmentation and extensive symmetrical atrophy of the choroid and outer retina. These manifestations were apparent both anatomically and functionally. A more severe phenotype was also associated with whole CHM gene deletion in a previous genotype-phenotype choroideremia study [72]. However, in this study, the phenotype was described as choroideremia plus, due to additional syndromic features. These additional symptoms, such as deafness, were attributed to the extent of the deletion encompassing several surrounding genes, which is not the case for the whole gene deletion patients described here.
Going forward with the Target 5000 study, more efforts will be focused on the detection of additional variants in unresolved IRD cases and also in the functional assessment of variants of unknown significance identified thus far. In the age of budding emerging gene therapies, it has never been more important to stringently identify the pathogenic variants that underpin a condition. It is therefore vital that the variants located outside of exonic regions causing disease pathologies are detected, and that these participants are also considered for treatment, should an applicable gene therapy become available in the future. A whole exome or whole genome approach will be more extensively employed in the future to increase the identification of novel variants. Recent studies have highlighted that molecular diagnostic yield is increased when WGS is employed by comparison with other methods [51]. It is expected that for many of the currently unresolved cases, pathogenic variants may be present in deep-intronic, promoter, enhancer regions or in genes that are not yet associated with an IRD [73,74]. Recent studies such as that in STGD1, highlighting the relative prevalence of deep intronic mutations in the ABCA4 gene, attest to the need for more extensive sequence analysis [13]. These regions have been very poorly covered in patients screened by target capture protocols, unless specifically designed to do so. Additionally, WGS data will offer higher resolution in terms of data associated with copy number abnormalities and large structural rearrangements. It has been reported that there are over 1300 CNVs associated with IRD genes reported in the literature [75]. Exon-based target capture methods have a significantly reduced capacity to detect CNVs and SVs. This is due in part to the range of read-depth at captured regions and the inability to detect breakpoints that occur outside of captured loci. Notably, similarly targeted methods, such as non-WES/WGS studies, which have specifically probed intronic and exonic regions of genes have been shown to successfully detect large structural variants [13]. This illustrates that the enormous data volumes generated by WGS studies may be surplus to requirement if the SVs/CNVs to be searched for are relevant to a small genomic region.
Table 2 illustrates that many variants with seemingly strong evidence of pathogenicity may still be classified as VUS. Variants that are currently of unknown significance likely to alter enzyme function, such as those in RPE65 [76], may be applicable to assessment in vitro by employing enzyme function assays. Alternatively, if predicted to perturb splicing having undergone in silico interrogation [77], these variants could be assessed by midigene analysis. Midigene analysis refers to the incorporation of variants of interest into vectors containing several exons of the relevant IRD gene, enabling the in vitro interrogation of the effect(s) of that variant directly at an RNA level. This approach is particularly effective for testing exonic and intronic variants that are likely to disrupt correct splicing, as these variants will have a distinct impact on the RNA product when compared to the wild-type equivalent. This method is also particularly useful for the interpretation of variants that are located within introns, yet outside of canonical splice sites, and additionally to examine rare synonymous exonic variants proximal to exon junctions [32,78].
A viable in vivo strategy for the assessment of novel missense variants is to introduce these VUS into model organisms with similar or conserved genes. The significant advantages of such model organisms is the ability to rapidly generate models of disease [79]. This is particularly relevant currently, as recent advances in genome editing technologies has made this approach more readily accessible and successful than ever before [80]. Although not all model organisms fully represent the visual system that exists in humans, many organisms can offer useful information ranging from protein mislocalisation [81] to optokinetic response [82]. By introducing a VUS into the appropriate system, additional evidence can be gained to better evaluate the functional impact of the VUS in human ocular pathologies.
