Draft Genomes of Two Artocarpus Plants, Jackfruit (A. heterophyllus) and Breadfruit (A. altilis)

 ,
,  , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection, NGS Library Construction, and Sequencing
2.2. Evaluation of Genome Size
2.3. De Novo Genome Assembly
2.4. Genome Assembly Evaluation
2.5. Repeat Annotation
2.6. Gene Prediction
2.7. Functional Annotation of Protein-Coding Genes
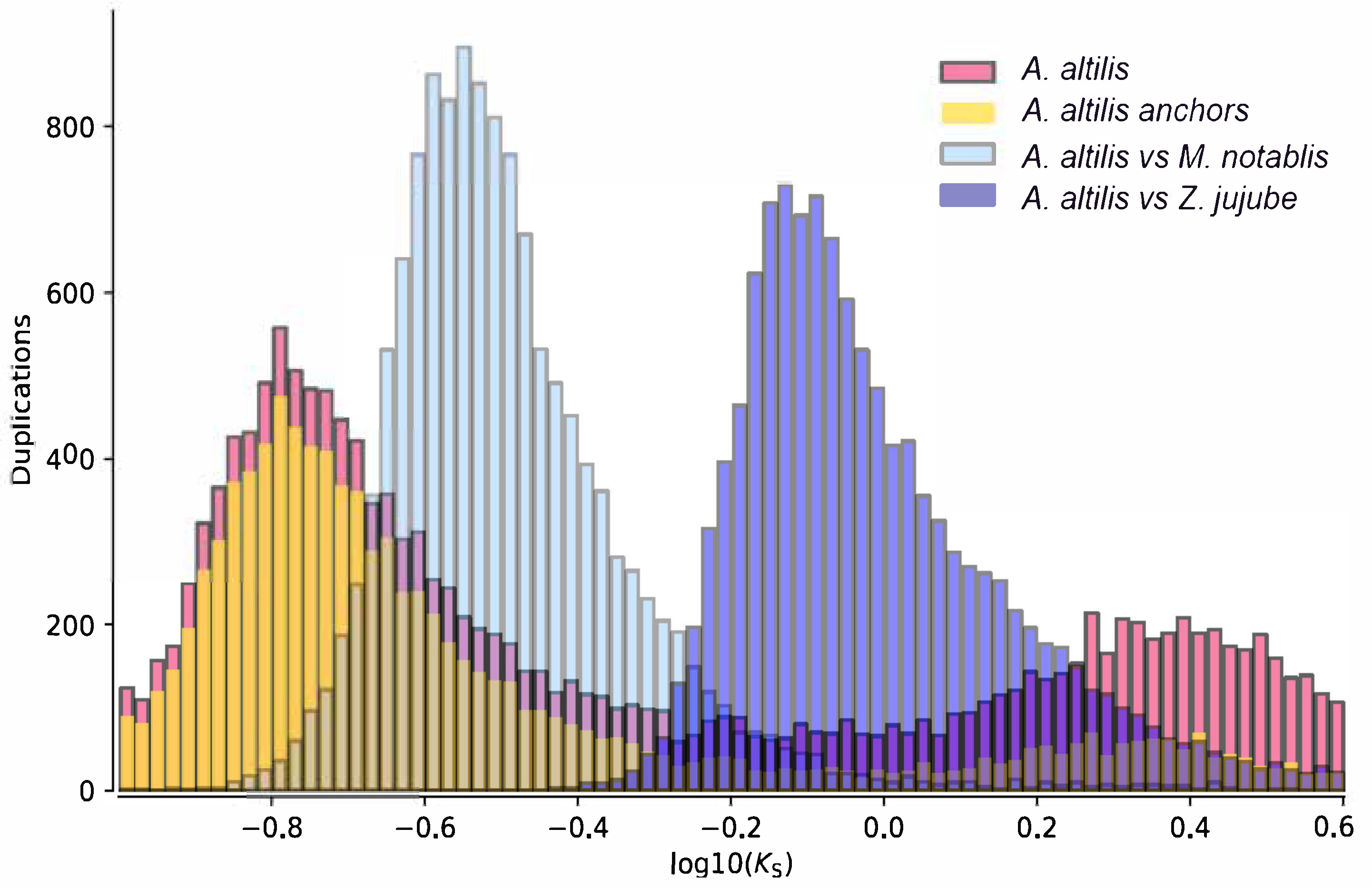
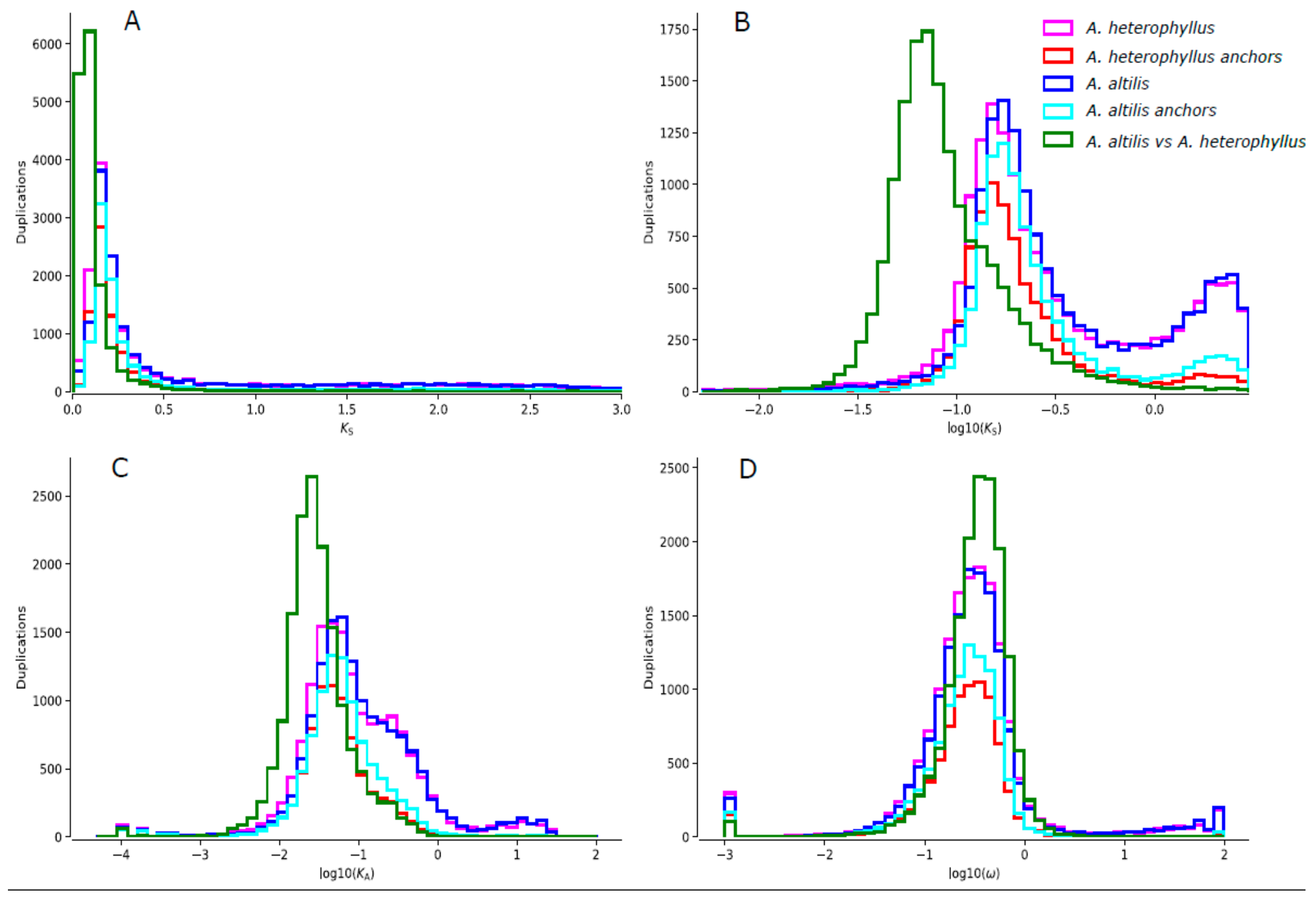
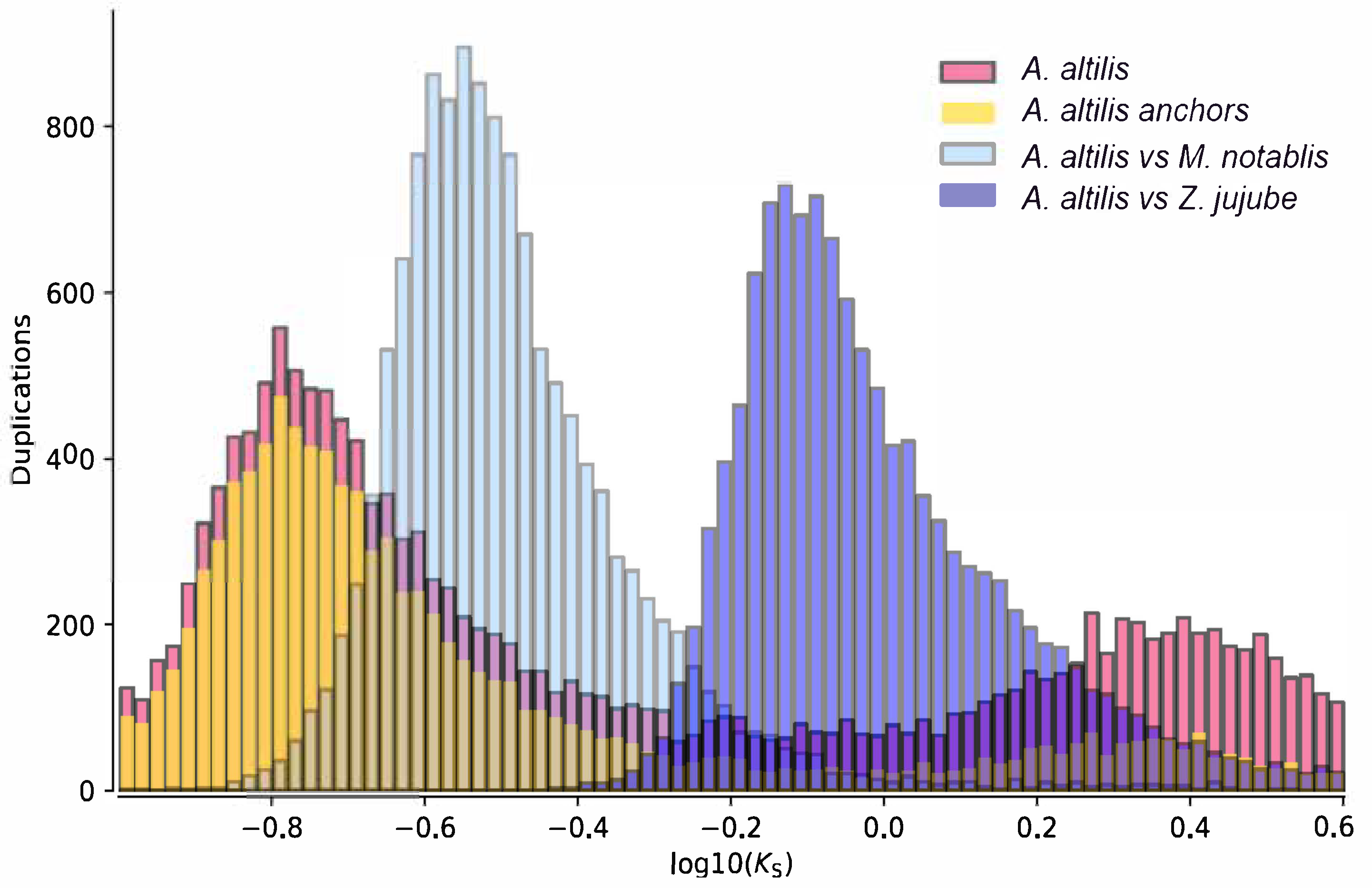
