Genome-Wide Analyses and Prediction of Resistance to MLN in Large Tropical Maize Germplasm

 ,
,  , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Trial Design
2.2. Viral Inoculum and Artificial Inoculation
2.3. Phenotypic and Genotypic Data Analysis
2.4. Population Structure, PCA, and Linkage Disequilibrium Analysis
2.5. Genomic Prediction
3. Results
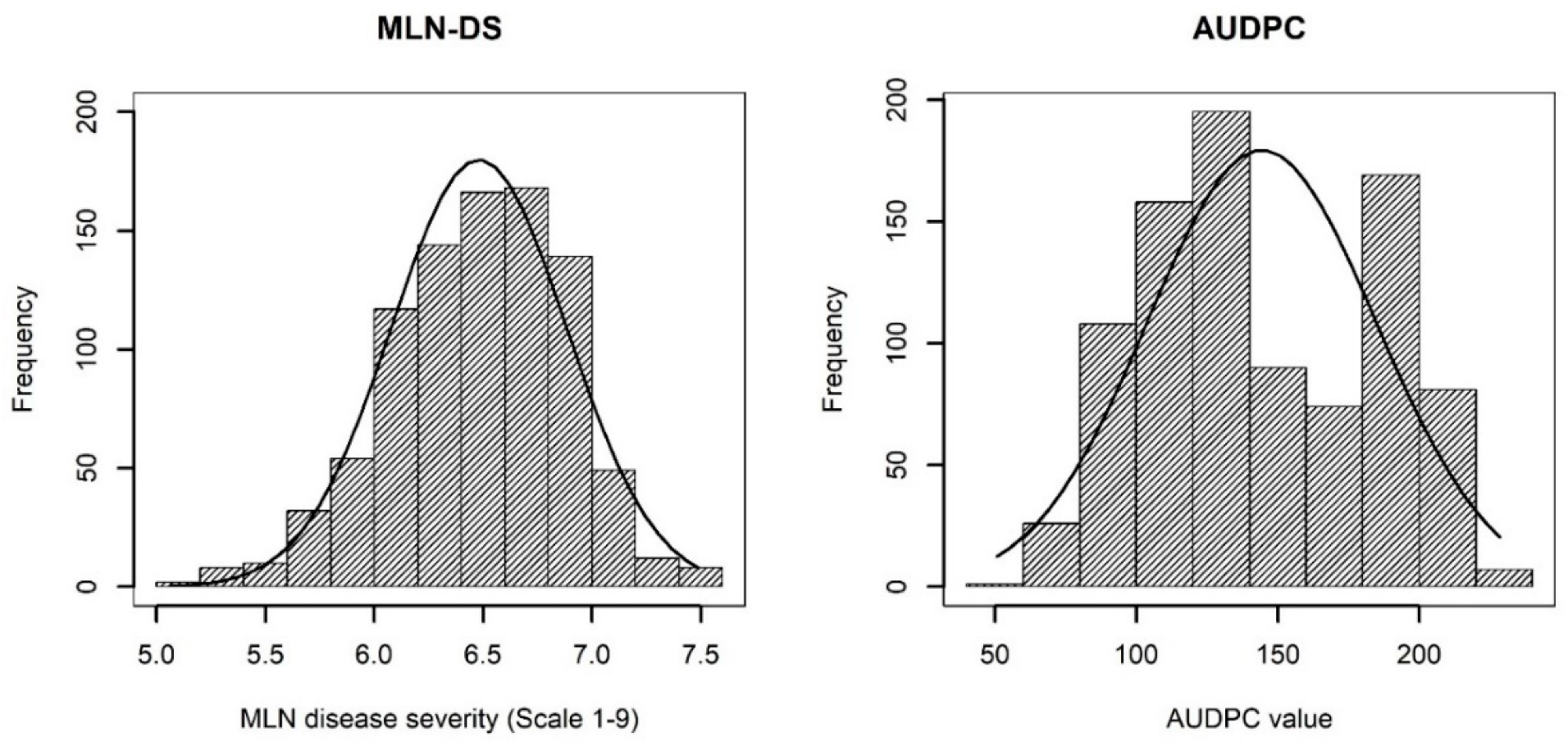
3.1. Phenotypic Analysis
3.2. PCA and Population Structure
3.3. Linkage Disequilibrium and GWAS
3.4. Genomic Prediction
4. Discussion and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mahuku, G.; Lockhart, B.; Wanjala, B.; Jones, M.; Kimunye, J.; Stewart, L.; Cassone, B.; Subramanian, S.; Nyasani, J.; Kusia, E.; et al. Maize lethal necrosis (MLN), an emerging threat to maize-based food security in Sub-Saharan Africa. Phytopathology 2015, 105, 956–965. [Google Scholar] [CrossRef] [Green Version]
- Beyene, Y.; Gowda, M.; Suresh, L.M.; Mugo, S.; Olsen, M.; Oikeh, S.O.; Juma, C.; Tarekegne, A.; Prasanna, B.M. Genetic analysis of tropical maize inbred lines for resistance to maize lethal necrosis disease. Euphytica 2017, 213, 224. [Google Scholar] [CrossRef] [Green Version]
- Redinbaugh, M.; Stewart, L. Maize lethal necrosis: An emerging, synergistic viral disease. Annu. Rev. Virol. 2018, 5, 301–322. [Google Scholar] [CrossRef]
- Mudde, B.; Olubayo, F.; Miano, D.; Asea, G.; Kilalo, D.; Kiggundu, A.; Kwemoi, D.; Adriko, J. Distribution, incidence and severity of maize lethal necrosis disease in major maize growing agro-ecological zones of Uganda. J. Agric. Sci. 2018, 10, 72–85. [Google Scholar] [CrossRef] [Green Version]
- Adams, I.; Harju, V.; Hodges, T.; Hany, U.; Skelton, A.; Rai, S.; Deka, M.K.; Smith, J.; Fox, A.; Uzayisenga, B.; et al. First report of maize lethal necrosis disease in Rwanda. New Dis. Rep. 2014, 29, 22. [Google Scholar] [CrossRef] [Green Version]
- Lukanda, M.; Owati, A.; Ogunsanya, P.; Valimunzigha, K.; Katsongo, K.; Ndemere, H.; Kumar, L. First report of Maize chlorotic mottle virus infecting maize (Zea mays L.) in the Democratic Republic of the Congo. Plant Dis. 2014, 98, 1448. [Google Scholar] [CrossRef] [PubMed]
- Jane Wamaitha, M.; Nigam, D.D.; Maina, S.; Stomeo, F.; Wangai, A.; Njoki Njuguna, J.; Holton, T.; Wanjala, B.; Wamalwa, M.; Lucas, T.; et al. Metagenomic analysis of viruses associated with maize lethal necrosis in Kenya. Virol. J. 2018, 15, 90. [Google Scholar] [CrossRef] [PubMed]
- Wangai, A.W.; Redinbaugh, M.G.; Kinyua, Z.M.; Miano, D.W.; Leley, P.K.; Kasina, M.; Mahuku, G.; Scheets, K.; Jeffers, D. First report of maize chlorotic mottle virus and maize lethal necrosis in Kenya. Plant Dis. 2012, 96, 1582. [Google Scholar] [CrossRef] [PubMed]
