The Complete Genome of an Endogenous Nimavirus (Nimav-1_LVa) From the Pacific Whiteleg Shrimp Penaeus (Litopenaeus) Vannamei


Abstract
1. Introduction
2. Materials and Methods
2.1. Nimav-1_LVa Virus Consensus Reconstruction
2.2. Viral Gene Prediction and Visualization
2.3. Homology Searches
2.4. Dataset
3. Results
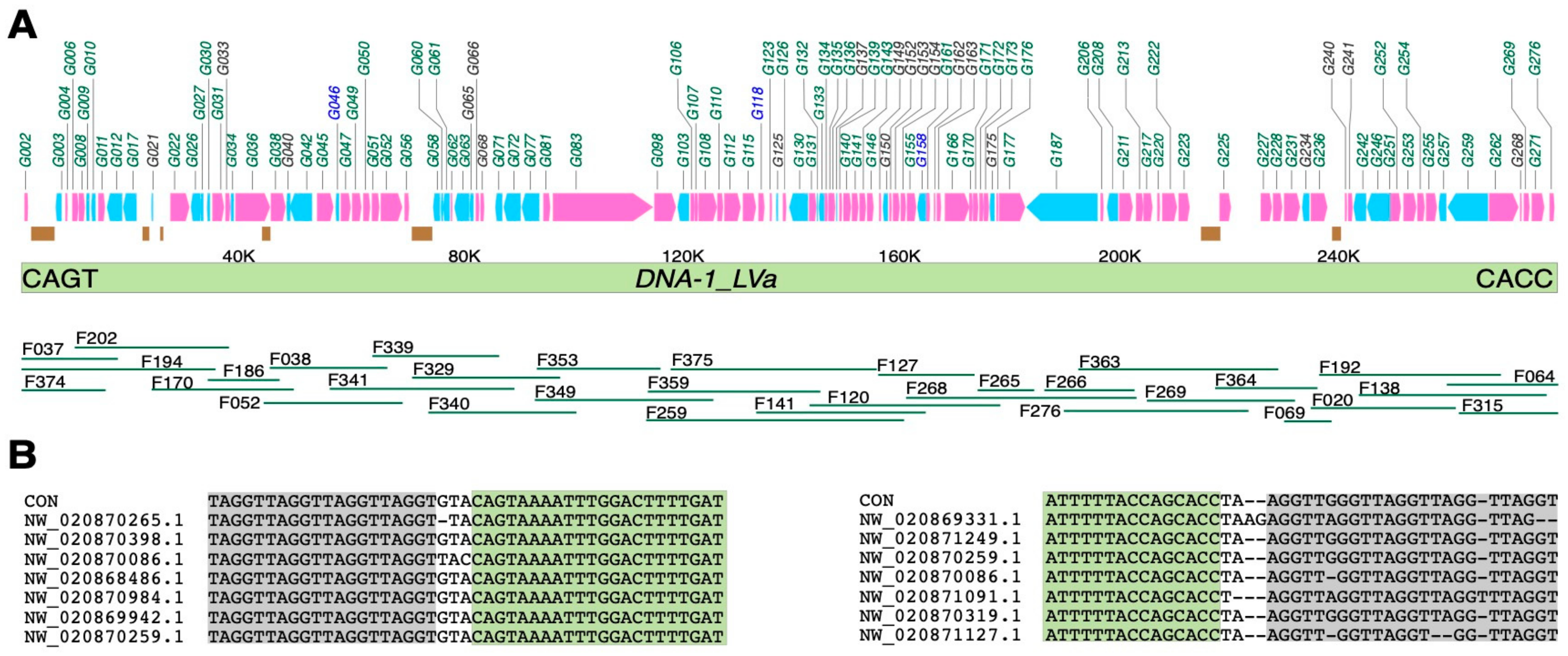
3.1. Building the Consensus of Nimav-1_LVa
3.2. The Integration Site of Nimav-1_LVa
3.3. Nimav-1_LVa Sequences in Other Penaeid Shrimps
3.4. Genes Encoded in Nimav-1_LVa
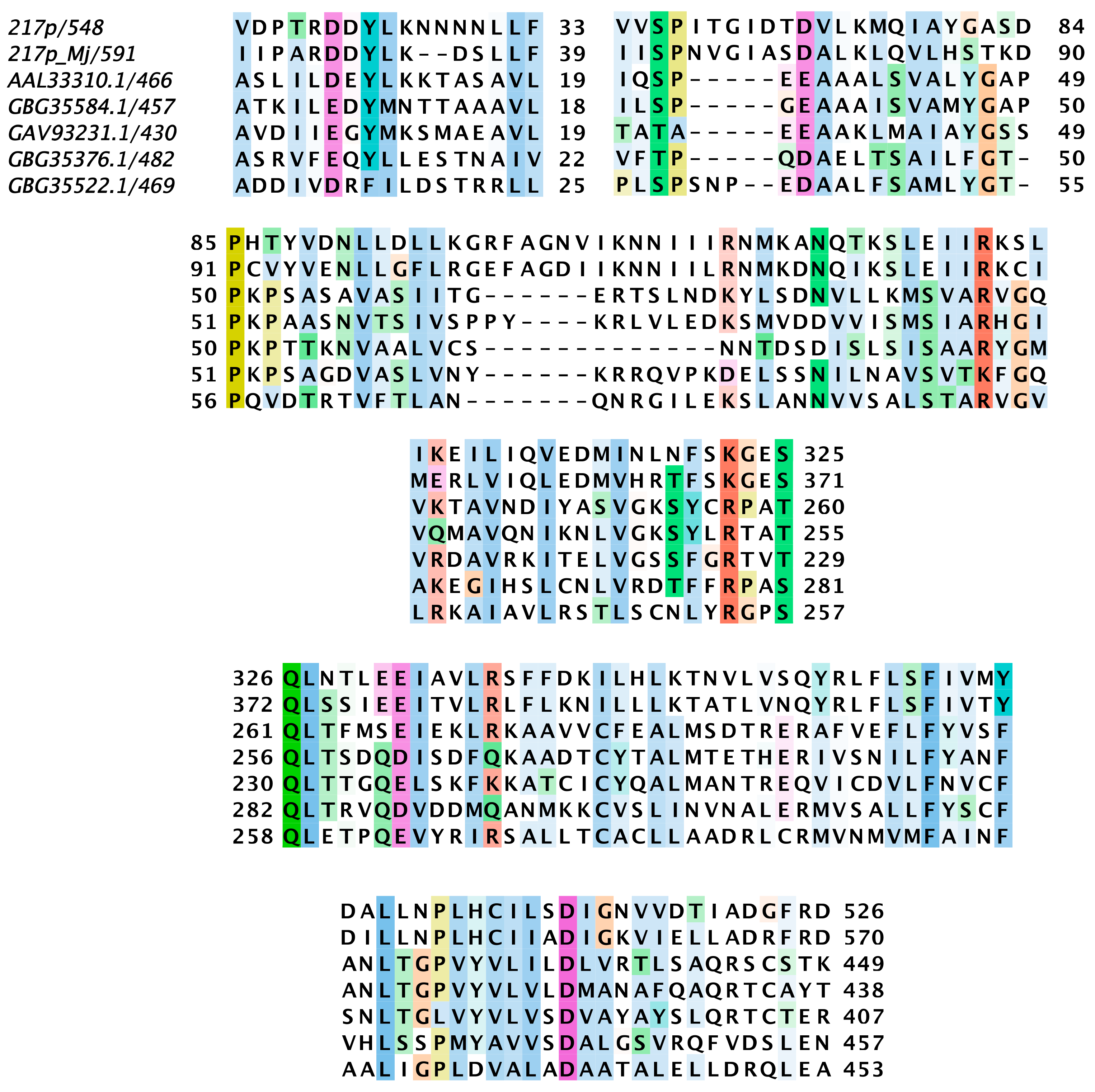
3.5. Nimav-1_LVa Genes Involved in Host-Pathogen Interaction
4. Discussion
4.1. Nimav-1_LVa Consensus Sequence
4.2. Endogenized or Free-Living Virus
4.3. Nimav-1_LVa Encoded Proteins
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alcivar-Warren, A. The Shrimp Genome and Epigenome: A Review of Genome Sizes, Transposable Elements, Simple Sequence Repeats, Integrated Viruses, and Epigenetic Components of Penaeids. J. Shellfish Res. 2020, in press. [Google Scholar]
- Lightner, D.V. Biosecurity in shrimp farming: Pathogen exclusion through use of SPF stock and routine surveillance. J. World Aquac. Soc. 2005, 36, 229–248. [Google Scholar] [CrossRef]
- Alday-Sanz, V.; Brock, J.; Flegel, T.W.; McIntosh, R.; Bondad-Reantaso, M.G.; Salazar, M.; Subasinghe, R. Facts, truths and myths about SPF shrimp in Aquaculture. Rev. Aquac. 2018. [Google Scholar] [CrossRef]
- Bao, W.; Bogden, R.; Tao, Q.; Iyer, S.; Mikhaylenko, G.; Wittendorp, J.; Mraz, A.; Hart, E.; Hatas, E.; Kujawa, S.; et al. Transposable Elements, Simple Sequence Repeats, and Integrated Viruses in Specific Pathogen-Free (SPF) Shrimp, Penaeus (Litopenaeus) Vannamei, Domesticated by the Breeding Program of the US Marine Shrimp Farming Program (USMSFP). Genes 2020, in press. [Google Scholar]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef]
- Zhang, X.; Yuan, J.; Sun, Y.; Li, S.; Gao, Y.; Yu, Y.; Liu, C.; Wang, Q.; Lv, X.; Zhang, X.; et al. Penaeid shrimp genome provides insights into benthic adaptation and frequent molting. Nat. Commun. 2019, 10, 356. [Google Scholar] [CrossRef]
- Yang, Q.; Dong, X.; Xie, G.; Fu, S.; Zou, P.; Sun, J.; Wang, Y.; Huang, J. Comparative genomic analysis unravels the transmission pattern and intra-species divergence of acute hepatopancreatic necrosis disease (AHPND)-causing Vibrio parahaemolyticus strains. Mol. Genet. Genom. 2019, 294, 1007–1022. [Google Scholar] [CrossRef]
- Feng, S.Y.; Liang, G.F.; Xu, Z.S.; Li, A.F.; Du, J.X.; Song, G.N.; Ren, S.Y.; Yang, Y.L.; Jiang, G. Meta-analysis of antiviral protection of white spot syndrome virus vaccine to the shrimp. Fish Shellfish Immunol. 2018, 81, 260–265. [Google Scholar] [CrossRef]