Thus far, genetic analysis of IRD patients has helped to resolve ambiguous phenotypes and to identify causative mutations in nearly 70% (495/710) of sequenced pedigrees. The continuous expansion of our cohort has enabled us to better interrogate the sequencing data and interpret the potential pathogenicity of novel variants when detected. In addition to this, the growing body of data from NGS studies of IRDs globally should facilitate better correlations between genotype and phenotype and further refine methods for diagnoses and prognoses. Furthermore, whole gene and WGS analyses are highlighting the significant role of non-coding variants as causative of some IRDs. Target 5000 aims to provide actionable outcomes empowering patients with genetic diagnoses and potentially future access to clinical trials or approved treatments where appropriate. Given the rapid development of the field of ocular therapeutics, it is clear that genetically characterising those affected by an IRD has become a diagnostic imperative of the utmost importance.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4425/11/1/105/s1, Table S1: Primers used to probe exonic regions of CHM (MIM: 300390; ChrX: 85861180-86047558) and the neighbouring genes, POF1B (MIM: 300603; ChrX: 85277396-85379709) and DACH2 (MIM: 300608; ChrX: 86148451-86832602) as well as the intergenic space between. Table S2: Tabular summary of the clinical presentation of 1004 recruited Target 5000 pedigrees. RP: Retinitis Pigmentosa, FFM: Fundus Flavimaculatus, MD: Macular Dystrophy, CRD: Cone-Rod Dystrophy, RCD: Rod-Cone Dystrophy, LCA: Leber Congenital Amaurosis, EOSRD: Early-Onset Retinal Degeneration, BBS: Bardet-Biedl Syndrome, CSNB: Congenital Stationary Night Blindness, Table S3: Tabular summary of the diagnostic yield rates for 710 Target 5000 pedigrees utilising target capture next-generation sequencing of the exonic regions of over 250 genes and previously identified pathogenic intronic variants, Table S4: Tabular summar of the genetic architecture of retinitis pigmentosa (RP) in Target 5000 participants, Table S5: Tabular summary of the genetic architecture of macular dystrophies in Target 5000 participants. CRD: Cone-Rod Dystrophy FA: Fundus Albipunctatus, Table S6: Tabular summary of the genetic architecture of Usher Syndrome (USH) in Target 5000 participants, Table S7: Tabular summary of the genetic architecture of less common phenotypes encountered in the Target 5000 study. LCA: Leber Congenital Amaurosis, LHON: Leber Hereditary Optic Neuropathy, Figure S1: The available age distribution of recruited Target 5000 participants, Figure S2: Sequencing coverage from three affected X-linked retinitis pigmentosa individuals (H41, P1 and K1) from three distinct pedigrees as seen on IGV software. Alligned to this region is sequencing primers (R4,R7,R8,R9,F10 and R5) and PCR primers (F3,R6 and P9) as shown by forward (blue) and reverse (red) orientations.
Author Contributions
Conceptualization, P.H., D.K., P.K. and G.J.F.; Data curation, L.W. and A.D.; Formal analysis, L.W. and A.D.; Funding acquisition, G.J.F.; Investigation, L.W., A.D., N.W., J.Z., K.S., G.S., J.T., J.J.O., D.K., P.F.K. and G.J.F.; Methodology, A.D., M.C. and G.J.F.; Project administration, G.J.F.; Resources, G.J.F.; Software, A.D. and M.C.; Supervision, A.D., M.C. and G.J.F.; Validation, L.W. and A.D.; Visualization, L.W. and A.D.; Writing—original draft, L.W. and A.D.; Writing—review & editing, L.W. and A.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by grant awards from Fighting Blindness Ireland (FB Irl; FB16FAR, FB18CRE), The Health Research Board of Ireland (HRB; POR/2010/97) in conjunction with Health Research Charities Ireland (HRCI; MRCG-2013-8, MRCG-2016-14), the Irish Research Council (IRC; GOIPG/2017/1631), and Science Foundation Ireland (SFI; 16/1A/4452).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mei, S.; Huang, X.; Cheng, L.; Peng, S.; Zhu, T.; Chen, L.; Wang, Y.; Zhao, J. A missense mutation in OPA1 causes dominant optic atrophy in a Chinese family. J. Ophthalmol. 2019, 2019, 1424928. [Google Scholar] [CrossRef]
- Gao, F.-J.; Zhang, S.-H.; Chen, J.-Y.; Xu, G.-Z.; Wu, J.-H. Digenic heterozygous mutations in EYS/LRP5 in a Chinese family with retinitis pigmentosa. Int. J. Ophthalmol. 2017, 10, 325–328. [Google Scholar]
- Bhattacharya, S.S.; Wright, A.F.; Clayton, J.F.; Price, W.H.; Phillips, C.I.; McKeown, C.M.; Jay, M.; Bird, A.C.; Pearson, P.L.; Southern, E.M. Close genetic linkage between X-linked retinitis pigmentosa and a restriction fragment length polymorphism identified by recombinant DNA probe L1.28. Nature 1984, 309, 253–255. [Google Scholar] [CrossRef]
- Farrar, G.J.; Kenna, P.; Jordan, S.A.; Kumar-Singh, R.; Humphries, M.M.; Sharp, E.M.; Sheils, D.M.; Humphries, P. A three-base-pair deletion in the peripherin–RDS gene in one form of retinitis pigmentosa. Nature 1991, 354, 478–480. [Google Scholar] [CrossRef]
- Kumar-Singh, R.; Bradley, D.G.; Farrar, G.J.; Lawler, M.; Jordan, S.A.; Humphries, P. Autosomal dominant retinitis pigmentosa: A new multi-allelic marker (D3S621) genetically linked to the disease locus (RP4). Hum. Genet. 1991, 86, 502–504. [Google Scholar] [CrossRef] [PubMed]
- RetNet: Summaries. Available online: https://sph.uth.edu/retnet/sum-dis.htm (accessed on 29 October 2019).
