Splicing Characteristics of Dystrophin Pseudoexons and Identification of a Novel Pathogenic Intronic Variant in the DMD Gene

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient and mRNA Analysis
2.2. Dystrophin Pseudoexons
2.3. In Silico Prediction
2.3.1. Splice Site and Branch Point
2.3.2. Numbers and Densities of Auxiliary Splicing Regulatory Elements
2.4. Statistical Analysis
3. Results
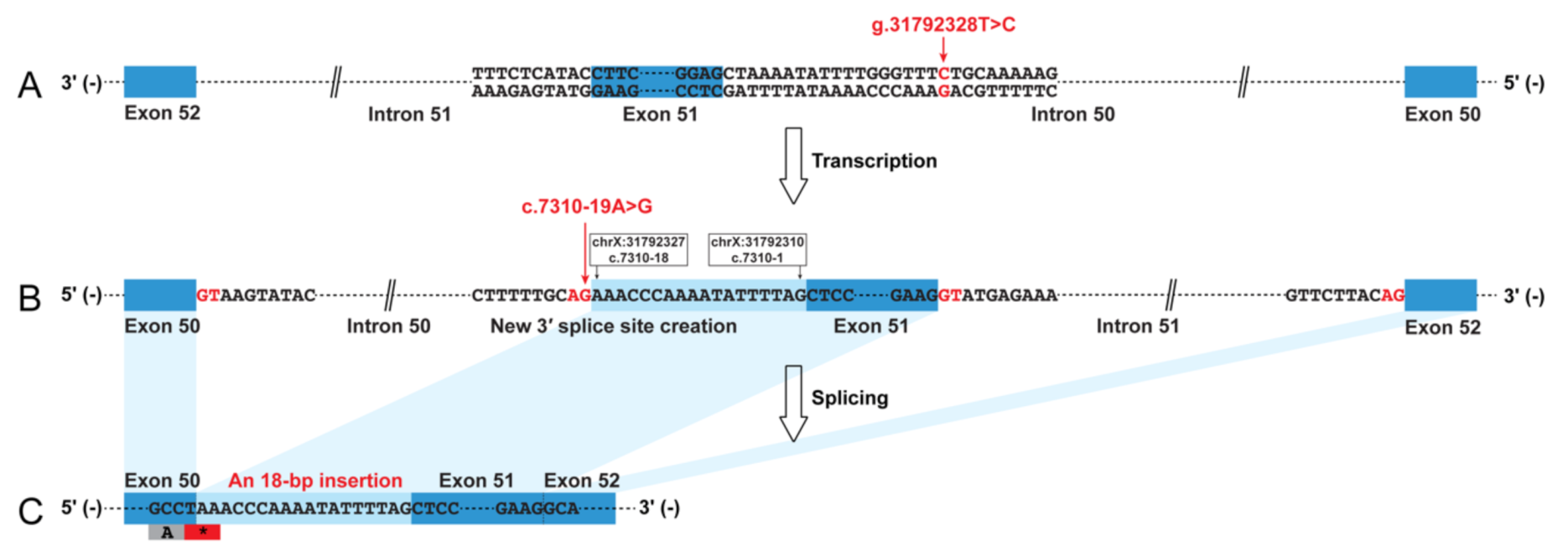
3.1. Dystrophin Protein and mRNA Analysis
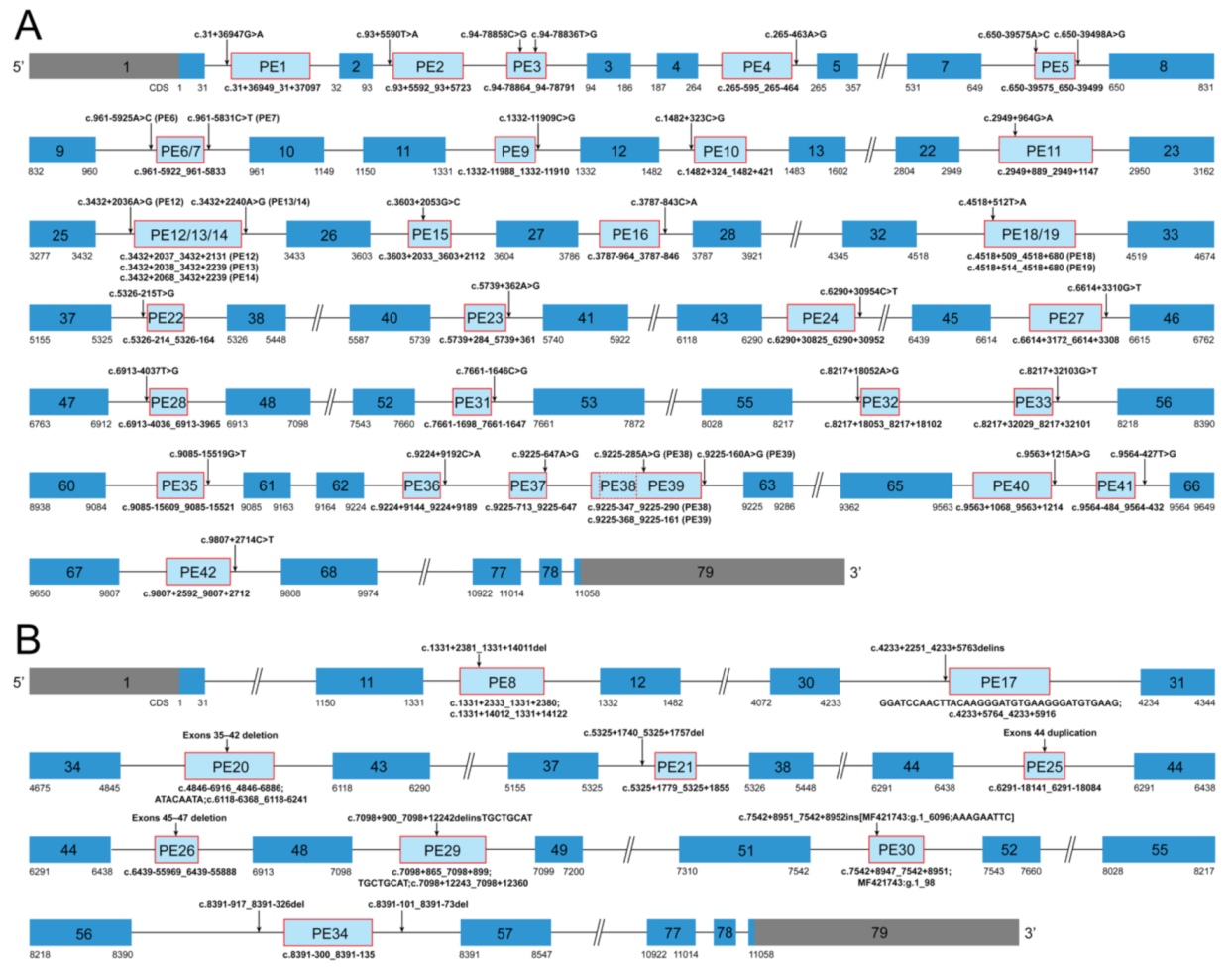
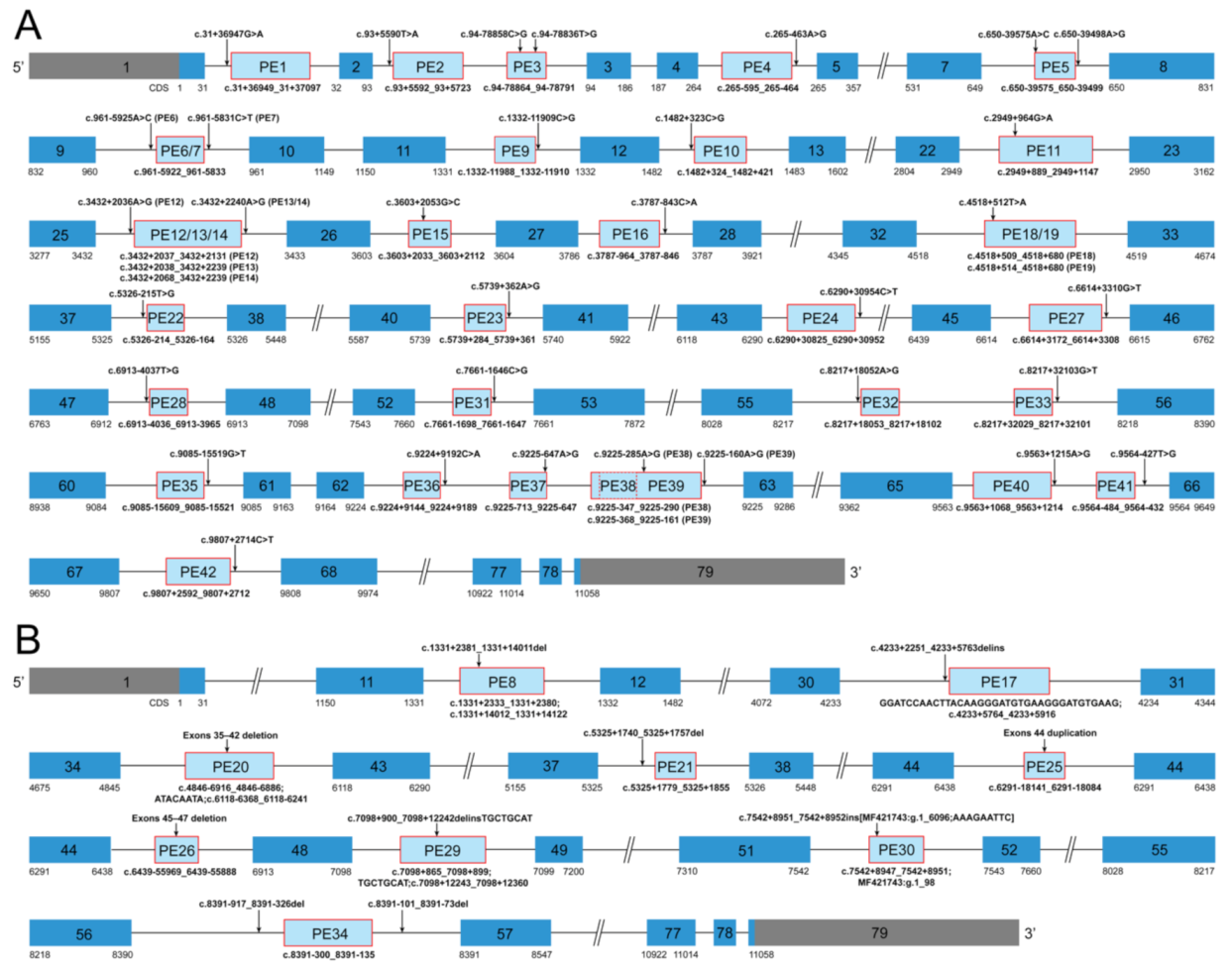
3.2. Summary of Dystrophin Pseudoexons
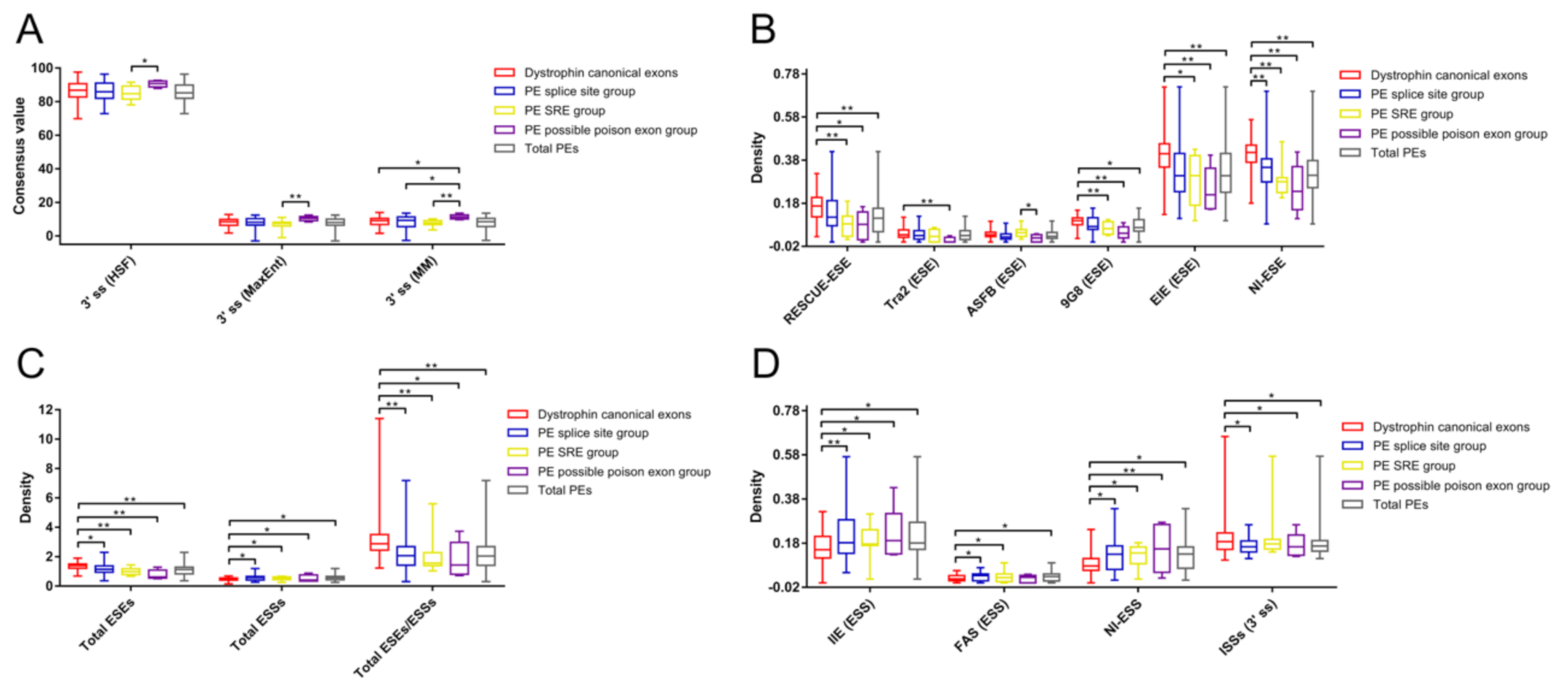
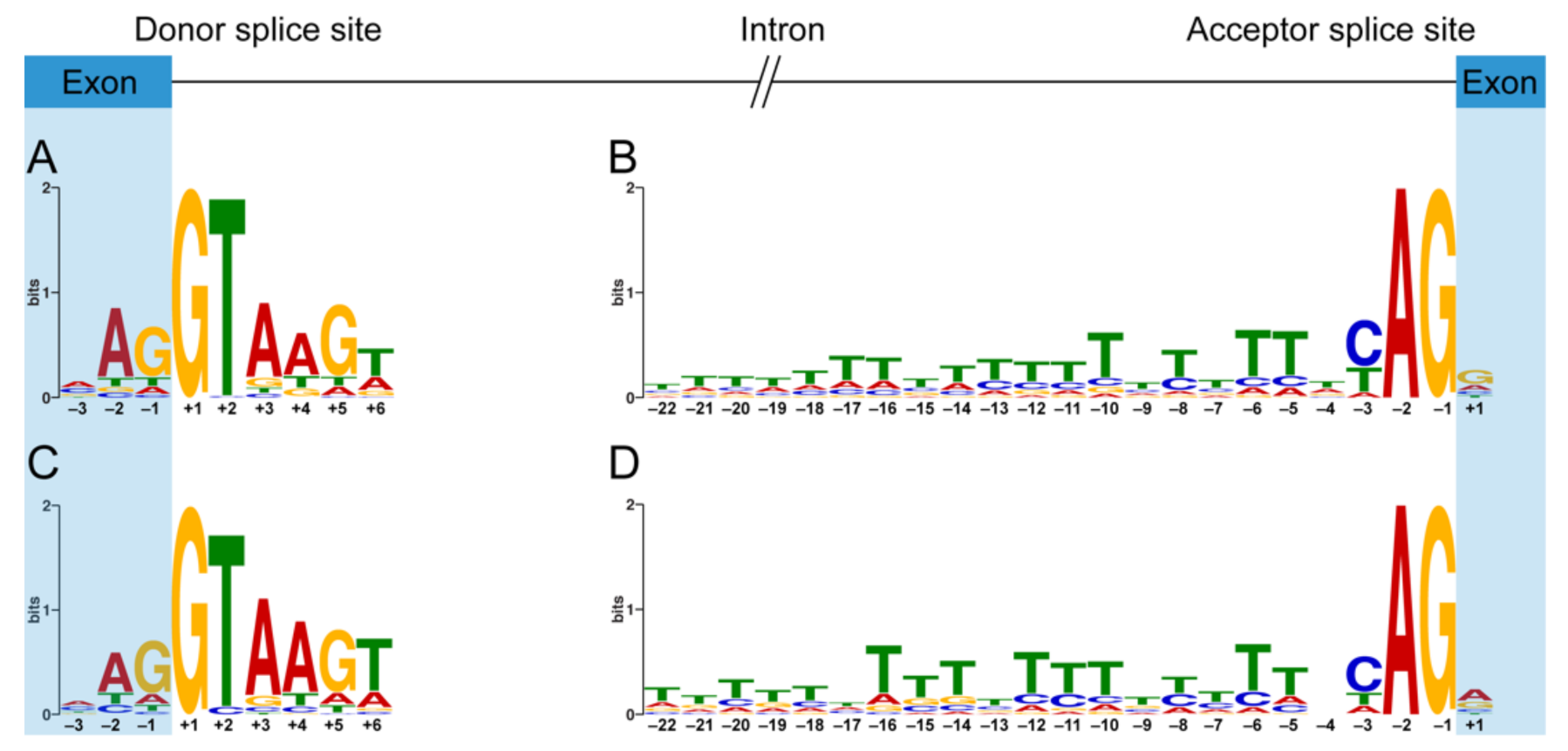
3.3. Comparative Analyses of Essential Splicing Signals
3.4. Comparative Analyses of Auxiliary Splicing Regulatory Elements
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003, 2, 731–740. [Google Scholar] [CrossRef]
- Bladen, C.L.; Salgado, D.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; Dawkins, H.; Lamont, L.; Roy, A.J.; Chamova, T.; et al. The TREAT-NMD DMD global database: Analysis of more than 7000 duchenne muscular dystrophy mutations. Hum. Mutat. 2015, 36, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Keegan, N.P. Pseudoexons of the DMD Gene. J. Neuromuscul. Dis. 2020, 7, 77–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, M.; Li, J.J.; Xu, G.R.; Wang, N.; Wang, Z.Q. Cryptic exon activation causes dystrophinopathy in two Chinese families. Eur. J. Hum. Genet. 2020, 28, 947–955. [Google Scholar] [CrossRef]