- Oakey, J.; Smith, C.; Underwood, D.; Afsharnasab, M.; Alday-Sanz, V.; Dhar, A.; Sivakumar, S.; Sahul Hameed, A.S.; Beattie, K.; Crook, A. Global distribution of white spot syndrome virus genotypes determined using a novel genotyping assay. Arch. Virol. 2019, 164, 2061–2082. [Google Scholar] [CrossRef]
- Stentiford, G.D.; Lightner, D.V. Cases of white spot disease (WSD) in European shrimp farms. Aquaculture 2011, 319, 302–306. [Google Scholar] [CrossRef]
- Zhan, W.; Wang, Y.; Fryer, J.L.; Yu, K.; Fukuda, H.; Meng, Q. White spot syndrome virus infection of cultured shrimp in China. J. Aquat. Anim. Health 1998, 10, 405–410. [Google Scholar] [CrossRef]
- Knibb, W.; Le, C.; Katouli, M.; Bar, I.; Lloyd, C. Assessment of the origin of white spot syndrome virus DNA sequences in farmed Penaeus monodon in Australia. Aquaculture 2018, 494, 26–29. [Google Scholar] [CrossRef]
- Mohan, C.V.; Shankar, K.M.; Kulkarni, S.; Sudha, P.M. Histopathology of cultured shrimp showing gross signs of yellow head syndrome and white spot syndrome during 1994 Indian epizootics. Dis. Aquat. Organ. 1998, 34, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Walker, P.J.; Mohan, C.V. Viral disease emergence in shrimp aquaculture: Origins, impact and the effectiveness of health management strategies. Rev. Aquac. 2009, 1, 125–154. [Google Scholar] [CrossRef]
- Tang, K.F.J.; Le Groumellec, M.; Lightner, D.V. Novel, closely related, white spot syndrome virus (WSSV) genotypes from Madagascar, Mozambique and the Kingdom of Saudi Arabia. Dis. Aquat. Org. 2013, 106, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Van Hulten, M.C.; Witteveldt, J.; Peters, S.; Kloosterboer, N.; Tarchini, R.; Fiers, M.; Sandbrink, H.; Lankhorst, R.K.; Vlak, J.M. The white spot syndrome virus DNA genome sequence. Virology 2001, 286, 7–22. [Google Scholar] [CrossRef]
- Sánchez-Paz, A. White spot syndrome virus: An overview on an emergent concern. Vet. Res. 2010, 41, 43. [Google Scholar] [CrossRef]
- Wang, H.C.; Hirono, I.; Maningas, M.B.B.; Somboonwiwat, K.; Stentiford, G.; Ictv, R.C. ICTV Virus Taxonomy Profile: Nimaviridae. J. Gen. Virol. 2019, 100, 1053–1054. [Google Scholar] [CrossRef]
- Stentiford, G.D.; Bonami, J.R.; Alday-Sanz, V. A critical review of susceptibility of crustaceans to Taura syndrome, Yellowhead disease and White Spot Disease and implications of inclusion of these diseases in European legislation. Aquaculture 2009, 291, 1–17. [Google Scholar] [CrossRef]
- Jiang, L.; Xiao, J.; Liu, L.; Pan, Y.; Yan, S.; Wang, Y. Characterization and prevalence of a novel white spot syndrome viral genotype in naturally infected wild crayfish, Procambarus clarkii, in Shanghai, China. Virusdisease 2017, 28, 250–261. [Google Scholar] [CrossRef]
- Parrilla-Taylor, D.P.; Vibanco-Pérez, N.; Durán-Avelar, M.J.; Gomez-Gil, B.; Llera-Herrera, R.; Vázquez-Juárez, R. Molecular variability and genetic structure of white spot syndrome virus strains from northwest Mexico based on the analysis of genomes. FEMS Microbiol. Lett. 2018, 365. [Google Scholar] [CrossRef] [PubMed]
