A Case Report of Left Atrial Isomerism in a Syndromic Context

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
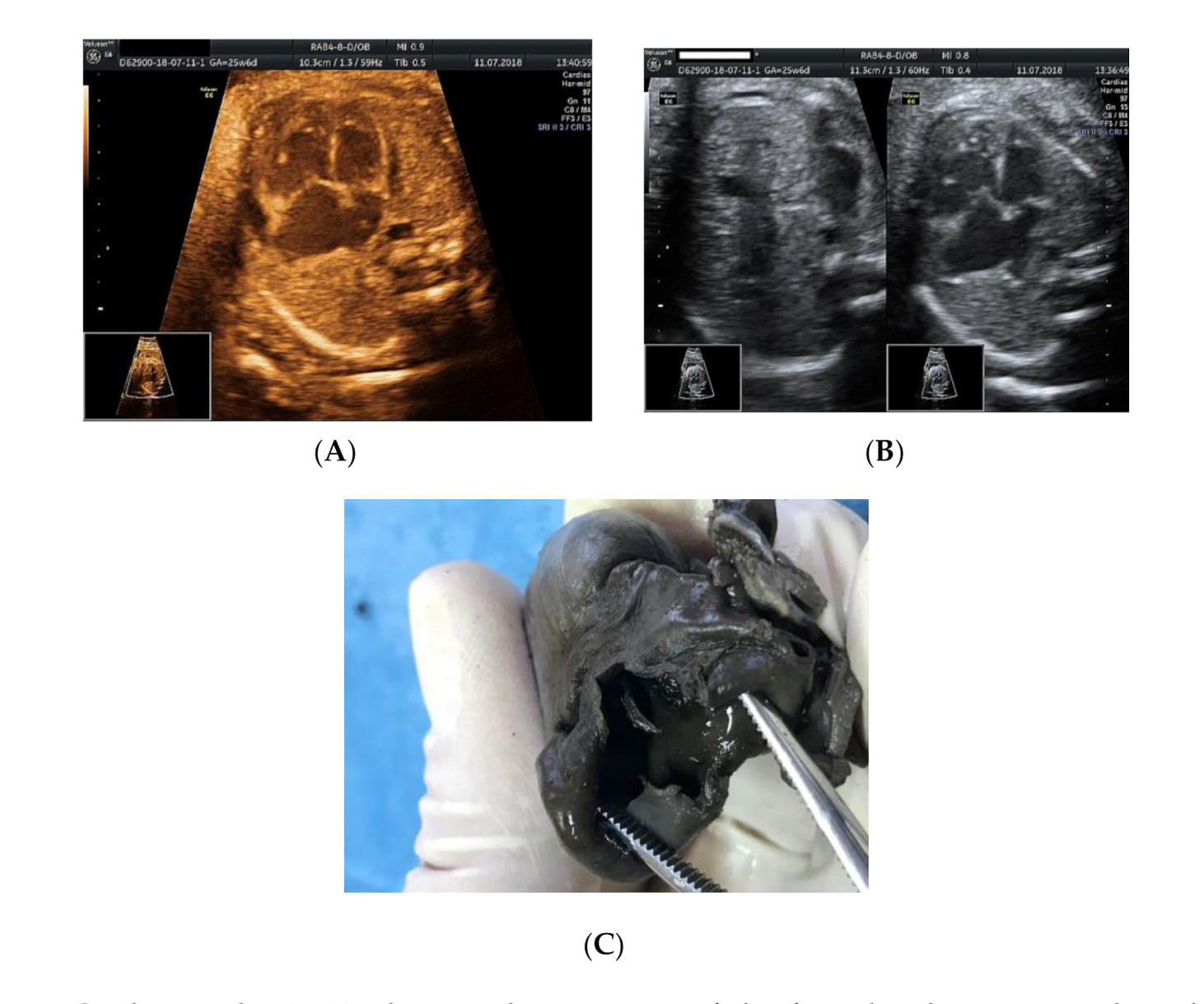
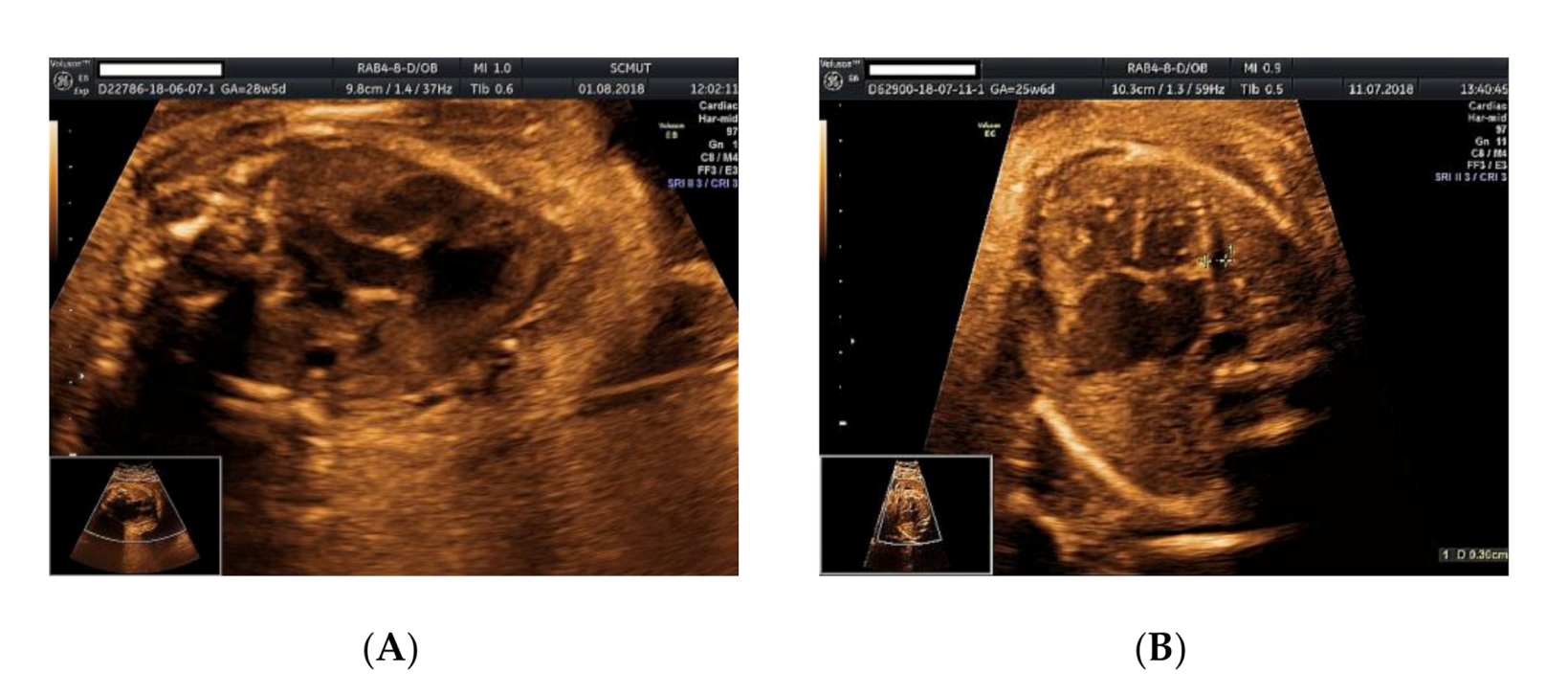