2.8. Ks-Distribution Analysis
2.9. One vs. One Synteny
2.10. Gene Family Construction
2.11. Collinearity Analysis
2.12. Phylogenetic Analysis and Divergence Time Estimation
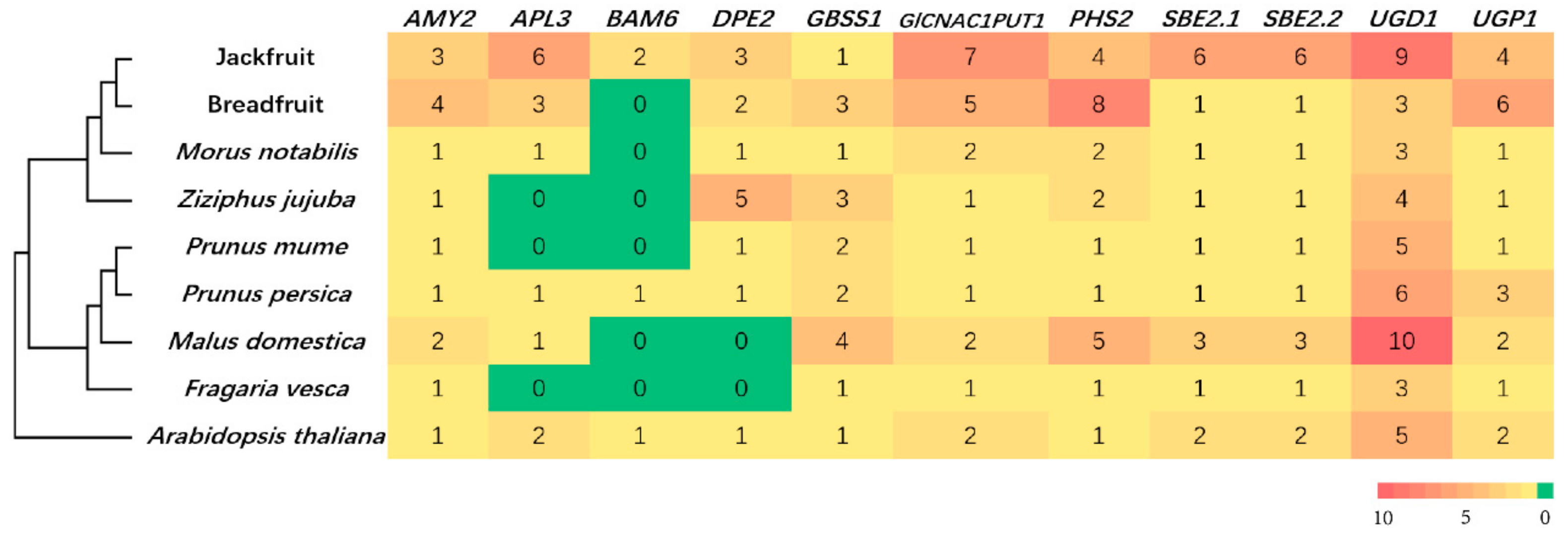
2.13. Identification of Starch Biosynthesis-Related Genes
3. Results and Discussion
3.1. Genome Sequencing and Assembly
3.2. Gene Annotation
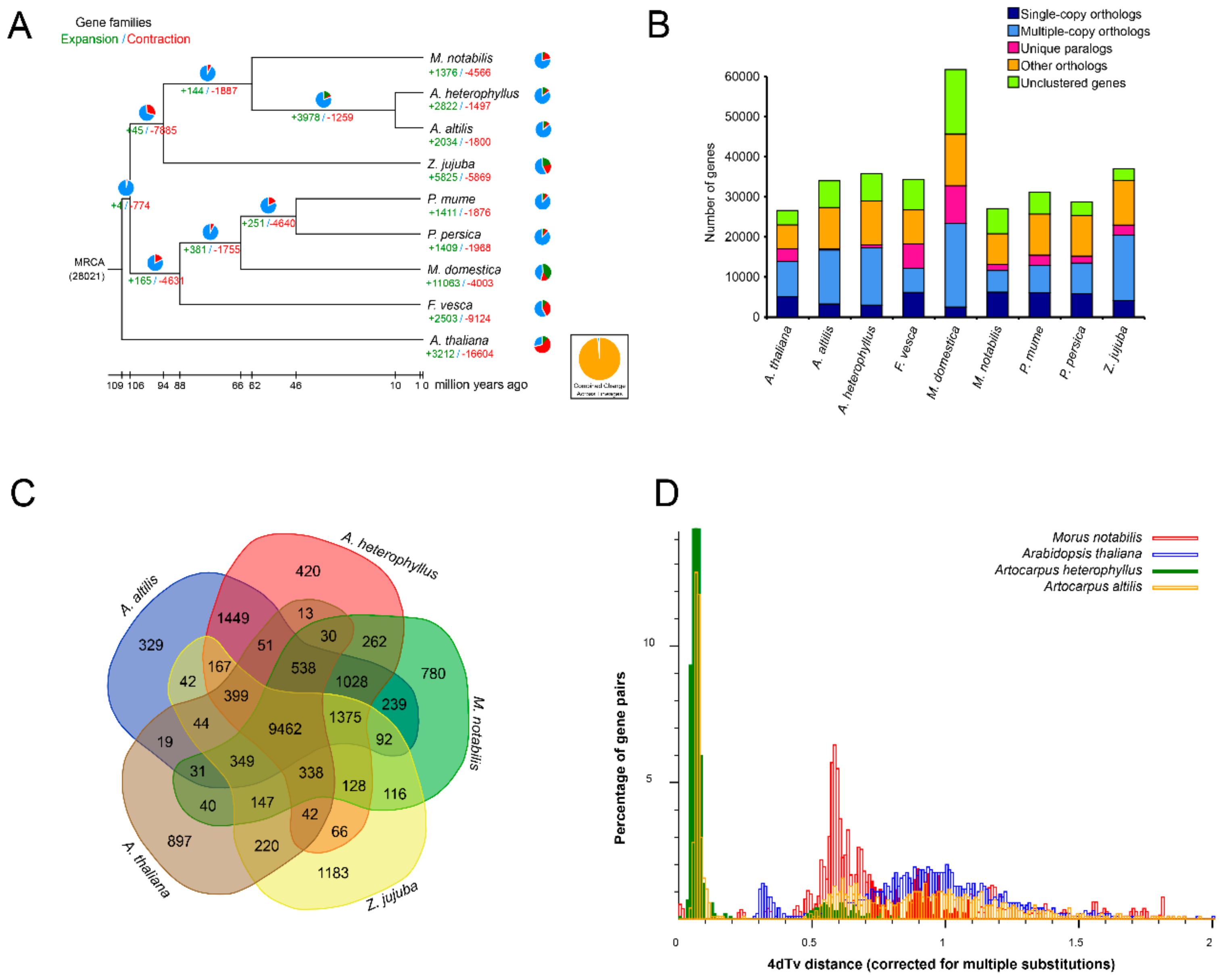
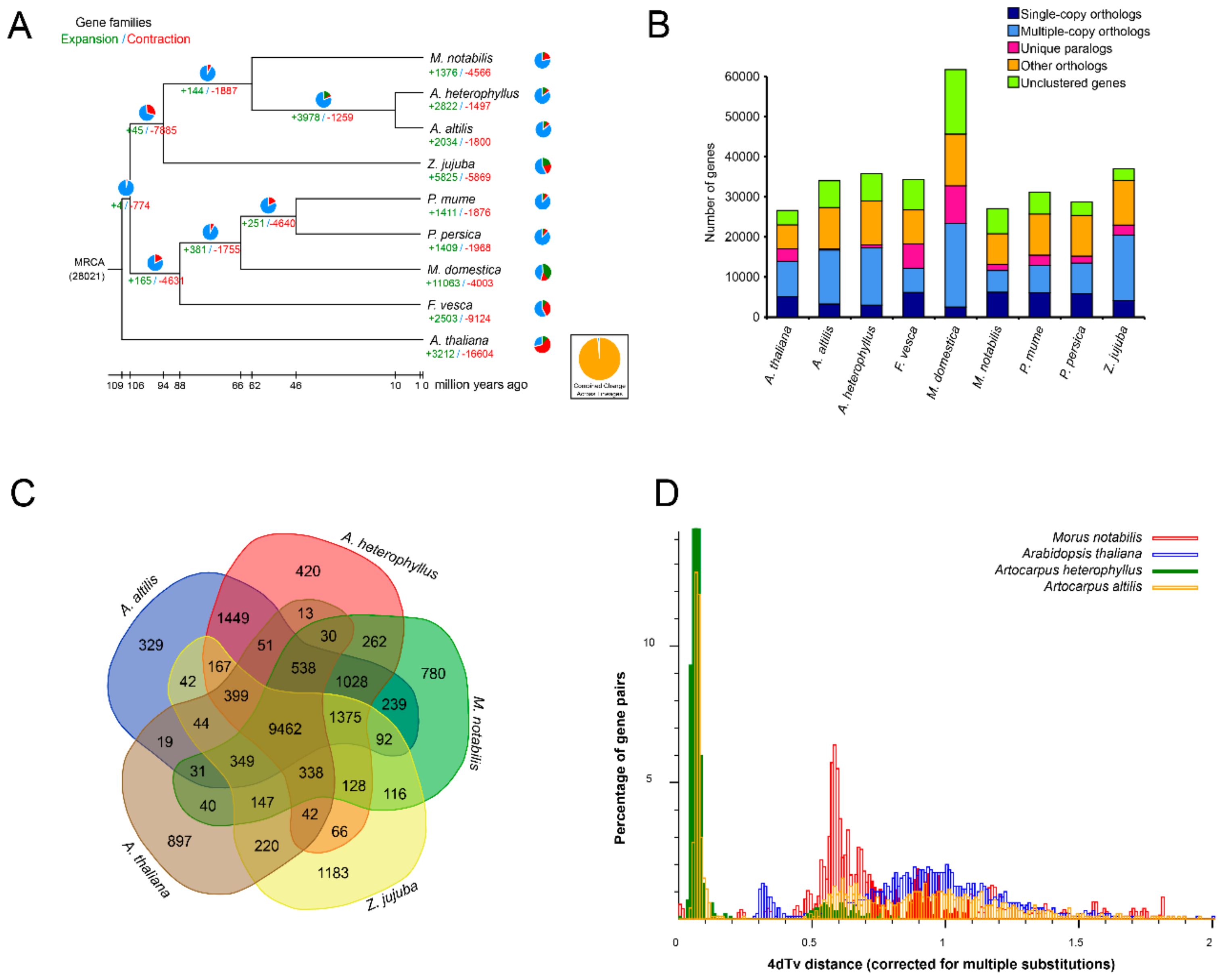
3.3. Gene Family Evolution and Comparison