- Utari, H.B.; Soowannayan, C.; Flegel, T.W.; Whityachumnarnkul, B.; Kruatrachue, M. Variable RNA expression from recently acquired, endogenous viral elements (EVE) of white spot syndrome virus (WSSV) in shrimp. Dev. Comp. Immunol. 2017, 76, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Taengchaiyaphum, S.; Srisala, J.; Bunphimpapha, P.; Supungul, P.; Tassanakajon, A.; Chaiyapechara, S.; Bowornpinyo, S.; Sritunyalucksana, K.; Flegel, T.W. Mendelian inheritance of endogenous viral elements (EVE) of white spot syndrome virus (WSSV) in shrimp. Dev. Comp. Immunol. 2019, 96, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Niu, G.J.; Wang, S.; Xu, J.D.; Yang, M.C.; Sun, J.J.; He, Z.H.; Zhao, X.F.; Wang, J.X. The polymeric immunoglobulin receptor-like protein from Marsupenaeus japonicus is a receptor for white spot syndrome virus infection. PLoS Pathog. 2019, 15, e1007558. [Google Scholar] [CrossRef]
- Cuéllar-Anjel, J.; White-Noble, B.; Schofield, P.; Chamorro, R.; Lightner, D.V. Report of significant WSSV-resistance in the Pacific white shrimp, Litopenaeus vannamei, from a Panamanian breeding program. Aquaculture 2012, 368, 36–39. [Google Scholar] [CrossRef]
- Trang, T.T.; Hung, N.H.; Ninh, N.H.; Knibb, W.; Nguyen, N.H. Genetic Variation in Disease Resistance Against White Spot Syndrome Virus (WSSV) in Liptopenaeus vannamei. Front. Genet. 2019, 10, 264. [Google Scholar] [CrossRef]
- Rozenberg, A.; Brand, P.; Rivera, N.; Leese, F.; Schubart, C.D. Characterization of fossilized relatives of the White Spot Syndrome Virus in genomes of decapod crustaceans. BMC Evol. Biol. 2015, 15, 142. [Google Scholar] [CrossRef]
- Kawato, S.; Shitara, A.; Wang, Y.; Nozaki, R.; Kondo, H.; Hirono, I. Crustacean Genome Exploration Reveals the Evolutionary Origin of White Spot Syndrome Virus. J. Virol. 2019, 93, e01144-18. [Google Scholar] [CrossRef]
- Bao, W. DNA viruses from the shrimp genome. Repbase Rep. 2018, 18, 1352. [Google Scholar]
- Bao, W.; Alcivar-Warren, A.; Bogden, R.; Tao, Q.; Iyer, S.; Mikhaylenko, G.; Wittendorp, J.; Mraz, A.; Hart, E.; Hatas, E.; et al. A fossilized white spot syndrome virus-like element (DNAV-1_LVa) in the genome of the original specific pathogen-free (SPF) shrimp Penaeus (Litopenaeus) vannamei domesticated by the breeding program of the US Marine Shrimp Farming Program (USMSFP) from Hawaii, USA. In Proceedings of the Aquaculture 2019, New Orleans, LA, USA, 7–11 March 2019; p. 80. [Google Scholar]
- Iranzo, J.; Krupovic, M.; Koonin, E.V. The Double-Stranded DNA Virosphere as a Modular Hierarchical Network of Gene Sharing. mBio 2016, 7, e00978-16. [Google Scholar] [CrossRef]
- Smit, A.F.A.; Hubley, R. RepeatModeler Open-1.0. 2008–2015. Available online: http://www.repeatmasker.org (accessed on 2 November 2019).