- Werdich, X.Q.; Place, E.M.; Pierce, E.A. Systemic diseases associated with retinal dystrophies. Semin. Ophthalmol. 2014, 29, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.; Di Iorio, E.; Barbaro, V.; Ponzin, D.; Sorrentino, F.S.; Parmeggiani, F. Retinitis pigmentosa: Genes and disease mechanisms. Curr. Genomics 2011, 12, 238–249. [Google Scholar] [PubMed]
- Farrar, G.J.; Carrigan, M.; Dockery, A.; Millington-Ward, S.; Palfi, A.; Chadderton, N.; Humphries, M.; Kiang, A.S.; Kenna, P.F.; Humphries, P. Toward an elucidation of the molecular genetics of inherited retinal degenerations. Hum. Mol. Genet. 2017, 26, R2–R11. [Google Scholar] [CrossRef]
- Cremers, F.P.; van de Pol, D.J.; van Driel, M.; den Hollander, A.I.; van Haren, F.J.; Knoers, N.V.; Tijmes, N.; Bergen, A.A.; Rohrschneider, K.; Blankenagel, A.; et al. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt’s disease gene ABCR. Hum. Mol. Genet. 1998, 7, 355–362. [Google Scholar] [CrossRef]
- Maugeri, A.; Klevering, B.J.; Rohrschneider, K.; Blankenagel, A.; Brunner, H.G.; Deutman, A.F.; Hoyng, C.B.; Cremers, F.P.M. Mutations in the ABCA4 (ABCR) gene are the major cause of autosomal recessive cone-rod dystrophy. Am. J. Hum. Genet. 2000, 67, 960–966. [Google Scholar] [CrossRef]
- Maugeri, A.; van Driel, M.A.; van de Pol, D.J.; Klevering, B.J.; van Haren, F.J.; Tijmes, N.; Bergen, A.A.; Rohrschneider, K.; Blankenagel, A.; Pinckers, A.J.; et al. The 2588G→C mutation in the ABCR gene is a mild frequent founder mutation in the Western European population and allows the classification of ABCR mutations in patients with Stargardt disease. Am. J. Hum. Genet. 1999, 64, 1024–1035. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Cornelis, S.S.; Pozo-Valero, M.; del Whelan, L.; Runhart, E.H.; Mishra, K.; Bults, F.; AlSwaiti, Y.; AlTabishi, A.; Baere, E.D.; et al. Resolving the dark matter of ABCA4 for 1054 Stargardt disease probands through integrated genomics and transcriptomics. bioRxiv 2019, 817767. [Google Scholar] [CrossRef]
- Kim, D.Y.; Mukai, S. X-linked Juvenile Retinoschisis (XLRS): A review of genotype-phenotype relationships. Semin. Ophthalmol. 2013, 28, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Dockery, A.; Stephenson, K.; Keegan, D.; Wynne, N.; Silvestri, G.; Humphries, P.; Kenna, P.F.; Carrigan, M.; Farrar, G.J. Target 5000: Target capture sequencing for inherited retinal degenerations. Genes 2017, 8, 304. [Google Scholar] [CrossRef] [PubMed]
- Maguire, A.M.; Russell, S.; Wellman, J.A.; Chung, D.C.; Yu, Z.-F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; Marshall, K.A.; et al. Efficacy, Safety, and Durability of Voretigene Neparvovec-rzyl in RPE65 Mutation-Associated Inherited Retinal Dystrophy: Results of Phase 1 and 3 Trials. Ophthalmology 2019, 126, 1273–1285. [Google Scholar] [CrossRef]
- Consugar, M.B.; Navarro-Gomez, D.; Place, E.M.; Bujakowska, K.M.; Sousa, M.E.; Fonseca-Kelly, Z.D.; Taub, D.G.; Janessian, M.; Wang, D.Y.; Au, E.D.; et al. Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genet. Med. 2015, 17, 253–261. [Google Scholar] [CrossRef]
- Carss, K.J.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive rare variant analysis via whole-genome sequencing to determine the molecular pathology of inherited retinal disease. Am. J. Hum. Genet. 2017, 100, 75–90. [Google Scholar] [CrossRef]
- Li, J.; Tang, J.; Feng, Y.; Xu, M.; Chen, R.; Zou, X.; Sui, R.; Chang, E.Y.; Lewis, R.A.; Zhang, V.W.; et al. Improved diagnosis of inherited retinal dystrophies by high-fidelity PCR of ORF15 followed by next-generation sequencing. J. Mol. Diagn. 2016, 18, 817–824. [Google Scholar] [CrossRef]