- Cabanas, D.; Watanabe, S.; Higashi, C.; Bressan, A. Dissecting the mode of maize chlorotic mottle virus transmission (Tombusviridae: Machlomovirus) by Frankliniella williamsi (Thysanoptera: Thripidae). J. Econ. Entomol. 2013, 106, 16–24. [Google Scholar] [CrossRef]
- Maldonado, C.; Mora, F.; Scapim, C.; Coan, M. Genome-wide haplotype-based association analysis of key traits of plant lodging and architecture of maize identifies major determinants for leaf angle: hapLA4. PLoS ONE 2019, 14, e0212925. [Google Scholar] [CrossRef] [Green Version]
- Crossa, J.; Pérez-Rodríguez, P.; Cuevas, J.; Montesinos-López, O.; Jarquín, D.; de los Campos, G.; Burgueño, J.; González-Camacho, J.M.; Pérez-Elizalde, S.; Beyene, Y.; et al. Genomic selection in plant breeding: Methods, models, and perspectives. Trends Plant Sci. 2017, 22, 961–975. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Gore, M.; Buckler, E.; Yu, J. Status and prospects of association mapping in plants. Plant Genome 2008, 1, 5–20. [Google Scholar] [CrossRef]
- Liu, N.; Xue, Y.; Guo, Z.; Li, W.; Tang, J. Genome-wide association study identifies candidate genes for starch content regulation in maize kernels. Front. Plant Sci. 2016, 7, 1046. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, H.; Li, L.; Lan, H.; Ren, Z.; Liu, D.; Wu, L.; Liu, H.; Jaqueth, J.; Li, B.; et al. Characterizing the population structure and genetic diversity of maize breeding germplasm in Southwest China using genome-wide SNP markers. BMC Genom. 2016, 17, 697. [Google Scholar] [CrossRef] [Green Version]
- Gowda, M.; Das, B.; Makumbi, D.; Babu, R.; Semagn, K.; Mahuku, G.; Olsen, M.; Bright, J.M.; Beyene, Y.; MPrasanna, B. Genome-wide association and genomic prediction of resistance to maize lethal necrosis disease in tropical maize germplasm. Theor. Appl. Genet. 2015, 128, 1957–1968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Xu, Y.; Hu, Z.; Xu, C. Genomic selection methods for crop improvement: Current status and prospects. Crop J. 2018, 6, 330–340. [Google Scholar] [CrossRef]
- Tian, C.; Gregersen, P.; Seldin, M.F. Accounting for ancestry: Population substructure and genome-wide association studies. Hum. Mol. Genet. 2008, 17, R143–R150. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Lv, X.; Weng, J.; Zhu, H.; Liu, C.; Hao, Z.; Zhou, Y.; Zhang, D.; Li, M.; Ci, X.; et al. Genetic characterization and linkage disequilibrium mapping of resistance to gray leaf spot in maize (Zea mays L.). Crop J. 2014, 2, 132–143. [Google Scholar] [CrossRef] [Green Version]