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Solovyev, V.; Kosarev, P.; Seledsov, I.; Vorobyev, D. Automatic annotation of eukaryotic genes, pseudogenes and promoters. Genome Biol. 2006, 7, S10.1–S10.12. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef]
- Kohany, O.; Gentles, A.J.; Hankus, L.; Jurka, J. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinform. 2006, 7, 474. [Google Scholar] [CrossRef]
- HMMER: Biosequence Analysis Using Profile Hidden Markov Models. Available online: http://hmmer.org/ (accessed on 2 November 2019).
- Alcivar-Warren, A.; Meehan-Meola, D.; Wang, Y.; Guo, X.; Zhou, L.; Xiang, J.; Moss, S.; Arce, S.; Warren, W.; Xu, Z.; et al. Isolation and mapping of telomeric pentanucleotide (TAACC)n repeats of the Pacific whiteleg shrimp, Penaeus vannamei, using fluorescence in situ hybridization. Mar. Biotechnol. (NY) 2006, 8, 467–480. [Google Scholar] [CrossRef]
- Arbuckle, J.H.; Medveczky, M.M.; Luka, J.; Hadley, S.H.; Luegmayr, A.; Ablashi, D.; Lund, T.C.; Tolar, J.; De Meirleir, K.; Montoya, J.G.; et al. The latent human herpesvirus-6A genome specifically integrates in telomeres of human chromosomes in vivo and in vitro. Proc. Natl. Acad. Sci. USA 2010, 107, 5563–5568. [Google Scholar] [CrossRef]
- Pantry, S.N.; Medveczky, P.G. Latency, Integration, and Reactivation of Human Herpesvirus-6. Viruses 2017, 9, 194. [Google Scholar] [CrossRef]
- Wood, M.L.; Royle, N.J. Chromosomally Integrated Human Herpesvirus 6: Models of Viral Genome Release from the Telomere and Impacts on Human Health. Viruses 2017, 9, 184. [Google Scholar] [CrossRef]
- Osterrieder, N.; Wallaschek, N.; Kaufer, B.B. Herpesvirus Genome Integration into Telomeric Repeats of Host Cell Chromosomes. Annu. Rev. Virol. 2014, 1, 215–235. [Google Scholar] [CrossRef]
- Kheimar, A.; Previdelli, R.L.; Wight, D.J.; Kaufer, B.B. Telomeres and Telomerase: Role in Marek’s Disease Virus Pathogenesis, Integration and Tumorigenesis. Viruses 2017, 9, 173. [Google Scholar] [CrossRef] [PubMed]
- Silke, J.; Vucic, D. IAP family of cell death and signaling regulators. In Methods in enzymology; Ashkenazi, A., Wells, J.A., Yuan, J., Eds.; Elsevier: Amsterdam, The Netherlands, 2014; Volume 545, pp. 35–65. [Google Scholar]
- Jin, H.S.; Lee, D.H.; Kim, D.H.; Chung, J.H.; Lee, S.J.; Lee, T.H. cIAP1, cIAP2, and XIAP act cooperatively via nonredundant pathways to regulate genotoxic stress-induced nuclear factor-kappaB activation. Cancer Res. 2009, 69, 1782–1791. [Google Scholar] [CrossRef] [PubMed]
- Galbán, S.; Duckett, C.S. XIAP as a ubiquitin ligase in cellular signaling. Cell Death Differ. 2010, 17, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Yang, F. Localization studies of two white spot syndrome virus structural proteins VP51 and VP76. Virol. J. 2006, 3, 76. [Google Scholar] [CrossRef]
- Wang, P.H.; Huang, T.; Zhang, X.; He, J.G. Antiviral defense in shrimp: From innate immunity to viral infection. Antivir. Res. 2014, 108, 129–141. [Google Scholar] [CrossRef]
- Radons, J. The human HSP70 family of chaperones: Where do we stand? Cell Stress Chaperones 2016, 21, 379–404. [Google Scholar] [CrossRef]
- Yan, F.; Xia, D.; Hu, J.; Yuan, H.; Zou, T.; Zhou, Q.; Liang, L.; Qi, Y.; Xu, H. Heat shock cognate protein 70 gene is required for prevention of apoptosis induced by WSSV infection. Arch. Virol. 2010, 155, 1077–1083. [Google Scholar] [CrossRef]
- Tassanakajon, A.; Somboonwiwat, K.; Supungul, P.; Tang, S. Discovery of immune molecules and their crucial functions in shrimp immunity. Fish Shellfish Immunol. 2013, 34, 954–967. [Google Scholar] [CrossRef]
- Valentim-Neto, P.A.; Moser, J.R.; Fraga, A.P.; Marques, M.R. Hsp70 expression in shrimp Litopenaeus vannamei in response to IHHNV and WSSV infection. Virusdisease 2014, 25, 437–440. [Google Scholar] [CrossRef]
- Janewanthanakul, S.; Supungul, P.; Tang, S.; Tassanakajon, A. Heat shock protein 70 from Litopenaeus vannamei (LvHSP70) is involved in the innate immune response against white spot syndrome virus (WSSV) infection. Dev. Comp. Immunol. 2020, 102, 103476. [Google Scholar] [CrossRef]