- Boyle, E.A.; O’Roak, B.J.; Martin, B.K.; Kumar, A.; Shendure, J. MIPgen: Optimized modeling and design of molecular inversion probes for targeted resequencing. Bioinformatics 2014, 30, 2670–2672. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Jarvik, G.P.; Browning, B.L. Consideration of cosegregation in the pathogenicity classification of genomic variants. Am. J. Hum. Genet. 2016, 98, 1077–1081. [Google Scholar] [CrossRef] [PubMed]
- Romanet, P.; Odou, M.-F.; North, M.-O.; Saveanu, A.; Coppin, L.; Pasmant, E.; Mohamed, A.; Goudet, P.; Borson-Chazot, F.; Calender, A.; et al. Proposition of adjustments to the ACMG-AMP framework for the interpretation of MEN1 missense variants. Hum. Mutat. 2019, 40, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Jian, X.; Liu, X. In silico prediction of deleteriousness for nonsynonymous and splice-altering single nucleotide variants in the human genome. In Vitro Mutagenesis; Reeves, A., Ed.; Springer: New York, NY, USA, 2017; Volume 1498, pp. 191–197. ISBN 978-1-4939-6470-3. [Google Scholar]
- Chen, X.; Schulz-Trieglaff, O.; Shaw, R.; Barnes, B.; Schlesinger, F.; Källberg, M.; Cox, A.J.; Kruglyak, S.; Saunders, C.T. Manta: Rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 2016, 32, 1220–1222. [Google Scholar] [CrossRef]
- Carrigan, M.; Duignan, E.; Malone, C.P.G.; Stephenson, K.; Saad, T.; McDermott, C.; Green, A.; Keegan, D.; Humphries, P.; Kenna, P.F.; et al. Panel-based population next-generation sequencing for inherited retinal degenerations. Sci. Rep. 2016, 6, 33248. [Google Scholar] [CrossRef]
- Sullivan, L.S.; Bowne, S.J.; Birch, D.G.; Hughbanks-Wheaton, D.; Heckenlively, J.R.; Lewis, R.A.; Garcia, C.A.; Ruiz, R.S.; Blanton, S.H.; Northrup, H.; et al. Prevalence of disease-causing mutations in families with autosomal dominant retinitis pigmentosa: A screen of known genes in 200 families. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3052–3064. [Google Scholar] [CrossRef]
- Lenassi, E.; Vincent, A.; Li, Z.; Saihan, Z.; Coffey, A.J.; Steele-Stallard, H.B.; Moore, A.T.; Steel, K.P.; Luxon, L.M.; Héon, E.; et al. A detailed clinical and molecular survey of subjects with nonsyndromic USH2A retinopathy reveals an allelic hierarchy of disease-causing variants. Eur. J. Hum. Genet. 2015, 23, 1318–1327. [Google Scholar] [CrossRef]
- Klevering, B.J.; Yzer, S.; Rohrschneider, K.; Zonneveld, M.; Allikmets, R.; van den Born, L.I.; Maugeri, A.; Hoyng, C.B.; Cremers, F.P.M. Microarray-based mutation analysis of the ABCA4 (ABCR) gene in autosomal recessive cone-rod dystrophy and retinitis pigmentosa. Eur. J. Hum. Genet. 2004, 12, 1024–1032. [Google Scholar] [CrossRef]
- Allikmets, R.; Singh, N.; Sun, H.; Shroyer, N.F.; Hutchinson, A.; Chidambaram, A.; Gerrard, B.; Baird, L.; Stauffer, D.; Peiffer, A.; et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat. Genet. 1997, 15, 236–246. [Google Scholar] [CrossRef]
- Sangermano, R.; Garanto, A.; Khan, M.; Runhart, E.H.; Bauwens, M.; Bax, N.M.; van den Born, L.I.; Khan, M.I.; Cornelis, S.S.; Verheij, J.B.G.M.; et al. Deep-intronic ABCA4 variants explain missing heritability in Stargardt disease and allow correction of splice defects by antisense oligonucleotides. Genet. Med. 2019, 21, 1751–1760. [Google Scholar] [CrossRef]
- Khan, M.; Cornelis, S.S.; Khan, M.I.; Elmelik, D.; Manders, E.; Bakker, S.; Derks, R.; Neveling, K.; Vorst, M.; van de Gilissen, C.; et al. Cost-effective molecular inversion probe-based ABCA4 sequencing reveals deep-intronic variants in Stargardt disease. Hum. Mutat. 2019, 40, 1749–1759. [Google Scholar] [CrossRef] [PubMed]
- GnomAD. Available online: https://gnomad.broadinstitute.org/ (accessed on 14 November 2019).