- Zila, C.T.; Samayoa, L.F.; Santiago, R.; Butrón, A.; Holland, J.B. A genome-wide association study reveals genes associated with Fusarium ear rot resistance in a maize core diversity panel. G3 (Bethesda) 2013, 3, 2095–2104. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.; Jiang, L.; Liu, Q.; Zhang, Y.; Zhang, R.; Roenn Ingvardsen, C.; Frei, U.; Wang, B.; Lai, J.; Lübberstedt, T.; et al. Combined linkage and association mapping reveals candidates for Scmv1, a major locus involved in resistance to sugarcane mosaic virus (SCMV) in maize. BMC Plant Biol. 2013, 13, 162. [Google Scholar] [CrossRef] [Green Version]
- Vlaming, R.; Groenen, P. The current and future use of ridge regression for prediction in quantitative genetics. Biomed Res. Int. 2015, 2015, 143712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massman, J.; Gordillo, A.; Lorenzana, R.E.; Bernardo, R. Genome wide predictions from maize single-cross data. Theor. Appl. Genet. 2012, 126, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Bhat, J.; Ali, S.; Salgotra, R.; Mir, Z.; Dutta, S.; Jadon, V.; Tyagi, A.; Mushtaq, M.; Jain, N.; Singh, P.K.; et al. Genomic selection in the era of next generation sequencing for complex traits in plant breeding. Front. Genet. 2016, 7, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jannink, J.-L.; Lorenz, A.J.; Iwata, H.; Jannink, J.-L.; Lorenz, A.J.; Iwata, H. Genomic selection in plant breeding: From theory to practice. Brief. Funct. Genom. 2010, 9, 166–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, B.; Singh, A.K. Marker-Assisted Plant Breeding: Principles and Practices; Springer: New Delhi, India, 2015. [Google Scholar]
- Sitonik, C.; Suresh, L.M.; Beyene, Y.; Olsen, M.S.; Makumbi, D.; Oliver, K.; Das, B.; Bright, J.M.; Mugo, S.; Crossa, J.; et al. Genetic architecture of maize chlorotic mottle virus and maize lethal necrosis through GWAS, linkage analysis and genomic prediction in tropical maize germplasm. Theor. Appl. Genet. 2019, 132, 2381–2399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Technow, F.; Pucher, A.; Melchinger, A.E. Genomic prediction of northern corn leaf blight resistance in maize with combined or separated training sets for heterotic groups. G3 (Bethesda) 2013, 3, 197–203. [Google Scholar] [CrossRef] [Green Version]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [Green Version]
- Bradbury, P.; Zhang, Z.; Kroon, D.E.; Casstevens, T.; Ramdoss, Y.; Buckler, E. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Endelman, J. Ridge regression and other kernels for genomic selection with R package rrBLUP. Plant Genome 2011, 4, 250–255. [Google Scholar] [CrossRef] [Green Version]
- Gowda, M.; Beyene, Y.; Makumbi, D.; Semagn, K.; Olsen, M.; Bright, J.M.; Das, B.; Mugo, S.; Suresh, L.M.; Prasanna, B.M.; et al. Discovery and validation of genomic regions associated with resistance to maize lethal necrosis in four biparental populations. Mol. Breed. 2018, 38, 6. [Google Scholar] [CrossRef] [Green Version]