- Dong, C.W.; Zhang, Y.B.; Zhang, Q.Y.; Gui, J.F. Differential expression of three Paralichthys olivaceus Hsp40 genes in responses to virus infection and heat shock. Fish Shellfish Immunol. 2006, 21, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Vidya, R.; Gireesh-Babu, P.; Pani Prasad, K. White spot syndrome virus Manipulates Ubiquitin Gene Expression in Penaeus monodon. Indian J. Virol. 2013, 24, 82–84. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yi, S.; Li, Y.; Shi, L.; Zhang, L. Novel Insights into Antiviral Gene Regulation of Red Swamp Crayfish, Procambarus clarkii, Infected with White Spot Syndrome Virus. Genes 2017, 8, 320. [Google Scholar] [CrossRef] [PubMed]
- Lertwimol, T.; Sangsuriya, P.; Phiwsaiya, K.; Senapin, S.; Phongdara, A.; Boonchird, C.; Flegel, T.W. Two new anti-apoptotic proteins of white spot syndrome virus that bind to an effector caspase (PmCasp) of the giant tiger shrimp Penaeus (Penaeus) monodon. Fish Shellfish Immunol. 2014, 38, 1–6. [Google Scholar] [CrossRef]
- Ventura-López, C.; Gómez-Anduro, G.; Arcos, F.G.; Llera-Herrera, R.; Racotta, I.S.; Ibarra, A.M. A novel CHH gene from the Pacific white shrimp Litopenaeus vannamei was characterized and found highly expressed in gut and less in eyestalk and other extra-eyestalk tissues. Gene 2016, 582, 148–160. [Google Scholar] [CrossRef]
- Ohira, T. Crustacean Hyperglycemic Hormone. In Handbook of Hormones; Takei, Y., Ando, H., Tsutsui, K., Eds.; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Zuo, H.; Yuan, J.; Niu, S.; Yang, L.; Weng, S.; He, J.; Xu, X. A molting-inhibiting hormone-like protein from Pacific white shrimp Litopenaeus vannamei is involved in immune responses. Fish Shellfish Immunol. 2018, 72, 544–551. [Google Scholar] [CrossRef]
- Wanlem, S.; Supamattaya, K.; Tantikitti, C.; Prasertsan, P.; Graidist, P. Expression and applications of recombinant crustacean hyperglycemic hormone from eyestalks of white shrimp (Litopenaeus vannamei) against bacterial infection. Fish Shellfish Immunol. 2011, 30, 877–885. [Google Scholar] [CrossRef]
- Xu, L.; Pan, L.; Zhang, X.; Wei, C. Crustacean hyperglycemic hormone (CHH) affects hemocyte intracellular signaling pathways to regulate exocytosis and immune response in white shrimp Litopenaeus vannamei. Peptides 2019, 116, 30–41. [Google Scholar] [CrossRef]
- Phelan, P.; Stebbings, L.A.; Baines, R.A.; Bacon, J.P.; Davies, J.A.; Ford, C. Drosophila Shaking-B protein forms gap junctions in paired Xenopus oocytes. Nature 1998, 391, 181–184. [Google Scholar] [CrossRef]
- Güiza, J.; Barría, I.; Sáez, J.C.; Vega, J.L. Innexins: Expression, Regulation, and Functions. Front. Physiol. 2018, 9, 1414. [Google Scholar] [CrossRef]
- Wang, S.P.; Chen, F.Y.; Dong, L.X.; Zhang, Y.Q.; Chen, H.Y.; Qiao, K.; Wang, K.J. A novel innexin2 forming membrane hemichannel exhibits immune responses and cell apoptosis in Scylla paramamosain. Fish Shellfish Immunol. 2015, 47, 485–499. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Li, M.; Zhang, Y.; Pang, Z.; Xiao, W.; Yang, Y.; Luo, K. A role for Innexin2 and Innexin3 proteins from Spodoptera litura in apoptosis. PLoS ONE 2013, 8, e70456. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-B.; Xiao, W.; Li, M.; Zhang, Y.; Yang, Y.; Hu, J.-S.; Luo, K.-J. N-terminally elongated SpliInx2 and SpliInx3 reduce baculovirus-triggered apoptosis via hemichannel closure. Arch. Insect Biochem. Physiol. 2016, 92, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, M.; Webb, B. Perspectives on polydnavirus origins and evolution. Adv. Virus Res. 2002, 58, 203–254. [Google Scholar] [PubMed]
- Tanaka, K.; Lapointe, R.; Barney, W.E.; Makkay, A.M.; Stoltz, D.; Cusson, M.; Webb, B.A. Shared and species-specific features among ichnovirus genomes. Virology 2007, 363, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Dupuy, C.; Huguet, E.; Drezen, J.M. Unfolding the evolutionary story of polydnaviruses. Virus Res. 2006, 117, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Chae, H.J.; Kim, H.R.; Xu, C.; Bailly-Maitre, B.; Krajewska, M.; Krajewski, S.; Banares, S.; Cui, J.; Digicaylioglu, M.; Ke, N.; et al. BI-1 regulates an apoptosis pathway linked to endoplasmic reticulum stress. Mol. Cell 2004, 15, 355–366. [Google Scholar] [CrossRef]
- Bultynck, G.; Kiviluoto, S.; Henke, N.; Ivanova, H.; Schneider, L.; Rybalchenko, V.; Luyten, T.; Nuyts, K.; De Borggraeve, W.; Bezprozvanny, I.; et al. The C Terminus of Bax Inhibitor-1 Forms a Ca2+-permeable Channel Pore. J. Biol. Chem. 2012, 287, 2544–2557. [Google Scholar] [CrossRef]
- Hückelhoven, R. BAX Inhibitor-1, an ancient cell death suppressor in animals and plants with prokaryotic relatives. Apoptosis 2004, 9, 299–307. [Google Scholar] [CrossRef]