- Bowne, S.J.; Humphries, M.M.; Sullivan, L.S.; Kenna, P.F.; Tam, L.C.S.; Kiang, A.S.; Campbell, M.; Weinstock, G.M.; Koboldt, D.C.; Ding, L.; et al. A dominant mutation in RPE65 identified by whole-exome sequencing causes retinitis pigmentosa with choroidal involvement. Eur. J. Hum. Genet. 2011, 19, 1074–1081. [Google Scholar] [CrossRef] [PubMed]
- Hull, S.; Mukherjee, R.; Holder, G.E.; Moore, A.T.; Webster, A.R. The clinical features of retinal disease due to a dominant mutation in RPE65. Mol. Vis. 2016, 22, 626–635. [Google Scholar]
- Li, S.; Xiao, X.; Yi, Z.; Sun, W.; Wang, P.; Zhang, Q. RPE65 mutation frequency and phenotypic variation according to exome sequencing in a tertiary centre for genetic eye diseases in China. Acta Ophthalmol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Jauregui, R.; Park, K.S.; Tsang, S.H. Two-year progression analysis of RPE65 autosomal dominant retinitis pigmentosa. Ophthalmic Genet. 2018, 39, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Chiabrando, D.; Castori, M.; di Rocco, M.; Ungelenk, M.; Gießelmann, S.; Di Capua, M.; Madeo, A.; Grammatico, P.; Bartsch, S.; Hübner, C.A.; et al. Mutations in the heme exporter FLVCR1 cause sensory neurodegeneration with loss of pain perception. PLoS Genet. 2016, 12, e1006461. [Google Scholar] [CrossRef]
- Castori, M.; Morlino, S.; Ungelenk, M.; Pareyson, D.; Salsano, E.; Grammatico, P.; Tolosano, E.; Kurth, I.; Chiabrando, D. Posterior column ataxia with retinitis pigmentosa coexisting with sensory-autonomic neuropathy and leukemia due to the homozygous p.Pro221Ser FLVCR1 mutation. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2017, 174, 732–739. [Google Scholar] [CrossRef]
- Ishiura, H.; Fukuda, Y.; Mitsui, J.; Nakahara, Y.; Ahsan, B.; Takahashi, Y.; Ichikawa, Y.; Goto, J.; Sakai, T.; Tsuji, S. Posterior column ataxia with retinitis pigmentosa in a Japanese family with a novel mutation in FLVCR1. Neurogenetics 2011, 12, 117–121. [Google Scholar] [CrossRef]
- Glöckle, N.; Kohl, S.; Mohr, J.; Scheurenbrand, T.; Sprecher, A.; Weisschuh, N.; Bernd, A.; Rudolph, G.; Schubach, M.; Poloschek, C.; et al. Panel-based next generation sequencing as a reliable and efficient technique to detect mutations in unselected patients with retinal dystrophies. Eur. J. Hum. Genet. 2014, 22, 99–104. [Google Scholar] [CrossRef]
- Yusuf, I.H.; Shanks, M.E.; Clouston, P.; MacLaren, R.E. A splice-site variant in FLVCR1 produces retinitis pigmentosa without posterior column ataxia. Ophthalmic Genet. 2018, 39, 263–267. [Google Scholar] [CrossRef]
- Tiwari, A.; Bahr, A.; Bähr, L.; Fleischhauer, J.; Zinkernagel, M.S.; Winkler, N.; Barthelmes, D.; Berger, L.; Gerth-Kahlert, C.; Neidhardt, J.; et al. Next generation sequencing based identification of disease-associated mutations in Swiss patients with retinal dystrophies. Sci. Rep. 2016, 6, 28755. [Google Scholar] [CrossRef] [PubMed]
- Dockery, A.; Carrigan, M.; Wynne, N.; Stephenson, K.; Keegan, D.; Kenna, P.F.; Farrar, G.J. A novel FLVCR1 variant implicated in retinitis pigmentosa. In Retinal Degenerative Diseases; Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2019; Volume 1185. [Google Scholar]
- Patel, A.; Hayward, J.D.; Tailor, V.; Nyanhete, R.; Ahlfors, H.; Gabriel, C.; Jannini, T.B.; Abbou-Rayyah, Y.; Henderson, R.; Nischal, K.K.; et al. The Oculome panel test: Next-generation sequencing to diagnose a diverse range of genetic developmental eye disorders. Ophthalmology 2019, 126, 888–907. [Google Scholar] [CrossRef] [PubMed]