- Corvelo, A.; Hallegger, M.; Smith, C.W.J.; Eyras, E. Genome-wide association between branch point properties and alternative splicing. PLoS Comput. Biol. 2010, 6, e1001016. [Google Scholar] [CrossRef] [Green Version]
- Xiong, H.Y.; Alipanahi, B.; Lee, L.J.; Bretschneider, H.; Merico, D.; Yuen, R.K.C.; Hua, Y.; Gueroussov, S.; Najafabadi, H.S.; Hughes, T.R.; et al. The human splicing code reveals new insights into the genetic determinants of disease. Science (80-) 2015, 347, 1254806. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Li, W.H.; Krainer, A.R.; Zhang, M.Q. RNA landscape of evolution for optimal exon and intron discrimination. Proc. Natl. Acad. Sci. USA 2008, 105, 5797–5802. [Google Scholar] [CrossRef] [Green Version]
- Juan-Mateu, J.; González-Quereda, L.; Rodríguez, M.J.; Verdura, E.; Lázaro, K.; Jou, C.; Nascimento, A.; Jiménez-Mallebrera, C.; Colomer, J.; Monges, S.; et al. Interplay between DMD Point mutations and splicing signals in dystrophinopathy phenotypes. PLoS ONE 2013, 8, e59916. [Google Scholar] [CrossRef] [Green Version]
- Gurvich, O.L.; Tuohy, T.M.; Howard, M.T.; Finkel, R.S.; Medne, L.; Anderson, C.B.; Weiss, R.B.; Wilton, S.D.; Flanigan, K.M. DMD pseudoexon mutations: Splicing efficiency, phenotype, and potential therapy. Ann. Neurol. 2008, 63, 81–89. [Google Scholar] [CrossRef]