- Roney, K.; Holl, E.; Ting, J. Immune plexins and semaphorins: Old proteins, new immune functions. Protein Cell 2013, 4, 17–26. [Google Scholar] [CrossRef]
- Nishide, M.; Kumanogoh, A. The role of semaphorins in immune responses and autoimmune rheumatic diseases. Nat. Rev. Rheumatol. 2018, 14, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, Y.; Takamatsu, H.; Okuno, T.; Kang, S.; Nojima, S.; Kimura, T.; Kataoka, T.R.; Ikawa, M.; Toyofuku, T.; Katayama, I.; et al. Identification of semaphorin 4B as a negative regulator of basophil-mediated immune responses. J. Immunol. 2011, 186, 2881–2888. [Google Scholar] [CrossRef] [PubMed]
- Takamatsu, H.; Kumanogoh, A. Diverse roles for semaphorin-plexin signaling in the immune system. Trends Immunol. 2012, 33, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Gui, J. Applications of genetic breeding biotechnologies in Chinese aquaculture. In Aquaculture in China: Success Stories and Modern Trends; Gui, J.-F., Tang, Q., Li, Z., Liu, J., De Silva, S.S., Eds.; Wiley Online Library: Hoboken, NJ, USA, 2018. [Google Scholar]
- Thézé, J.; Leclercq, S.; Moumen, B.; Cordaux, R.; Gilbert, C. Remarkable Diversity of Endogenous Viruses in a Crustacean Genome. Genome Biol. Evol. 2014, 6, 2129–2140. [Google Scholar] [CrossRef]
- Orosco, F.L.; Lluisma, A.O. Variation in virome diversity in wild populations of Penaeus monodon (Fabricius 1798) with emphasis on pathogenic viruses. Virusdisease 2017, 28, 262–271. [Google Scholar] [CrossRef]
- Drezen, J.M.; Provost, B.; Espagne, E.; Cattolico, L.; Dupuy, C.; Poirié, M.; Periquet, G.; Huguet, E. Polydnavirus genome: Integrated vs. free virus. J. Insect Physiol. 2003, 49, 407–417. [Google Scholar] [CrossRef]
- Whitfield, J.B.; Asgari, S. Virus or not? Phylogenetics of polydnaviruses and their wasp carriers. J. Insect Physiol. 2003, 49, 397–405. [Google Scholar] [CrossRef]
- He, Y.; Yang, K.; Zhang, X. Viral microRNAs targeting virus genes promote virus infection in shrimp in vivo. J. Virol. 2014, 88, 1104–1112. [Google Scholar] [CrossRef]
- Wang, P.H.; He, J.G. Nucleic Acid Sensing in Invertebrate Antiviral Immunity. Int. Rev. Cell Mol. Biol. 2019, 345, 287–360. [Google Scholar]
- Peruzza, L.; Shekhar, M.S.; Kumar, K.V.; Swathi, A.; Karthic, K.; Hauton, C.; Vijayan, K.K. Temporal changes in transcriptome profile provide insights of White Spot Syndrome Virus infection in Litopenaeus vannamei. Sci. Rep. 2019, 9, 13509. [Google Scholar] [CrossRef]
- Verbruggen, B.; Bickley, L.; van Aerle, R.; Bateman, K.; Stentiford, G.; Santos, E.; Tyler, C. Molecular Mechanisms of White Spot Syndrome Virus Infection and Perspectives on Treatments. Viruses 2016, 8, 23. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Nimaviruses | GenBank Accessions | Size (Kb) 1 |
|---|---|---|
| White spot syndrome virus (WSSV) | AF332093.3 | 305.119 |
| C. opilio bacilliform virus (CoBV) | BDLS01000001 and BDLS01000002 | 237.1 |
| M. japonicus endogenous nimavirus (Mj) | BFCD01000001 and AP010878 | ~220 |
| P. monodon endogenous nimavirus (Pm) | BFCF01000001 to BFCF01000003 | 191.8 |
| H. takanoi endogenous nimavirus (Ht) | BFCC01000001 to BFCC01000006 | 218.1 |
| M. ensis endogenous nimavirus (Me) | BFCE01000001 to BFCE01000010 | 232.4 |
| S. intermedium endogenous nimavirus (Si) | BFCG01000001 to BFCG01000014 | 189 |
| Nimavirus Type | Length (Identity 1) | |
|---|---|---|
| P. monodon | M. japonicus | |
| Nimav-1_LVa | >141 Kb (>99%) | >33 Kb (>99%) |
| Mj-like | >200 Kb (>91%) | >49 Kb (>88%) |
| Pm-like | >226 Kb (>88%) | >199 Kb (>88%) |
| Genes 1 | CDS start | CDS end | Direction | Protein (AA) | Viral Homolog 2 | Comment 3 |
|---|---|---|---|---|---|---|
| g002 | 1388 | 1990 | d | 201 | g009 | PF1 |
| g003 | 7002 | 8099 | r | 217 | AKS10635.1 | PF2, 4 exons, 2 BIR domains (cd00022) |
| g004 | 8792 | 9187 | d | 132 | BFCD01000001.1 (98,829–99,209) | |
| g006 | 10,037 | 11,149 | d | 371 | g008 | PF1 |
| g008 | 11,219 | 12,316 | d | 366 | g006 | PF1 |
| g009 | 12,527 | 13,123 | r | 199 | g002 | PF1 |
| g010 | 13,467 | 14,258 | r | 264 | g161 | PF1 |
| g011 | 14,749 | 15,966 | d | 406 | g006 | PF1 |
| g012 | 16,319 | 19,114 | r | 725 | AKS10635.1 | PF2, 4 exons, 3 BIR domains (cd00022), 1 RING-HC_BIRC2_3_7 (cd16713) |
| g017 | 19,256 | 21,711 | r | 710 | AKS10635.1 | PF2, 4 exons, 3 BIR domains (cd00022), 1 RING-HC_BIRC2_3_7 (cd16713) |
| g021 | 24,393 | 24,710 | r | 106 | ||
| g022 | 27,849 | 31,305 | d | 782 | AP010878.1 (14,919–16,765) | 5 exons |