- Stone, E.M.; Andorf, J.L.; Whitmore, S.S.; DeLuca, A.P.; Giacalone, J.C.; Streb, L.M.; Braun, T.A.; Mullins, R.F.; Scheetz, T.E.; Sheffield, V.C.; et al. Clinically focused molecular investigation of 1000 consecutive families with inherited retinal disease. Ophthalmology 2017, 124, 1314–1331. [Google Scholar] [CrossRef] [PubMed]
- Riera, M.; Navarro, R.; Ruiz-Nogales, S.; Méndez, P.; Burés-Jelstrup, A.; Corcóstegui, B.; Pomares, E. Whole exome sequencing using Ion Proton system enables reliable genetic diagnosis of inherited retinal dystrophies. Sci. Rep. 2017, 7, 42078. [Google Scholar] [CrossRef]
- Sharon, D.; Ben-Yosef, T.; Goldenberg-Cohen, N.; Pras, E.; Gradstein, L.; Soudry, S.; Mezer, E.; Zur, D.; Abbasi, A.H.; Zeitz, C.; et al. A nationwide genetic analysis of inherited retinal diseases in Israel as assessed by the Israeli inherited retinal disease consortium (IIRDC). Hum. Mutat. 2020, 41, 140–149. [Google Scholar] [CrossRef]
- Haer-Wigman, L.; van Zelst-Stams, W.A.; Pfundt, R.; van den Born, L.I.; Klaver, C.C.; Verheij, J.B.; Hoyng, C.B.; Breuning, M.H.; Boon, C.J.; Kievit, A.J.; et al. Diagnostic exome sequencing in 266 Dutch patients with visual impairment. Eur. J. Hum. Genet. 2017, 25, 591–599. [Google Scholar] [CrossRef]
- Ellingford, J.M.; Barton, S.; Bhaskar, S.; Williams, S.G.; Sergouniotis, P.I.; O’Sullivan, J.; Lamb, J.A.; Perveen, R.; Hall, G.; Newman, W.G.; et al. Whole genome sequencing increases molecular diagnostic yield compared with current diagnostic testing for inherited retinal disease. Ophthalmology 2016, 123, 1143–1150. [Google Scholar] [CrossRef]
- Allikmets, R.; Shroyer, N.F.; Singh, N.; Seddon, J.M.; Lewis, R.A.; Bernstein, P.S.; Peiffer, A.; Zabriskie, N.A.; Li, Y.; Hutchinson, A.; et al. Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science 1997, 277, 1805–1807. [Google Scholar] [CrossRef]
- Strauss, R.W.; Ho, A.; Muñoz, B.; Cideciyan, A.V.; Sahel, J.-A.; Sunness, J.S.; Birch, D.G.; Bernstein, P.S.; Michaelides, M.; Traboulsi, E.I.; et al. The natural history of the progression of atrophy secondary to Stargardt disease (ProgStar) studies: Design and baseline characteristics: ProgStar Report No. 1. Ophthalmology 2016, 123, 817–828. [Google Scholar] [CrossRef]
- Fishman, G.A.; Farbman, J.S.; Alexander, K.R. Delayed rod dark adaptation in patients with Stargardt’s disease. Ophthalmology 1991, 98, 957–962. [Google Scholar] [CrossRef]
- Cornelis, S.S.; Bax, N.M.; Zernant, J.; Allikmets, R.; Fritsche, L.G.; den Dunnen, J.T.; Ajmal, M.; Hoyng, C.B.; Cremers, F.P.M. In silico functional meta-analysis of 5,962 ABCA4 variants in 3,928 retinal dystrophy cases. Hum. Mutat. 2017, 38, 400–408. [Google Scholar] [CrossRef]
- Albert, S.; Garanto, A.; Sangermano, R.; Khan, M.; Bax, N.M.; Hoyng, C.B.; Zernant, J.; Lee, W.; Allikmets, R.; Collin, R.W.J.; et al. Identification and rescue of splice defects caused by two neighboring deep-intronic ABCA4 mutations underlying Stargardt disease. Am. J. Hum. Genet. 2018, 102, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Edwards, P.C.; Burghammer, M.; Villa, C.; Schertler, G.F.X. Structure of bovine rhodopsin in a trigonal crystal form. J. Mol. Biol. 2004, 343, 1409–1438. [Google Scholar] [CrossRef] [PubMed]