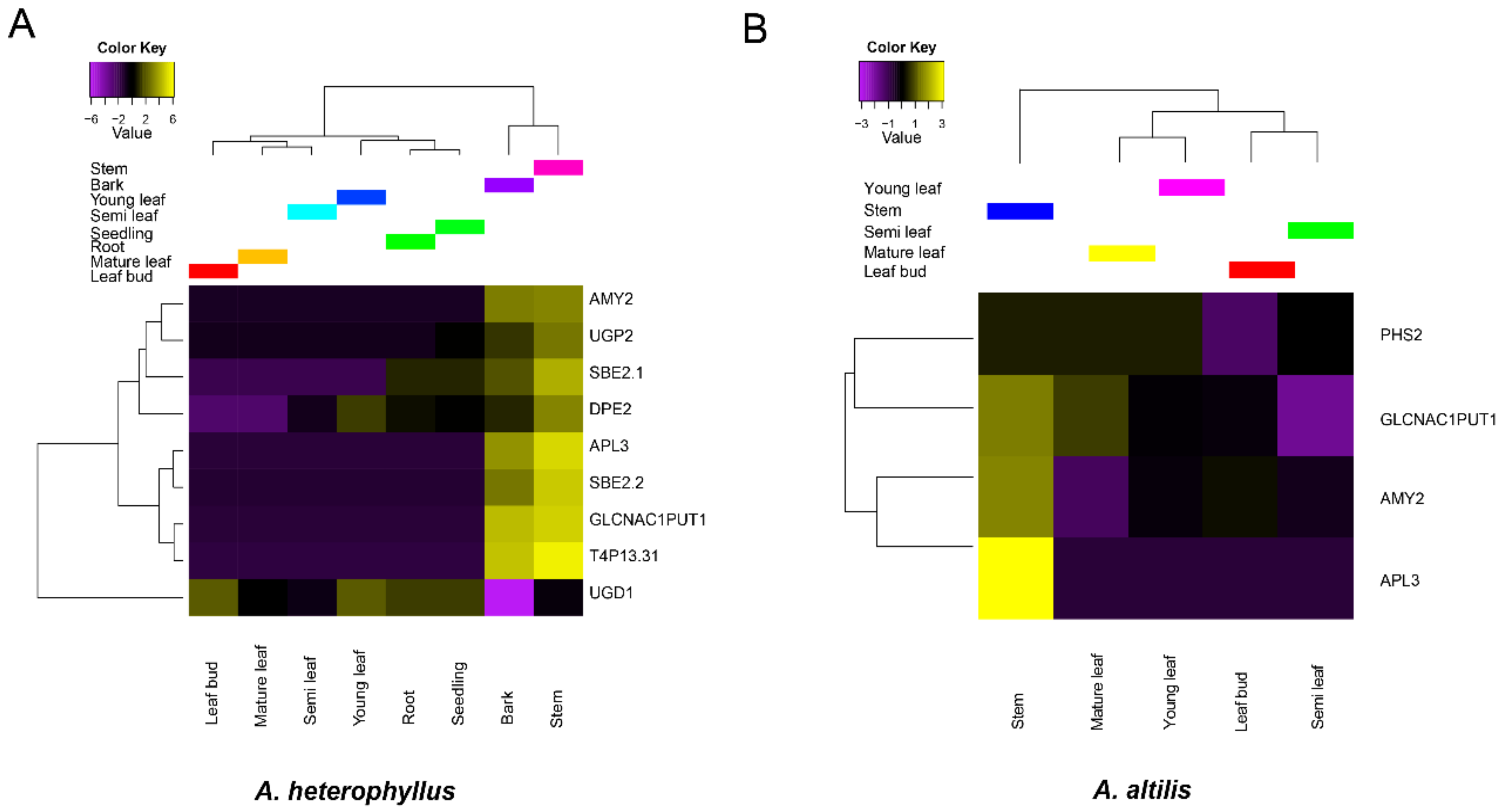
3.4. Gene Family Expansion and Tissue Specific Expression of Starch Synthesis-Related Genes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Availability of Supporting Data
References
- Zerega, N.J.; Supardi, N.; Motley, T.J. Phylogeny and recircumscription of Artocarpeae (Moraceae) with a focus on Artocarpus. Syst. Bot. 2010, 35, 766–782. [Google Scholar] [CrossRef] [Green Version]
- Williams, E.W.; Gardner, E.M.; Harris, R., III; Chaveerach, A.; Pereira, J.T.; Zerega, N.J. Out of Borneo: Biogeography, phylogeny and divergence date estimates of Artocarpus (Moraceae). Ann. Bot. 2017, 119, 611–627. [Google Scholar] [CrossRef] [Green Version]
- Zerega, N.J.; Gardner, E.M. Delimitation of the new tribe Parartocarpeae (Moraceae) is supported by a 333-gene phylogeny and resolves tribal level Moraceae taxonomy. Phytotaxa 2019, 388, 253–265. [Google Scholar] [CrossRef]
- The Plant List. Available online: http://www.theplantlist.org/ (accessed on 24 April 2019).