- Stich, B.; Möhring, J.; Piepho, H.-P.; Heckenberger, M.; Buckler, E.S.; Melchinger, A.E. Comparison of mixed-model approaches for association mapping. Genetics 2008, 178, 1745–1754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.; Cairns, J.; Babu, R.; Gowda, M.; Makumbi, D.; Magorokosho, C.; Zhang, A.; Liu, Y.; Wang, N.; Hao, Z.; et al. Genome-wide association mapping and genomic prediction analyses reveal the genetic architecture of grain yield and flowering time under drought and heat stress conditions in maize. Front. Plant Sci. 2019, 9, 1919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Pérez-Rodríguez, P.; Semagn, K.; Beyene, Y.; Babu, R.; Lopez-Cruz, M.; San Vicente, F.; Olsen, M.; Buckler, E.; Jannink, J.-L.; et al. Genomic prediction in biparental tropical maize populations in water-stressed and well-watered environments using low-density and GBS SNPs. Heredity (Edinb) 2014, 114, 291–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, S.; Loladze, A.; Yuan, Y.; Wu, Y.; Zhang, A.; Chen, J.; Huestis, G.; Cao, J.; Chaikam, V.; Olsen, M.; et al. Genome-wide analysis of tar spot complex resistance in maize using gbs SNPs and whole genome prediction. Plant Genome 2017, 10. [Google Scholar] [CrossRef] [Green Version]
- Chaikam, V.; Gowda, M.; Nair, S.; Melchinger, A.; Prasanna, B. Genome-wide association study to identify genomic regions influencing spontaneous fertility in maize haploids. Euphytica 2019, 215, 138. [Google Scholar] [CrossRef] [Green Version]
- Riedelsheimer, C.; Brotman, Y.; Méret, M.; Melchinger, A.E.; Willmitzer, L. The maize leaf lipidome shows multilevel genetic control and high predictive value for agronomic traits. Sci. Rep. 2013, 3, 2479. [Google Scholar] [CrossRef] [Green Version]
- Sekhon, R.; Lin, H.; Childs, K.L.; Hansey, C.N.; Robin Buell, C.; de Leon, N.; Kaeppler, S. Genome-wide atlas of transcription during maize development. Plant J. 2011, 66, 553–563. [Google Scholar] [CrossRef]
- Lin, F.; Jiang, L.; Liu, Y.; Lv, Y.; Dai, H.; Zhao, H. Genome-wide identification of housekeeping genes in maize. Plant Mol. Biol. 2014, 86, 543–554. [Google Scholar] [CrossRef]
- Salvo, S.A.G.D.; Hirsch, C.N.; Buell, C.R.; Kaeppler, S.M.; Kaeppler, H.F. Whole transcriptome profiling of maize during early somatic embryogenesis reveals altered expression of stress factors and embryogenesis-related genes. PLoS ONE 2014, 9, e111407. [Google Scholar] [CrossRef] [Green Version]