- Zhang, Z.; Habara, Y.; Nishiyama, A.; Oyazato, Y.; Yagi, M.; Takeshima, Y.; Matsuo, M. Identification of seven novel cryptic exons embedded in the dystrophin gene and characterization of 14 cryptic dystrophin exons. J. Hum. Genet. 2007, 52, 607–617. [Google Scholar] [CrossRef]
- Santos, R.; Gonçalves, A.; Oliveira, J.; Vieira, E.; Vieira, J.P.; Evangelista, T.; Moreno, T.; Santos, M.; Fineza, I.; Bronze-Da-Rocha, E. New variants, challenges and pitfalls in DMD genotyping: Implications in diagnosis, prognosis and therapy. J. Hum. Genet. 2014, 59, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Sun, C.; Liu, Y.; Yu, M.; Zheng, Y.; Meng, L.; Wang, G.; Cornejo, D.M.; Bharadwaj, T.; Yan, J.; et al. Practical approach to the genetic diagnosis of unsolved dystrophinopathies: A stepwise strategy in the genomic era. J. Med. Genet. 2020. [Google Scholar] [CrossRef] [PubMed]
- den Dunnen, J.T.; Dalgleish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; Mcgowan-Jordan, J.; Roux, A.F.; Smith, T.; Antonarakis, S.E.; Taschner, P.E.M. HGVS recommendations for the description of sequence variants: 2016 update. Hum. Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davydov, E.V.; Goode, D.L.; Sirota, M.; Cooper, G.M.; Sidow, A.; Batzoglou, S. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol. 2010, 6, e1001025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME Suite: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, w202–w208. [Google Scholar] [CrossRef]