- Jarrett, F. The syncarp of Artocarpus—A unique biological phenomenon [tropical fruits, tropical Asia]. Gard. Bull. 1977, 29, 35–39. [Google Scholar]
- Wang, M.M.; Gardner, E.M.; Chung, R.C.; Chew, M.Y.; Milan, A.R.; Pereira, J.T.; Zerega, N.J. Origin and diversity of an underutilized fruit tree crop, cempedak (Artocarpus integer, Moraceae). Am. J. Bot. 2018, 105, 898–914. [Google Scholar] [CrossRef] [PubMed]
- Gardner, E.M.; Gagné, R.J.; Kendra, P.E.; Montgomery, W.S.; Raguso, R.A.; McNeil, T.T.; Zerega, N.J. A flower in fruit’s clothing: Pollination of jackfruit (Artocarpus heterophyllus, Moraceae) by a new species of gall midge, Clinodiplosis ultracrepidata sp. nov. (Diptera: Cecidomyiidae). Int. J. Plant Sci. 2018, 179, 350–367. [Google Scholar] [CrossRef]
- Witherup, C.; Ragone, D.; Wiesner-Hanks, T.; Irish, B.; Scheffler, B.; Simpson, S.; Zee, F.; Zuberi, M.I.; Zerega, N.J. Development of microsatellite loci in Artocarpus altilis (Moraceae) and cross-amplification in congeneric species. Appl. Plant Sci. 2013, 1, 1200423. [Google Scholar] [CrossRef] [PubMed]
- Campbell, R.J.; Ledesma, N. The Exotic Jackfruit; Fairchild Tropical Botanic Garden: Coral Gables, FL, USA, 2003; p. 72. [Google Scholar]
- Morton, J.F.; Dowling, C.F. Fruits of Warm Climates; JF Morton: Miami, FL, USA, 1987; Volume 20534. [Google Scholar]
- Simon, L.; Shyamalamma, S.; Narayanaswamy, P. Morphological and molecular analysis of genetic diversity in jackfruit. J. Hortic. Sci. Biotechnol. 2007, 82, 764–768. [Google Scholar] [CrossRef]
- Odoemelam, S. Functional properties of raw and heat processed jackfruit (Artocarpus heterophyllus) flour. Pak. J. Nutri. 2005, 4, 366–370. [Google Scholar]
- Zerega, N.J.; Ragone, D.; Motley, T.J. Complex origins of breadfruit (Artocarpus altilis, Moraceae): Implications for human migrations in Oceania. Am. J. Bot. 2004, 91, 760–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragone, D. Breadfruit—Artocarpus altilis (Parkinson) Fosberg. In Exotic Fruits; Rodrigues, S., de Oliveira Silva, E., de Brito, E.S., Eds.; Academic Press: New York, NY, USA, 2018; pp. 53–60. Available online: https://doi.org/10.1016/B978-0-12-803138-4.00009-5 (accessed on 26 June 2019).
- Lincoln, N.K.; Ragone, D.; Zerega, N.; Roberts-Nkrumah, L.B.; Merlin, M.; Jones, A. Grow us our daily bread: A review of breadfruit cultivation in traditional and contemporary systems. Hortic. Rev. 2018, 46, 299–384. [Google Scholar]
- Murch, S.J.; Ragone, D.; Shi, W.L.; Alan, A.R.; Saxena, P.K. In vitro conservation and sustained production of breadfruit (Artocarpus altilis, Moraceae): Modern technologies for a traditional tropical crop. Naturwissenschaften 2008, 95, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Moo-Young, M. Comprehensive Biotechnology; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Zerega, N.; Ragone, D.; Motley, T. Species limits and a taxonomic treatment of breadfruit (Artocarpus, Moraceae). Syst. Bot. 2005, 30, 603–615. [Google Scholar] [CrossRef]
- Zerega, N.; Wiesner-Hanks, T.; Ragone, D.; Irish, B.; Scheffler, B.; Simpson, S.; Zee, F. Diversity in the breadfruit complex (Artocarpus, Moraceae): Genetic characterization of critical germplasm. Tree Genet. Genomes 2015, 11, 4. [Google Scholar] [CrossRef]
- Laricchia, K.M.; Johnson, M.G.; Ragone, D.; Williams, E.W.; Zerega, N.J.; Wickett, N.J. A transcriptome screen for positive selection in domesticated breadfruit and its wild relatives (Artocarpus spp.). Am. J. Bot. 2018, 105, 915–926. [Google Scholar] [CrossRef] [Green Version]
- Zerega, N.J.; Ragone, D. Toward a global view of breadfruit genetic diversity. Trop. Agric. 2016, 93, 77–91. [Google Scholar]
- Chang, Y.; Liu, M.; Liu, X. The draft genomes of five agriculturally important African orphan crops. GigaScience 2018, 8. [Google Scholar] [CrossRef]
- Hendre, P.S.; Muthemba, S.; Kariba, R.; Muchugi, A.; Fu, Y.; Chang, Y.; Song, B.; Liu, H.; Liu, M.; Liao, X. African Orphan Crops Consortium (AOCC): Status of developing genomic resources for African orphan crops. Planta 2019, 250, 989–1003. [Google Scholar] [CrossRef] [Green Version]
- Jones, A.M.P.; Ragone, D.; Aiona, K.; Lane, W.A.; Murch, S.J. Nutritional and morphological diversity of breadfruit (Artocarpus, Moraceae): Identification of elite cultivars for food security. J. Food Compos. Anal. 2011, 24, 1091–1102. [Google Scholar] [CrossRef]
- Liu, Y.; Ragone, D.; Murch, S.J. Breadfruit (Artocarpus altilis): A source of high-quality protein for food security and novel food products. Amino Acids 2015, 47, 847–856. [Google Scholar] [CrossRef]
- Jones, A.M.P.; Baker, R.; Ragone, D.; Murch, S.J. Identification of pro-vitamin A carotenoid-rich cultivars of breadfruit (Artocarpus, Moraceae). J. Food Compos. Anal. 2013, 31, 51–61. [Google Scholar] [CrossRef]