- Gullner, G.; Komives, T.; Király, L.; Schröder, P. Glutathione S-transferase enzymes in plant-pathogen interactions. Front. Plant Sci. 2018, 9, 1836. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.; Lu, X.; Zhang, P.; Wang, G.; Wei, L.; Wang, T. Systematic analysis of differentially expressed maize ZmbZIP genes between drought and rewatering transcriptome reveals bZIP family members involved in abiotic stress responses. Int. J. Mol. Sci. 2019, 20, 4103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Büttner, M.; Singh, K.B. Arabidopsis thaliana ethylene-responsive element binding protein (AtEBP), an ethylene-inducible, GCC box DNA-binding protein interacts with an ocs element binding protein. Proc. Natl. Acad. Sci. USA 1997, 94, 5961–5966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, X.; Lv, W.; Zhang, D.; Jiang, S.; Zhang, S.; Li, D. Genome-wide identification and analysis of expression profiles of maize mitogen-activated protein kinase kinase kinase. PLoS ONE 2013, 8, e57714. [Google Scholar] [CrossRef] [PubMed]
- Jagodzik, P.; Tajdel-Zielinska, M.; Ciesla, A.; Marczak, M.; Ludwikow, A. Mitogen-activated protein kinase cascades in plant hormone signaling. Front. Plant Sci. 2018, 9, 1387. [Google Scholar] [CrossRef] [PubMed]
- Lanubile, A.; Ferrarini, A.; Maschietto, V.; Delledonne, M.; Marocco, A.; Bellin, D. Functional genomic analysis of constitutive and inducible defense responses to Fusarium verticillioides infection in maize genotypes with contrasting ear rot resistance. BMC Genom. 2014, 15, 710. [Google Scholar] [CrossRef] [Green Version]
- Vogel, J.P.; Raab, T.K.; Somerville, C.R.; Somerville, S.C. Mutations in PMR5 result in powdery mildew resistance and altered cell wall composition. Plant J. 2004, 40, 968–978. [Google Scholar] [CrossRef]
- Crossa, J.; Pérez, P.; Hickey, J.; Burgueño, J.; Ornella, L.; Cerón-Rojas, J.; Zhang, X.; Dreisigacker, S.; Babu, R.; Li, Y.; et al. Genomic prediction in CIMMYT maize and wheat breeding programs. Heredity (Edinb) 2013, 112, 48. [Google Scholar] [CrossRef] [Green Version]
- Zhang, A.; Wang, H.; Beyene, Y.; Semagn, K.; Liu, Y.; Cao, S.; Cui, Z.; Ruan, Y.; Burgueño, J.; San Vicente, F.; et al. Effect of Trait heritability, training population size and marker density on genomic prediction accuracy estimation in 22 bi-parental tropical maize populations. Front. Plant Sci. 2017, 8, 1916. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DS (1–9) | AUDPC | |
|---|---|---|
| Mean | 6.25 | 135.28 |
| Minimum | 5.30 | 58.69 |
| Maximum | 7.12 | 216.05 |
| LSD | 0.93 | 20.43 |
| σ2G | 0.55 ** | 2303.16 ** |
| σ2GxE | 0.78 ** | 143.36 * |
| σ2e | 1.40 | 1267.99 |
| h2 | 0.42 | 0.86 |
| SNP | Chr | Position (bp) | MLM P-Values | R2 | MAF * | Allele | MAE ** | Putative Candidate Genes | Predicted Function of Candidate Gene |
|---|---|---|---|---|---|---|---|---|---|
| MLN disease severity | |||||||||
| S1_44539940 | 1 | 44539940 | 9.66E-09 | 0.05 | 0.10 | C/T | 0.42 | GRMZM2G024159 | protein YIPF5 homolog |