- Desmet, F.-O.; Hamroun, D.; Lalande, M.; Collod-Bëroud, G.; Claustres, M.; Béroud, C. Human splicing finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [Green Version]
- Yeo, G.; Burge, C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef]
- Fairbrother, W.G.; Yeh, R.F.; Sharp, P.A.; Burge, C.B. Predictive identification of exonic splicing enhancers in human genes. Science (80-) 2002, 297, 1007–1013. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Rolish, M.E.; Yeo, G.; Tung, V.; Mawson, M.; Burge, C.B. Systematic identification and analysis of exonic splicing silencers. Cell 2004, 119, 831–845. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.H.F.; Chasin, L.A. Computational definition of sequence motifs governing constitutive exon splicing. Genes Dev. 2004, 18, 1241–1250. [Google Scholar] [CrossRef] [Green Version]
- Vorechovsky, I. Transposable elements in disease-associated cryptic exons. Hum. Genet. 2010, 127, 135–154. [Google Scholar] [CrossRef] [PubMed]
- Sironi, M.; Menozzi, G.; Riva, L.; Cagliani, R.; Comi, G.P.; Bresolin, N.; Giorda, R.; Pozzoli, U. Silencer elements as possible inhibitors of pseudoexon splicing. Nucleic Acids Res. 2004, 32, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.J.; Zhang, C.; Wang, J.; Chew, S.L.; Zhang, M.Q.; Krainer, A.R. An increased specificity score matrix for the prediction of SF2/ASF-specific exonic splicing enhancers. Hum. Mol. Genet. 