| g026 | 31,556 | 33,238 | r | 561 | AP010878.1 (4458-6026) | |
| g027 | 33,243 | 33,857 | r | 205 | AP010878.1 (3787–4395) | |
| g030 | 34,488 | 35,048 | r | 187 | AKS10635.1 | PF2, 1 BIR domain (cd00022) |
| g031 | 3,5478 | 37,592 | d | 705 | GBG35399.1 | wsv220-like, capsid protein |
| g033 | 37,838 | 38,707 | d | 290 | ||
| g034 | 38,729 | 39,364 | r | 212 | GBG35402.1 | wsv206-like, PF6, containing macro domain (cd02749), a high-affinity ADP-ribose binding module, as shown in GBG35398. |
| g036 | 39,656 | 45,952 | d | 2099 | BFCD01000001.1 (62,195–69,574) | |
| g038 | 46,087 | 48,771 | d | 895 | BFCD01000001.1 (57,879–62,171) | |
| g040 | 48,899 | 49,639 | r | 247 | ||
| g042 | 49,557 | 53,573 | r | 1339 | GBG35397.1 | wsv026-like |
| g045 | 54,485 | 57,460 | d | 1138 | GBG35396.1 | wsv115-like, envelope protein |
| g046 | 57,833 | 58,502 | r | 123 | 3 exons, CHH-like, containing crust_neurohorm domain (pfam01147) | |
| g047 | 58,782 | 60,584 | d | 448 | AKS10635.1 | PF2, 4 exons, 2 BIR domains (cd00022), 1 RING-HC_BIRC4_8 (cd16714) |
| g049 | 60,851 | 62,533 | d | 412 | AKS10635.1 | PF2, 5 exons, 2 BIR domains (cd00022), 1 RING-HC_BIRC4_8 (cd16714) |
| g050 | 62,862 | 64,124 | d | 421 | g051 | PF3 |
| g051 | 64,435 | 65,805 | d | 457 | g050 | PF3 |
| g052 | 66,042 | 69,944 | d | 1301 | BFCD01000001.1 (167,202–171,290) | PF3 |
| g056 | 70,367 | 71,194 | d | 276 | g269 | PF5 |
| g058 | 75,545 | 76,801 | r | 419 | BFCD01000001.1 (79,831–81,120) | |
| g060 | 76,972 | 77,379 | r | 136 | BFCD01000001.1 (79,176–79,601) | |
| g061 | 77,382 | 78,608 | r | 409 | GBG35401.1 | wsv415-like, capsid protein |
| g062 | 78,779 | 79,048 | d | 90 | BFCD01000001.1 (77,744–77,460) | |
| g063 | 79,405 | 82,122 | r | 906 | GBG35400.1 | wsv216-like, envelope protein |
| g065 | 82,204 | 82,896 | r | 231 | ||
| g066 | 83,301 | 83,948 | d | 216 | ||
| g068 | 84,162 | 84,830 | d | 223 | ||
| g071 | 86,831 | 88,126 | r | 432 | GBG35404.1 | wsv161-like |
| g072 | 88,344 | 91,454 | r | 1037 | GBG35405.1 | wsv011-like, envelope protein |
| g077 | 91,612 | 94,842 | r | 1077 | GBG35406.1 | wsv313-like |
| g081 | 95,577 | 96,821 | d | 415 | GBG35407.1 | wsv282-like |
| g083 | 97,302 | 115,505 | d | 6068 | GBG35408.1 | wsv360-like, capsid protein |
| g098 | 115,730 | 119,659 | d | 1310 | GBG35428.1 | wsv037-like, capsid protein |
| g103 | 119,938 | 122,058 | r | 707 | GBG35427.1 | molecular chaperone DnaK (HSP70) protein domain (COG0443) |
| g106 | 122,293 | 123,042 | d | 250 | BFCD01000001.1 (191,561–192,277) | |
| g107 | 123,054 | 123,677 | d | 208 | GBG35426.1 | wsv021-like, envelope protein |
| g108 | 123,758 | 127,078 | d | 1107 | GBG35425.1 | wsv139-like |
| g110 | 127,163 | 128,206 | d | 348 | GBG35424.1 | wsv137-like |
| g112 | 128,419 | 131,400 | d | 994 | GBG35423.1 | wsv192-like |
| g115 | 131,798 | 134,191 | d | 798 | GBG35356.1 | SCV_095-like, ATP-dependent DNA ligase I (dnl1) domain (TIGR00574) and Poly (ADP-ribose) polymerase and DNA-Ligase Zn-finger (pfam00645) |
| g118 | 134,634 | 135,728 | d | 365 | DnaJ/Hsp40 protein, containing DnaJ-class molecular chaperone with C-terminal Zn finger domain (COG0484) | |
| g123 | 136,705 | 137,067 | d | 121 | GBG35422.1 | wsv136-like |
| g125 | 137,840 | 138,184 | r | 115 | ||
| g126 | 139,067 | 139,678 | d | 204 | BFCD01000001.1 (181,829–182,488) | |
| g130 | 140,199 | 143,633 | r | 1145 | GBG35421.1 | wsv271-like, capsid protein |
| g131 | 143,809 | 145,152 | d | 448 | GBG35420.1 | wsv131-like |
| g132 | 145,203 | 145,433 | r | 77 | BFCD01000001.1 (176,011–176,340) | ubiquitin-like (Ubl) domain (cd01803) found in ubiquitin |
| g133 | 145,548 | 146,687 | r | 380 | GBG35419.1 | wsv325-like, envelope protein |
| g134 | 146,697 | 147,203 | d | 169 | BFCD01000001.1 (172,895–173,647) | |
| g135 | 147,318 | 148,061 | d | 248 | GBG35417.1 | wsv133-like |
| g136 | 147,643 | 148,566 | d | 308 | GBG35418.1 | wsv134-like |
| g137 | 148,715 | 148,927 | r | 71 | ||
| g139 | 149,321 | 149,815 | d | 165 | GBG35402.1 | wsv206-like, PF6, containing macro domain (cd02749), a high-affinity ADP-ribose binding module, as shown in GBG35398. |
| g140 | 150,049 | 151,434 | d | 462 | BFCC01000003.1 (801–2426) | wsv112-like, dUTPase, containing deoxyuridine 5’-triphosphate nucleotidohydrolase (dut) domain (TIGR00576) |
| g141 | 151,587 | 152,777 | d | 397 | g143 | PF1 |
| g143 | 152,974 | 154,158 | d | 395 | g141 | PF1 |
| g146 | 154,360 | 155,547 | d | 396 | g006 | PF1 |