- Keel, S.B.; Doty, R.T.; Yang, Z.; Quigley, J.G.; Chen, J.; Knoblaugh, S.; Kingsley, P.D.; De Domenico, I.; Vaughn, M.B.; Kaplan, J.; et al. A heme export protein is required for red blood cell differentiation and iron homeostasis. Science 2008, 319, 825–828. [Google Scholar] [CrossRef] [PubMed]
- Tailor, C.S.; Willett, B.J.; Kabat, D. A putative cell surface receptor for anemia-inducing feline leukemia virus subgroup C is a member of a transporter superfamily. J. Virol. 1999, 73, 6500–6505. [Google Scholar] [CrossRef] [PubMed]
- Quigley, J.G.; Yang, Z.; Worthington, M.T.; Phillips, J.D.; Sabo, K.M.; Sabath, D.E.; Berg, C.L.; Sassa, S.; Wood, B.L.; Abkowitz, J.L. Identification of a human heme exporter that is essential for erythropoiesis. Cell 2004, 118, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, S.R.; Fitzpatrick, Z.; Harris, A.F.; Maitland, S.A.; Ferreira, J.S.; Zhang, Y.; Ma, S.; Sharma, R.B.; Gray-Edwards, H.L.; Johnson, J.A.; et al. In vivo selection yields AAV-B1 capsid for central nervous system and muscle gene therapy. Mol. Ther. 2016, 24, 1247–1257. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, A.-K.; Grosse, S.; Börner, K.; Krämer, C.; Wiedtke, E.; Gunkel, M.; Grimm, D. Impact of the assembly-activating protein (AAP) on molecular evolution of synthetic Adeno-associated virus (AAV) capsids. Hum. Gene Ther. 2018, 30, 21–35. [Google Scholar] [CrossRef]
- Tordo, J.; O’Leary, C.; Antunes, A.S.L.M.; Palomar, N.; Aldrin-Kirk, P.; Basche, M.; Bennett, A.; D’Souza, Z.; Gleitz, H.; Godwin, A.; et al. A novel adeno-associated virus capsid with enhanced neurotropism corrects a lysosomal transmembrane enzyme deficiency. Brain J. Neurol. 2018, 141, 2014–2031. [Google Scholar] [CrossRef]
- Smith, J.K.; Agbandje-McKenna, M. Creating an arsenal of Adeno-associated virus (AAV) gene delivery stealth vehicles. PLoS Pathog. 2018, 14, e1006929. [Google Scholar] [CrossRef]
- Paulk, N.K.; Pekrun, K.; Zhu, E.; Nygaard, S.; Li, B.; Xu, J.; Chu, K.; Leborgne, C.; Dane, A.P.; Haft, A.; et al. Bioengineered AAV capsids with combined high human liver transduction in vivo and unique humoral seroreactivity. Mol. Ther. 2018, 26, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Marlhens, F.; Bareil, C.; Griffoin, J.-M.; Zrenner, E.; Amalric, P.; Eliaou, C.; Liu, S.-Y.; Harris, E.; Redmond, T.M.; Arnaud, B.; et al. Mutations in RPE65 cause Leber’s congenital amaurosis. Nat. Genet. 1997, 17, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Marlhens, F.; Griffoin, J.-M.; Bareil, C.; Arnaud, B.; Claustres, M.; Hamel, C.P. Autosomal recessive retinal dystrophy associated with two novel mutations in the RPE65 gene. Eur. J. Hum. Genet. 1998, 6, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Hamel, C.P.; Griffoin, J.-M.; Lasquellec, L.; Bazalgette, C.; Arnaud, B. Retinal dystrophies caused by mutations in RPE65: Assessment of visual functions. Br. J. Ophthalmol. 2001, 85, 424–427. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Choi, E.H.; Suh, S.; Sander, C.L.; Hernandez, C.J.O.; Bulman, E.R.; Khadka, N.; Dong, Z.; Shi, W.; Palczewski, K.; Kiser, P.D. Insights into the pathogenesis of dominant retinitis pigmentosa associated with a D477G mutation in RPE65. Hum. Mol. Genet. 2018, 27, 2225–2243. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Izumi, T.; Hu, J.; Jin, H.H.; Siddiqui, A.-A.A.; Jacobson, S.G.; Bok, D.; Jin, M. Rescue of enzymatic function for disease-associated RPE65 proteins containing various missense mutations in non-active sites. J. Biol. Chem. 2014, 289, 18943–18956. [Google Scholar] [CrossRef]