2006, 15, 2490–2508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadler, M.B.; Shomron, N.; Yeo, G.W.; Schneider, A.; Xiao, X.; Burge, C.B. Inference of splicing regulatory activities by sequence neighborhood analysis. PLoS Genet. 2006, 2, e191. [Google Scholar] [CrossRef] [PubMed]
- Suyama, M.; Harrington, E.D.; Vinokourova, S.; Doeberitz, M.V.K.; Ohara, O.; Bork, P. A network of conserved co-occurring motifs for the regulation of alternative splicing. Nucleic Acids Res. 2010, 38, 7916–7926. [Google Scholar] [CrossRef] [Green Version]
- Churbanov, A.; Vořechovský, I.; Hicks, C. Computational prediction of splicing regulatory elements shared by Tetrapoda organisms. BMC Genom. 2009, 10, 508. [Google Scholar] [CrossRef] [Green Version]
- Piva, F.; Giulietti, M.; Nocchi, L.; Principato, G. SpliceAid: A database of experimental RNA target motifs bound by splicing proteins in humans. Bioinformatics 2009, 25, 1211–1213. [Google Scholar] [CrossRef]
- Carvill, G.L.; Engel, K.L.; Ramamurthy, A.; Cochran, J.N.; Roovers, J.; Stamberger, H.; Lim, N.; Schneider, A.L.; Hollingsworth, G.; Holder, D.H.; et al. Aberrant inclusion of a poison exon causes dravet syndrome and related SCN1A-associated genetic epilepsies. Am. J. Hum. Genet. 2018, 103, 1022–1029. [Google Scholar] [CrossRef] [Green Version]
- Burset, M.; Seledtsov, I.A.; Solovyev, V.V. SpliceDB: Database of canonical and non-canonical mammalian splice sites. Nucleic Acids Res. 2001, 29, 255–259. [Google Scholar] [CrossRef] [Green Version]