- Ranasinghe, R.; Maduwanthi, S.; Marapana, R. Nutritional and Health Benefits of Jackfruit (Artocarpus heterophyllus Lam.): A Review. Int. J. Food Sci. 2019, 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bellis, F.; Malapa, R.; Kagy, V.; Lebegin, S.; Billot, C.; Labouisse, J.P. New development and validation of 50 SSR markers in breadfruit (Artocarpus altilis, Moraceae) by next-generation sequencing. Appl. Plant Sci. 2016, 4, 1600021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witherup, C.; Zuberi, M.I.; Hossain, S.; Zerega, N.J. Genetic Diversity of Bangladeshi Jackfruit (Artocarpus heterophyllus) over Time and Across Seedling Sources. Econ. Bot. 2019, 73, 233–248. [Google Scholar] [CrossRef]
- Gardner, E.M.; Laricchia, K.M.; Murphy, M.; Ragone, D.; Scheffler, B.E.; Simpson, S.; Williams, E.W.; Zerega, N.J. Chloroplast microsatellite markers for Artocarpus (Moraceae) developed from transcriptome sequences. Appl. Plant Sci. 2015, 3, 1500049. [Google Scholar] [CrossRef]
- Gardner, E.M. Evolutionary Transitions: Phylogenomics and Pollination of Artocarpus (Moraceae); Northwestern University: Evanston, IL, USA, 2017. [Google Scholar]
- Gardner, E.M.; Johnson, M.G.; Ragone, D.; Wickett, N.J.; Zerega, N.J. Low-coverage, whole-genome sequencing of Artocarpus camansi (Moraceae) for phylogenetic marker development and gene discovery. Appl. Plant Sci. 2016, 4, 1600017. [Google Scholar] [CrossRef] [Green Version]
- DNA Extraction for Plant Samples by CTAB. Available online: https://www.protocols.io/view/dna-extraction-for-plant-samples-by-ctab-pzqdp5w/metadata (accessed on 24 December 2019).
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2012, 1, 18. [Google Scholar] [CrossRef]
- Teh, B.T.; Lim, K.; Yong, C.H.; Ng, C.C.Y.; Rao, S.R.; Rajasegaran, V.; Lim, W.K.; Ong, C.K.; Chan, K.; Cheng, V.K.Y. The draft genome of tropical fruit durian (Durio zibethinus). Nat. Genet. 2017, 49, 1633. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Shi, Y.; Yuan, J.; Hu, X.; Zhang, H.; Li, N.; Li, Z.; Chen, Y.; Mu, D.; Fan, W. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. arXiv 2013, arXiv:1308.2012. [Google Scholar]
- Kajitani, R.; Yoshimura, D.; Okuno, M.; Minakuchi, Y.; Kagoshima, H.; Fujiyama, A.; Kubokawa, K.; Kohara, Y.; Toyoda, A.; Itoh, T. Platanus-allee is a de novo haplotype assembler enabling a comprehensive access to divergent heterozygous regions. Nat. Commun. 2019, 10, 1702. [Google Scholar] [CrossRef] [Green Version]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Z.; Li, G.; Liu, J.; Zhang, Y.; Ashby, C.; Liu, D.; Cramer, C.L.; Huang, X. Bridger: A new framework for de novo transcriptome assembly using RNA-seq data. Genome Biol. 2015, 16, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, W.J. BLAT--the BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Yu, C.; Li, Y.; Lam, T.W.; Yiu, S.M.; Kristiansen, K.; Wang, J. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 2009, 25, 1966–1967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2009, 25, 4–10. [Google Scholar] [CrossRef]
- Han, Y.; Wessler, S.R. MITE-Hunter: A program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res. 2010, 38, e199. [Google Scholar] [CrossRef] [Green Version]
- Ellinghaus, D.; Kurtz, S.; Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinform. 2008, 9, 18. [Google Scholar] [CrossRef] [Green Version]
- Gremme, G.; Steinbiss, S.; Kurtz, S. GenomeTools: A comprehensive software library for efficient processing of structured genome annotations. IEEE/ACM Trans. Comput. Biol. Bioinform. 2013, 10, 645–656. [Google Scholar] [CrossRef]
- Steinbiss, S.; Willhoeft, U.; Gremme, G.; Kurtz, S. Fine-grained annotation and classification of de novo predicted LTR retrotransposons. Nucleic Acids Res. 2009, 37, 7002–7013. [Google Scholar] [CrossRef]
- Chan, P.P.; Lowe, T.M. GtRNAdb 2.0: An expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 2016, 44, D184–D189. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Campbell, M.S.; Holt, C.; Moore, B.; Yandell, M. Genome Annotation and Curation Using MAKER and MAKER-P. Curr. Protoc. Bioinform. 2014, 48, 4–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanke, M.; Schoffmann, O.; Morgenstern, B.; Waack, S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinform. 2006, 7, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lomsadze, A.; Ter-Hovhannisyan, V.; Chernoff, Y.O.; Borodovsky, M. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 2005, 33, 6494–6506. [Google Scholar] [CrossRef] [PubMed]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef] [Green Version]
- Nawrocki, E.P.; Burge, S.W.; Bateman, A.; Daub, J.; Eberhardt, R.Y.; Eddy, S.R.; Floden, E.W.; Gardner, P.P.; Jones, T.A.; Tate, J.; et al. Rfam 12.0: Updates to the RNA families database. Nucleic Acids Res. 2015, 43, D130–D137. [Google Scholar] [CrossRef]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Tanabe, M.; Kanehisa, M. Using the KEGG database resource. Curr. Protoc. Bioinform. 2012, 38, 1–12. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Koonin, E.V.; Lipman, D.J. A Genomic Perspective on Protein Families. Science 1997, 278, 631–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boeckmann, B.; Bairoch, A.; Apweiler, R.; Blatter, M.C.; Estreicher, A.; Gasteiger, E.; Martin, M.J.; Michoud, K.; O’Donovan, C.; Phan, I.; et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003, 31, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.D.; Mistry, J.; Tate, J.; Coggill, P.; Heger, A.; Pollington, J.E.; Gavin, O.L.; Gunasekaran, P.; Ceric, G.; Forslund, K.; et al. The Pfam protein families database. Nucleic Acids Res. 2010, 38, D211–D222. [Google Scholar] [CrossRef]