| S1_281333891 | 1 | 281333891 | 8.46E-08 | 0.04 | 0.17 | C/G | 0.07 | GRMZM2G177046 | bZIP transcription factor |
| S3_147938951 | 3 | 147938951 | 8.93E-08 | 0.04 | 0.12 | C/G | −0.22 | GRMZM2G044867 | unknown |
| S3_149313702 | 3 | 149313702 | 2.50E-07 | 0.04 | 0.22 | A/G | −0.02 | GRMZM2G428168 | S-glutathionylation and deglutathionylation |
| S3_161574458 | 3 | 161574458 | 9.25E-07 | 0.04 | 0.09 | C/G | −1.97 | GRMZM2G145346 | PAK-box/P21-Rho-binding |
| S3_161574468 | 3 | 161574468 | 4.40E-07 | 0.03 | 0.11 | T/A | 0.73 | GRMZM2G145346 | PAK-box/P21-Rho-binding |
| S3_161574470 | 3 | 161574470 | 1.25E-06 | 0.03 | 0.09 | A/G | 0.75 | GRMZM2G145346 | PAK-box/P21-Rho-binding |
| S3_161574471 | 3 | 161574471 | 2.69E-07 | 0.04 | 0.09 | C/A | 0.8 | GRMZM2G145346 | PAK-box/P21-Rho-binding |
| S4_199711804 | 4 | 199711804 | 4.76E-07 | 0.04 | 0.29 | C/T | −0.11 | GRMZM2G134857 | uncharacterized protein |
| S7_140411743 | 7 | 140411743 | 4.84E-07 | 0.04 | 0.07 | C/T | 1.49 | GRMZM2G071015 | BAG-associated GRAM protein |
| S7_143109798 | 7 | 143109798 | 9.81E-07 | 0.03 | 0.37 | C/T | 0.71 | GRMZM2G179021 | RNA.regulation of transcription. |
| S7_166270242 | 7 | 166270242 | 5.95E-07 | 0.04 | 0.11 | G/C | 0.54 | GRMZM2G520980 | unknown |
| S9_9599125 | 9 | 9599125 | 5.21E-07 | 0.03 | 0.08 | A/C | 1.25 | GRMZM2G159402 | transcriptional activation |
| S9_149758216 | 9 | 149758216 | 4.94E-08 | 0.04 | 0.25 | T/C | 0.11 | GRMZM2G540298 | unknown |
| S10_3189860 | 10 | 3189860 | 4.97E-07 | 0.04 | 0.05 | A/G | −1.16 | GRMZM5G862857 | uncharacterized protein |
| S10_138075442 | 10 | 138075442 | 7.56E-07 | 0.03 | 0.26 | G/C | 0.77 | GRMZM2G117667 | GDSL-like Lipase/Acylhydrolase superfamily protein |
| S10_138075445 | 10 | 138075445 | 7.56E-07 | 0.03 | 0.26 | C/T | 0.77 | GRMZM2G117668 | unknown |
| S10_140985097 | 10 | 140985097 | 3.94E-07 | 0.04 | 0.08 | T/C | 0.06 | GRMZM2G109753 | scramblase family protein |
| Total R2 | 17.05 | ||||||||
| Area under disease progress curve | |||||||||
| S1_44539940 | 1 | 44539940 | 6.92E-16 | 0.1 | 0.1 | C/T | 10.15 | GRMZM2G024159 | Yip1 domain containing protein |
| S1_253798682 | 1 | 253798682 | 7.08E-07 | 0.03 | 0.33 | G/A | 23.06 | GRMZM2G043127 | translocase of the outer mitochondrial membrane |
| S2_28895383 | 2 | 28895383 | 1.86E-07 | 0.03 | 0.42 | A/G | 7.3 | GRMZM2G077420 | unknown |
| S3_33757503 | 3 | 33757503 | 7.85E-07 | 0.03 | 0.13 | T/C | −18.42 | GRMZM2G563119 | unknown |
| S3_55239348 | 3 | 55239348 | 5.33E-07 | 0.03 | 0.37 | C/G | 4.69 | GRMZM2G520940 | protein coding |
| S3_56468811 | 3 | 56468811 | 5.63E-09 | 0.04 | 0.43 | A/G | 3.02 | GRMZM2G409309 | powdery mildew resistant protein5 |