- Wahl, M.C.; Will, C.L.; Lührmann, R. The spliceosome: Design principles of a dynamic RNP machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef] [Green Version]
- McNaughton, J.C.; Hughes, G.; Jones, W.A.; Stockwell, P.A.; Klamut, H.J.; Petersen, G.B. The evolution of an intron: Analysis of a long, deletion-prone intron in the human dystrophin gene. Genomics 1997, 40, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- De Conti, L.; Baralle, M.; Buratti, E. Exon and intron definition in pre-mRNA splicing. Wiley Interdiscip. Rev. RNA 2013, 4, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, M.H.; Wu, X.; Kodani, A.; Fan, J.; Doan, R.; Ozawa, M.; Ma, J.; Yoshida, N.; Reiter, J.F.; et al. Cell-type-specific alternative splicing governs cell fate in the developing cerebral cortex. Cell 2016, 166, 1147–1162.e5. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ma, M.; Xiao, X.; Wang, Z. Intronic splicing enhancers, cognate splicing factors and context-dependent regulation rules. Nat. Struct. Mol. Biol. 2012, 19, 1044–1053. [Google Scholar] [CrossRef] [Green Version]
- Ke, S.; Zhang, X.H.F.; Chasin, L.A. Positive selection acting on splicing motifs reflects compensatory evolution. Genome Res. 2008, 18, 533–543. [Google Scholar] [CrossRef] [Green Version]