- Letunic, I.; Doerks, T.; Bork, P. SMART 6: Recent updates and new developments. Nucleic Acids Res. 2009, 37, D229–D232. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Casagrande, J.T.; Thomas, P.D. Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 2013, 8, 1551–1566. [Google Scholar] [CrossRef]
- Attwood, T.K.; Bradley, P.; Flower, D.R.; Gaulton, A.; Maudling, N.; Mitchell, A.L.; Moulton, G.; Nordle, A.; Paine, K.; Taylor, P. PRINTS and its automatic supplement, prePRINTS. Nucleic Acids Res. 2003, 31, 400–402. [Google Scholar] [CrossRef] [Green Version]
- Corpet, F.; Servant, F.; Gouzy, J.; Kahn, D. ProDom and ProDom-CG: Tools for protein domain analysis and whole genome comparisons. Nucleic Acids Res. 2000, 28, 267–269. [Google Scholar] [CrossRef]
- He, N.; Zhang, C.; Qi, X.; Zhao, S.; Tao, Y.; Yang, G.; Lee, T.-H.; Wang, X.; Cai, Q.; Li, D. Draft genome sequence of the mulberry tree Morus notabilis. Nat. Commun. 2013, 4, 2445. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.-J.; Zhao, J.; Cai, Q.-L.; Liu, G.-C.; Wang, J.-R.; Zhao, Z.-H.; Liu, P.; Dai, L.; Yan, G.; Wang, W.-J. The complex jujube genome provides insights into fruit tree biology. Nat. Commun. 2014, 5, 5315. [Google Scholar] [CrossRef] [Green Version]
- Van Bel, M.; Diels, T.; Vancaester, E.; Kreft, L.; Botzki, A.; Van de Peer, Y.; Coppens, F.; Vandepoele, K. PLAZA 4.0: An integrative resource for functional, evolutionary and comparative plant genomics. Nucleic Acids Res. 2017, 46, D1190–D1196. [Google Scholar] [CrossRef] [PubMed]
- Zwaenepoel, A.; Van de Peer, Y. wgd—Simple command line tools for the analysis of ancient whole-genome duplications. Bioinformatics 2018, 35, 2153–2155. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dongen, S.M. Graph Clustering by Flow Simulation. Ph.D. Thesis, Utrecht University, Utrecht, The Netherlands, 2000. [Google Scholar]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Proost, S.; Fostier, J.; De Witte, D.; Dhoedt, B.; Demeester, P.; Van de Peer, Y.; Vandepoele, K. i-ADHoRe 3.0—Fast and sensitive detection of genomic homology in extremely large data sets. Nucleic Acids Res. 2011, 40, e11. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.-h.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Bowers, J.E.; Wang, X.; Ming, R.; Alam, M.; Paterson, A.H. Synteny and collinearity in plant genomes. Science 2008, 320, 486–488. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- De Bie, T.; Cristianini, N.; Demuth, J.P.; Hahn, M.W. CAFE: A computational tool for the study of gene family evolution. Bioinformatics 2006, 22, 1269–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Q.; Chen, L.-L.; Ruan, X.; Chen, D.; Zhu, A.; Chen, C.; Bertrand, D.; Jiao, W.-B.; Hao, B.-H.; Lyon, M.P. The draft genome of sweet orange (Citrus sinensis). Nat. Genet. 2013, 45, 59. [Google Scholar] [CrossRef] [PubMed]
- Verde, I.; Abbott, A.G.; Scalabrin, S.; Jung, S.; Shu, S.; Marroni, F.; Zhebentyayeva, T.; Dettori, M.T.; Grimwood, J.; Cattonaro, F. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat. Genet. 2013, 45, 487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ming, R.; VanBuren, R.; Wai, C.M.; Tang, H.; Schatz, M.C.; Bowers, J.E.; Lyons, E.; Wang, M.-L.; Chen, J.; Biggers, E. The pineapple genome and the evolution of CAM photosynthesis. Nat. Genet. 2015, 47, 1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Min, J.; Lai, R.; Wu, Z.; Chen, Y.; Yu, L.; Cheng, C.; Jin, Y.; Tian, Q.; Liu, Q. Genome-wide sequencing of longan (Dimocarpus longan Lour.) provides insights into molecular basis of its polyphenol-rich characteristics. Gigascience 2017, 6, gix023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Oka, T.; Jigami, Y. Reconstruction of de novo pathway for synthesis of UDP-glucuronic acid and UDP-xylose from intrinsic UDP-glucose in Saccharomyces cerevisiae. FEBS J. 2006, 273, 2645–2657. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A. altilis | A. heterophyllus | |||||||
|---|---|---|---|---|---|---|---|---|
| Parameters | Contig | Scaffold | Contig | Scaffold | ||||
| Length (bp) | Number | Length (bp) | Number | Length (bp) | Number | Length (bp) | Number | |
| N90 | 3361 | 52,085 | 183,851 | 637 | 4902 | 39,073 | 77,281 | 2115 |
| N50 | 16,898 | 13,662 | 1,536,010 | 151 | 26,681 | 9516 | 547,861 | 527 |
| N10 | 47,070 | 1284 | 5,076,803 | 14 | 82,850 | 846 | 1,422,119 | 54 |
| Total length | 803,695,923 | 833,038,871 | 930,343,435 | 982,020,585 | ||||
| Maximum length | 174,221 | 7,444,155 | 255,416 | 3,088,173 | ||||
| Total number ≥ 100 bp | 180,971 | 98,152 | 162,440 | 108,267 | ||||
| Total number ≥ 2000 bp | 61,693 | 4338 | 52,444 | 7263 | ||||
| N content (%) | 3.52 | 5.26 | ||||||
| BUSCOs | A. altilis | A. heterophyllus | ||