| g149 | 156,541 | 156,915 | d | 125 | ||
| g150 | 157,204 | 158,148 | r | 315 | ||
| g152 | 158,385 | 158,825 | d | 147 | ||
| g153 | 159,042 | 160,274 | d | 411 | ||
| g154 | 160,465 | 161,412 | d | 316 | ||
| g155 | 161,589 | 163,290 | d | 428 | BFCD01000001.1 (51,019–52,483) | 4 exons, innexin domain (pfam00876) |
| g158 | 163,455 | 165,615 | r | 274 | 5 exons, Bax inhibitor (BI)-1 domain (cd10430). | |
| g161 | 165,152 | 165,772 | d | 207 | g010 | PF1 |
| g162 | 166,492 | 166,767 | d | 92 | ||
| g163 | 167,017 | 167,787 | d | 257 | ||
| g166 | 168,473 | 172,924 | d | 1484 | BBD20107.1 | wsv209-like, envelope protein |
| g170 | 172,897 | 173,529 | d | 211 | AP010878.1 (53,502–54,038) | |
| g171 | 173,590 | 174,540 | d | 317 | BBD20108.1 | wsv267-like, anti-apoptotic protein |
| g172 | 174,735 | 175,523 | d | 263 | AP010878.1 (55,291–56,157) | PF4 |
| g173 | 175,556 | 176,485 | d | 310 | AP010878.1 (55,291–56,157) | PF4 |
| g175 | 176,584 | 177,510 | r | 309 | ||
| g176 | 177,844 | 178,152 | d | 103 | BBD20109.1 | wsv293a-like, envelope protein |
| g177 | 178,301 | 183,058 | d | 1586 | GBG35554.1 | wsv289-like, capsid protein |
| g187 | 183,225 | 196,220 | r | 4332 | BBD20111.1 | wsv343-like |
| g206 | 196,741 | 197,286 | d | 182 | BFCG01000002.1 (22,541–22,065) | SCV_028-like |
| g208 | 197,905 | 199,944 | r | 680 | BBD20112.1 | wsv327-like, envelope protein |
| g211 | 200,119 | 202,590 | d | 824 | BBD20113.1 | wsv332-like |
| g213 | 203,182 | 204,525 | d | 448 | BBD20114.1 | wsv306-like, tegument protein |
| g217 | 204,533 | 206,176 | d | 548 | AP010878.1 (81,486–83,258) | wsv308-like, capsid protein |
| g220 | 207,060 | 207,515 | d | 152 | AP010878.1 (84,507–85,430) | |
| g222 | 207,948 | 210,626 | d | 893 | BBD20115.1 | wsv285-like |
| g223 | 210,916 | 212,928 | d | 671 | AP010878.1 (89,194–91,152) | |
| g225 | 218,360 | 220,426 | d | 689 | GBG35515.1 | wsv226-like |
| g227 | 225,839 | 227,851 | d | 671 | AP010878.1 (111,284–114,223) | semaphorin 1A (Sema_1A) domain (cd11237) |
| g228 | 228,056 | 229,738 | d | 561 | AP010878.1 (109,235–110,863) | |
| g231 | 230,064 | 232,973 | d | 970 | GBG35403.1 | wsv035-like, envelope protein |
| g234 | 233,341 | 234,681 | r | 447 | In GenBank, part of 234p is computationally predicted as high mobility group protein DSP1-like (XP_027238145.1), 184 AA. | |
| g236 | 234,925 | 237,936 | d | 1004 | BFCD01000001.1 (86,470–89,643) | |
| g240 | 241,130 | 241,501 | d | 124 | ||
| g241 | 241,716 | 242,537 | d | 274 | ||
| g242 | 242,655 | 244,988 | r | 778 | GBG35414.1 | wsv303-like |
| g246 | 245,095 | 249,063 | r | 1323 | GBG35413.1 | wsv433-like |
| g251 | 249,059 | 249,457 | r | 133 | GBG35412.1 | wsv432-like |
| g252 | 249,459 | 251,285 | d | 609 | GBG35411.1 | wsv427-like |
| g253 | 251,758 | 254,091 | d | 778 | BFCD01000001.1 (134,943–137,015) | |
| g254 | 254,266 | 255,603 | d | 446 | GBG35410.1 | wsv423-like, Protein kinase 1 |
| g255 | 255,856 | 257,829 | d | 658 | GBG35409.1 | wsv440-like |
| g257 | 258,137 | 259,543 | r | 469 | g050 | PF3 |
| g259 | 259,757 | 267,121 | r | 2455 | GBG35416.1 | wsv514-like, DNA polymerase |
| g262 | 267,305 | 272,629 | d | 1775 | GBG35415.1 | wsv447-like |
| g268 | 272,942 | 273,223 | d | 94 | ||
| g269 | 273,601 | 274,653 | d | 351 | g271 | PF5 |
| g271 | 275,034 | 277,334 | d | 767 | g269 | PF5 |
| g276 | 278,291 | 279,160 | d | 290 | AP010878.1 (55,291–56,157) | PF4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bao, W.; Tang, K.F.J.; Alcivar-Warren, A. The Complete Genome of an Endogenous Nimavirus (Nimav-1_LVa) From the Pacific Whiteleg Shrimp Penaeus (Litopenaeus) Vannamei. Genes 2020, 11, 94. https://doi.org/10.3390/genes11010094
Bao W, Tang KFJ, Alcivar-Warren A. The Complete Genome of an Endogenous Nimavirus (Nimav-1_LVa) From the Pacific Whiteleg Shrimp Penaeus (Litopenaeus) Vannamei. Genes. 2020; 11(1):94. https://doi.org/10.3390/genes11010094
Chicago/Turabian StyleBao, Weidong, Kathy F. J. Tang, and Acacia Alcivar-Warren. 2020. "The Complete Genome of an Endogenous Nimavirus (Nimav-1_LVa) From the Pacific Whiteleg Shrimp Penaeus (Litopenaeus) Vannamei" Genes 11, no. 1: 94. https://doi.org/10.3390/genes11010094
APA StyleBao, W., Tang, K. F. J., & Alcivar-Warren, A. (2020). The Complete Genome of an Endogenous Nimavirus (Nimav-1_LVa) From the Pacific Whiteleg Shrimp Penaeus (Litopenaeus) Vannamei. Genes, 11(1), 94. https://doi.org/10.3390/genes11010094