- Thomas, J.D.; Polaski, J.T.; Feng, Q.; De Neef, E.J.; Hoppe, E.R.; McSharry, M.V.; Pangallo, J.; Gabel, A.M.; Belleville, A.E.; Watson, J.; et al. RNA isoform screens uncover the essentiality and tumor-suppressor activity of ultraconserved poison exons. Nat. Genet. 2020, 52, 84–94. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, Z.; Tang, L.; Xie, Z.; Sun, C.; Shuai, H.; Zhou, C.; Liu, Y.; Yu, M.; Zheng, Y.; Meng, L.; et al. Splicing Characteristics of Dystrophin Pseudoexons and Identification of a Novel Pathogenic Intronic Variant in the DMD Gene. Genes 2020, 11, 1180. https://doi.org/10.3390/genes11101180
Xie Z, Tang L, Xie Z, Sun C, Shuai H, Zhou C, Liu Y, Yu M, Zheng Y, Meng L, et al. Splicing Characteristics of Dystrophin Pseudoexons and Identification of a Novel Pathogenic Intronic Variant in the DMD Gene. Genes. 2020; 11(10):1180. https://doi.org/10.3390/genes11101180
Chicago/Turabian StyleXie, Zhiying, Liuqin Tang, Zhihao Xie, Chengyue Sun, Haoyue Shuai, Chao Zhou, Yilin Liu, Meng Yu, Yiming Zheng, Lingchao Meng, and et al. 2020. "Splicing Characteristics of Dystrophin Pseudoexons and Identification of a Novel Pathogenic Intronic Variant in the DMD Gene" Genes 11, no. 10: 1180. https://doi.org/10.3390/genes11101180
APA StyleXie, Z., Tang, L., Xie, Z., Sun, C., Shuai, H., Zhou, C., Liu, Y., Yu, M., Zheng, Y., Meng, L., Zhang, W., Leal, S. M., Wang, Z., Schrauwen, I., & Yuan, Y. (2020). Splicing Characteristics of Dystrophin Pseudoexons and Identification of a Novel Pathogenic Intronic Variant in the DMD Gene. Genes, 11(10), 1180. https://doi.org/10.3390/genes11101180