|---|---|---|---|---|
| N | P (%) | N | P (%) | |
| Complete BUSCOs | 1371 | 95.20 | 1369 | 95.00 |
| Complete single-copy | 988 | 68.60 | 932 | 64.70 |
| Complete duplicated | 383 | 26.60 | 437 | 30.30 |
| Fragmented | 14 | 1.00 | 15 | 1.00 |
| Missing | 55 | 3.80 | 56 | 4.00 |
| Species | Dataset | Number | Total Length (bp) | Base Coverage by Assembly (%) | Sequence Coverage by Assembly (%) |
|---|---|---|---|---|---|
| A. altilis | All | 141,626 | 165,794,671 | 87.7 | 97.92 |
| >200 bp | 141,626 | 165,794,671 | 87.7 | 97.92 | |
| >500 bp | 79,410 | 146,265,291 | 86.81 | 97.62 | |
| >1000 bp | 49,485 | 125,138,638 | 85.97 | 96.99 | |
| A. heterophyllus | All | 14,858 | 6,364,445 | 90.39 | 98.89 |
| >200 bp | 14,858 | 6,364,445 | 90.39 | 98.89 | |
| >500 bp | 2949 | 2,853,909 | 84.41 | 96.74 | |
| >1000 bp | 765 | 1,386,949 | 74.83 | 92.16 |
| A. altilis | A. heterophyllus | |||
|---|---|---|---|---|
| Repeat Type | in Genome (%) | Length (bp) | in Genome (%) | Length (bp) |
| SINE | 0 | 1187 | 0.03 | 384,983 |
| LINE | 0.14 | 1,214,650 | 0.99 | 9,775,316 |
| LTR | 45.95 | 382,841,531 | 36.99 | 363,293,617 |
| DNA | 2.95 | 24,608,939 | 3.76 | 36,982,825 |
| Satellite | 0 | 34,585 | 0.3 | 3,001,478 |
| Simple repeat | 0.03 | 253,818 | 0.04 | 485,582 |
| Unknown | 5.4 | 45,013,282 | 12.23 | 120,128,962 |
| Total | 52.04 | 433,486,547 | 51.01 | 500,968,186 |
| A. altilis | A. heterophyllus | F. vesca | M. domestica | M. notabilis | P. persica | Z. jujuba | |
|---|---|---|---|---|---|---|---|
| Protein-coding gene number | 34,010 | 35,858 | 34,301 | 61,721 | 27,085 | 28,701 | 37,526 |
| Mean gene length (bp) | 3545.4 | 3472.2 | 2824.6 | 2692.5 | 2866.8 | 2464.8 | 3313.5 |
| Mean cds length (bp) | 1252.6 | 1241.5 | 1174.7 | 1141.4 | 1086.9 | 1210.8 | 1353.0 |
| Mean exons per gene | 5.4 | 5.5 | 5.1 | 4.8 | 4.6 | 4.9 | 5.5 |
| Mean exon length (bp) | 227.8 | 226.5 | 232.5 | 236.7 | 236.4 | 243.6 | 246.0 |
| Mean intron length (bp) | 509.6 | 497.7 | 407.1 | 405.8 | 494.6 | 315.8 | 435.7 |
| Species | Type | Copy (w) | Average Length (bp) | Total Length (bp) | % of Genome |
|---|---|---|---|---|---|
| A. altilis | miRNA | 159 | 126.7 | 20,145 | 0.002418 |
| tRNA | 713 | 75.3 | 53,705 | 0.006447 | |
| rRNA | 466 | 183.2 | 85,353 | 0.010246 | |
| 18S | 76 | 551.4 | 41,907 | 0.005031 | |
| 28S | 98 | 125.5 | 12,296 | 0.001476 | |
| 5.8S | 32 | 135.6 | 4338 | 0.000521 | |
| 5S | 260 | 103.1 | 26,812 | 0.003219 | |
| snRNA | 1554 | 105.4 | 163,744 | 0.019656 | |
| CD-box | 1410 | 102.6 | 144,676 | 0.017367 | |
| HACA-box | 52 | 130.1 | 6765 | 0.000812 | |
| splicing | 92 | 133.7 | 12,303 | 0.001477 | |
| A. heterophyllus | miRNA | 168 | 126.3 | 21,227 | 0.002162 |
| tRNA | 689 | 75.2 | 51,813 | 0.005276 | |
| rRNA | 2706 | 268.2 | 725,709 | 0.073900 | |
| 18S | 654 | 737.5 | 482,306 | 0.049114 | |
| 28S | 920 | 123.6 | 113,667 | 0.011575 | |
| 5.8S | 242 | 151.6 | 36,699 | 0.003737 | |
| 5S | 890 | 104.5 | 93,037 | 0.009474 | |
| snRNA | 1005 | 108.2 | 108,724 | 0.011071 | |
| CD-box | 814 | 102.5 | 83,426 | 0.008495 | |
| HACA-box | 68 | 127.4 | 8665 | 0.000882 | |
| splicing | 123 | 135.2 | 16,633 | 0.001694 |
| A. altilis | A. heterophyllus | |||
|---|---|---|---|---|
| Values | Number | Percentage | Number | Percentage |
| Total | 34,010 | 100.0% | 35,858 | 100.0% |
| Nr | 33,353 | 98.1% | 35,076 | 97.8% |
| Swissprot | 26,689 | 78.5% | 27,741 | 77.4% |
| KEGG | 24,860 | 73.1% | 25,804 | 72.0% |
| COG | 12,875 | 37.9% | 13,408 | 37.4% |
| TrEMBL | 33,240 | 97.7% | 34,968 | 97.5% |
| Interpro | 26,422 | 77.7% | 27,632 | 77.1% |
| GO | 17,428 | 51.2% | 18,336 | 51.1% |
| Overall | 33,394 | 98.2% | 35,109 | 97.9% |
| Unannotated | 616 | 1.8% | 749 | 2.1% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sahu, S.K.; Liu, M.; Yssel, A.; Kariba, R.; Muthemba, S.; Jiang, S.; Song, B.; Hendre, P.S.; Muchugi, A.; Jamnadass, R.; et al. Draft Genomes of Two Artocarpus Plants, Jackfruit (A. heterophyllus) and Breadfruit (A. altilis). Genes 2020, 11, 27. https://doi.org/10.3390/genes11010027
Sahu SK, Liu M, Yssel A, Kariba R, Muthemba S, Jiang S, Song B, Hendre PS, Muchugi A, Jamnadass R, et al. Draft Genomes of Two Artocarpus Plants, Jackfruit (A. heterophyllus) and Breadfruit (A. altilis). Genes. 2020; 11(1):27. https://doi.org/10.3390/genes11010027
Chicago/Turabian StyleSahu, Sunil Kumar, Min Liu, Anna Yssel, Robert Kariba, Samuel Muthemba, Sanjie Jiang, Bo Song, Prasad S. Hendre, Alice Muchugi, Ramni Jamnadass, and et al. 2020. "Draft Genomes of Two Artocarpus Plants, Jackfruit (A. heterophyllus) and Breadfruit (A. altilis)" Genes 11, no. 1: 27. https://doi.org/10.3390/genes11010027
APA StyleSahu, S. K., Liu, M., Yssel, A., Kariba, R., Muthemba, S., Jiang, S., Song, B., Hendre, P. S., Muchugi, A., Jamnadass, R., Kao, S.-M., Featherston, J., Zerega, N. J. C., Xu, X., Yang, H., Van Deynze, A., de Peer, Y. V., Liu, X., & Liu, H. (2020). Draft Genomes of Two Artocarpus Plants, Jackfruit (A. heterophyllus) and Breadfruit (A. altilis). Genes, 11(1), 27. https://doi.org/10.3390/genes11010027