| S3_136082606 | 3 | 136082606 | 2.87E-07 | 0.04 | 0.39 | G/C | 32.3 | GRMZM2G092169 | uncharacterized protein |
| S3_147938951 | 3 | 147938951 | 1.04E-07 | 0.04 | 0.12 | C/G | −0.22 | GRMZM2G044867 | unknown |
| S3_190890553 | 3 | 190890553 | 9.37E-07 | 0.03 | 0.15 | G/A | 1.6 | GRMZM2G563190 | mitochondrial electron transport/ATP synthesis. |
| S4_199711804 | 4 | 199711804 | 1.89E-19 | 0.12 | 0.29 | C/T | −2.87 | GRMZM2G134857 | uncharacterized protein |
| S4_200034077 | 4 | 200034077 | 3.06E-07 | 0.03 | 0.16 | A/G | 12.07 | GRMZM2G465165 | ATP binding/amino acid phosphorylation |
| S5_182091386 | 5 | 182091386 | 8.77E-07 | 0.03 | 0.25 | T/A | 39.18 | GRMZM2G137426 | protein dimerization activity |
| S6_99770682 | 6 | 99770682 | 1.04E-06 | 0.03 | 0.39 | T/G | 22.51 | GRMZM2G112337 | MAP65-2 microtubule-associated protein |
| S6_148513637 | 6 | 148513637 | 8.16E-07 | 0.03 | 0.07 | A/G | 47.5 | GRMZM2G020856 | O-fucosyltransferase family protein |
| S7_8677545 | 7 | 8677545 | 1.16E-08 | 0.05 | 0.24 | C/G | −10.84 | GRMZM2G107408 | uncharacterized protein |
| S7_168745410 | 7 | 168745410 | 3.55E-07 | 0.03 | 0.4 | A/G | 1.59 | GRMZM2G039757 | tolB protein-related |
| S8_150798179 | 8 | 150798179 | 5.92E-07 | 0.03 | 0.43 | A/C | −7.05 | GRMZM2G531490 | unknown |
| S10_138075442 | 10 | 138075442 | 2.75E-07 | 0.03 | 0.26 | G/C | 0.77 | GRMZM2G117667 | GDSL-like Lipase/Acylhydrolase superfamily protein |
| S10_138075445 | 10 | 138075445 | 2.75E-07 | 0.03 | 0.26 | C/T | 0.77 | GRMZM2G117668 | unknown |
| Total R2 | 24.75 | ||||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nyaga, C.; Gowda, M.; Beyene, Y.; Muriithi, W.T.; Makumbi, D.; Olsen, M.S.; Suresh, L.M.; Bright, J.M.; Das, B.; Prasanna, B.M. Genome-Wide Analyses and Prediction of Resistance to MLN in Large Tropical Maize Germplasm. Genes 2020, 11, 16. https://doi.org/10.3390/genes11010016
Nyaga C, Gowda M, Beyene Y, Muriithi WT, Makumbi D, Olsen MS, Suresh LM, Bright JM, Das B, Prasanna BM. Genome-Wide Analyses and Prediction of Resistance to MLN in Large Tropical Maize Germplasm. Genes. 2020; 11(1):16. https://doi.org/10.3390/genes11010016
Chicago/Turabian StyleNyaga, Christine, Manje Gowda, Yoseph Beyene, Wilson T. Muriithi, Dan Makumbi, Michael S. Olsen, L. M. Suresh, Jumbo M. Bright, Biswanath Das, and Boddupalli M. Prasanna. 2020. "Genome-Wide Analyses and Prediction of Resistance to MLN in Large Tropical Maize Germplasm" Genes 11, no. 1: 16. https://doi.org/10.3390/genes11010016
APA StyleNyaga, C., Gowda, M., Beyene, Y., Muriithi, W. T., Makumbi, D., Olsen, M. S., Suresh, L. M., Bright, J. M., Das, B., & Prasanna, B. M. (2020). Genome-Wide Analyses and Prediction of Resistance to MLN in Large Tropical Maize Germplasm. Genes, 11(1), 16. https://doi.org/10.3390/genes11010016