- Li, Y.; Furhang, R.; Ray, A.; Duncan, T.; Soucy, J.; Mahdi, R.; Chaitankar, V.; Gieser, L.; Poliakov, E.; Qian, H.; et al. Aberrant RNA splicing is the major pathogenic effect in a knock-in mouse model of the dominantly inherited c.1430A>G human RPE65 mutation. Hum. Mutat. 2019, 40, 426–443. [Google Scholar] [CrossRef]
- Simunovic, M.P.; Jolly, J.K.; Xue, K.; Edwards, T.L.; Groppe, M.; Downes, S.M.; MacLaren, R.E. The spectrum of CHM gene mutations in choroideremia and their relationship to clinical phenotype. Investig. Ophthalmol. Vis. Sci. 2016, 57, 6033–6039. [Google Scholar] [CrossRef]
- Bauwens, M.; Garanto, A.; Sangermano, R.; Naessens, S.; Weisschuh, N.; De Zaeytijd, J.; Khan, M.; Sadler, F.; Balikova, I.; Van Cauwenbergh, C.; et al. ABCA4-associated disease as a model for missing heritability in autosomal recessive disorders: Novel noncoding splice, cis-regulatory, structural, and recurrent hypomorphic variants. Genet. Med. 2019, 21, 1761–1771. [Google Scholar] [CrossRef]
- Radziwon, A.; Arno, G.K.; Wheaton, D.; McDonagh, E.M.; Baple, E.L.; Webb-Jones, K.G.; Birch, D.; Webster, A.R.; MacDonald, I.M. Single-base substitutions in the CHM promoter as a cause of choroideremia. Hum. Mutat. 2017, 38, 704–715. [Google Scholar] [CrossRef]
- Van Schil, K.; Naessens, S.; Van de Sompele, S.; Carron, M.; Aslanidis, A.; Van Cauwenbergh, C.; Kathrin Mayer, A.; Van Heetvelde, M.; Bauwens, M.; Verdin, H.; et al. Mapping the genomic landscape of inherited retinal disease genes prioritizes genes prone to coding and noncoding copy-number variations. Genet. Med. 2018, 20, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Uppal, S.; Liu, T.; Poliakov, E.; Gentleman, S.; Redmond, T.M. The dual roles of RPE65 S-palmitoylation in membrane association and visual cycle function. Sci. Rep. 2019, 9, 5218. [Google Scholar] [CrossRef] [PubMed]
- Ellingford, J.M.; Thomas, H.B.; Rowlands, C.; Arno, G.; Beaman, G.; Gomes-Silva, B.; Campbell, C.; Gossan, N.; Hardcastle, C.; Webb, K.; et al. Functional and in-silico interrogation of rare genomic variants impacting RNA splicing for the diagnosis of genomic disorders. bioRxiv 2019, 781088. [Google Scholar] [CrossRef]
- Verbakel, S.K.; Fadaie, Z.; Klevering, B.J.; van Genderen, M.M.; Feenstra, I.; Cremers, F.P.M.; Hoyng, C.B.; Roosing, S. The identification of a RNA splice variant in TULP1 in two siblings with early-onset photoreceptor dystrophy. Mol. Genet. Genomic Med. 2019, 7, e660. [Google Scholar] [CrossRef]
- Dickinson, D.J.; Goldstein, B. CRISPR-based methods for Caenorhabditis elegans genome engineering. Genetics 2016, 202, 885–901. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Dam, T.J.P.; van Kennedy, J.; Lee, R.; van der Vrieze, E.; de Wunderlich, K.A.; Rix, S.; Dougherty, G.W.; Lambacher, N.J.; Li, C.; Jensen, V.L.; et al. CiliaCarta: An integrated and validated compendium of ciliary genes. PLoS ONE 2019, 14, e0216705. [Google Scholar]
- Messchaert, M.; Dona, M.; Broekman, S.; Peters, T.A.; Corral-Serrano, J.C.; Slijkerman, R.W.N.; van Wijk, E.; Collin, R.W.J. Eyes shut homolog is important for the maintenance of photoreceptor morphology and visual function in zebrafish. PLoS ONE 2018, 13, e0200789. [Google Scholar] [CrossRef]












© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).