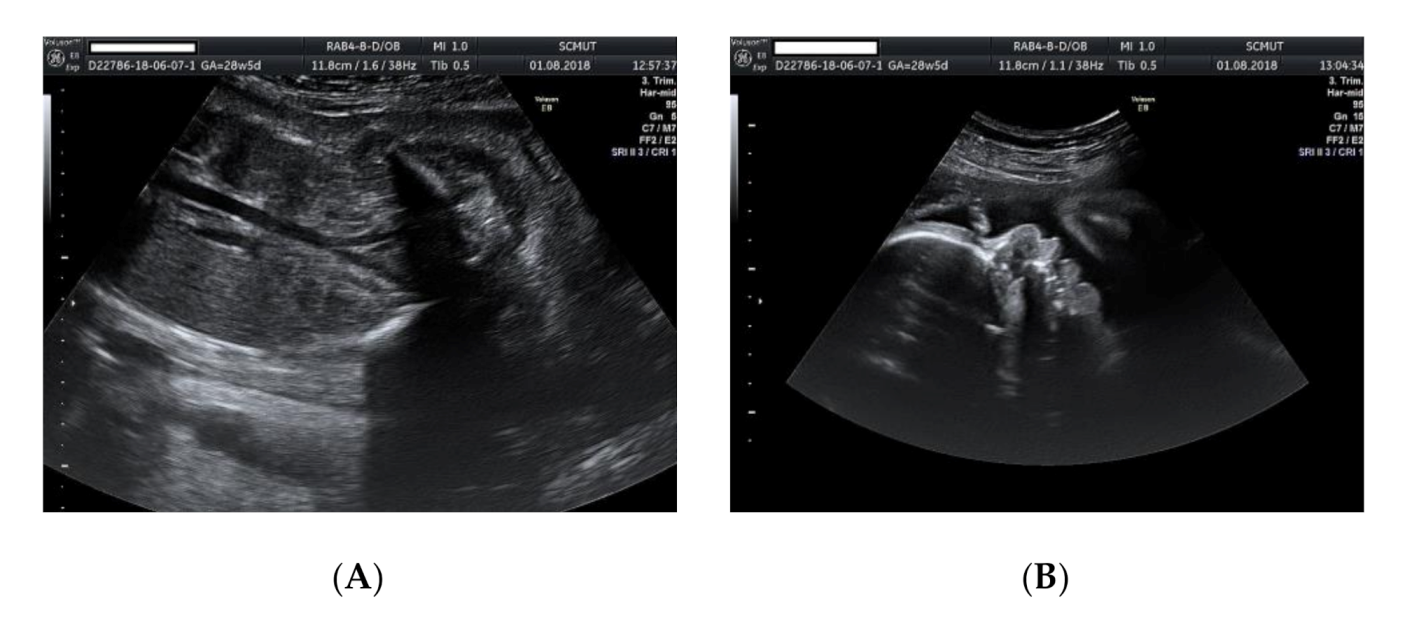
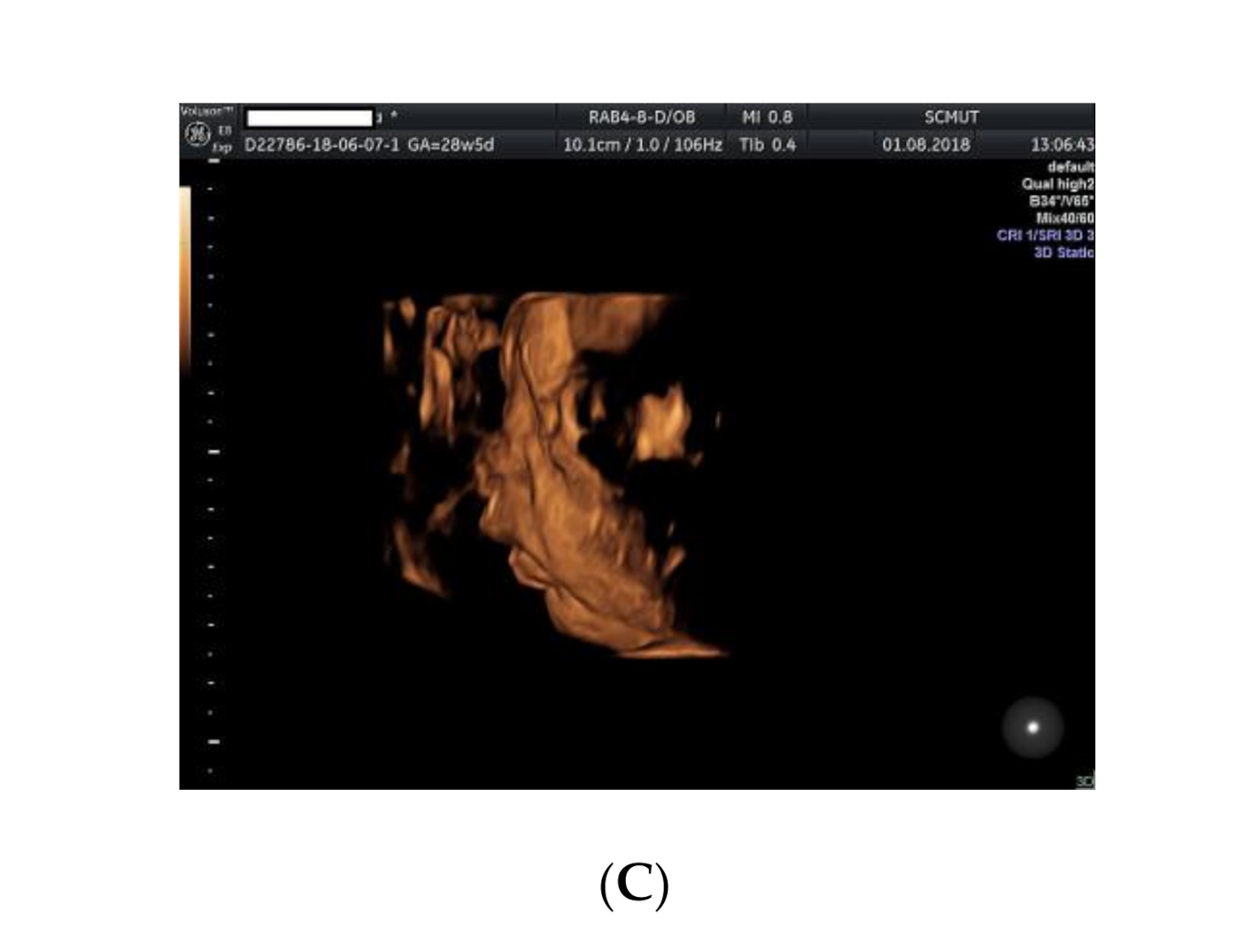
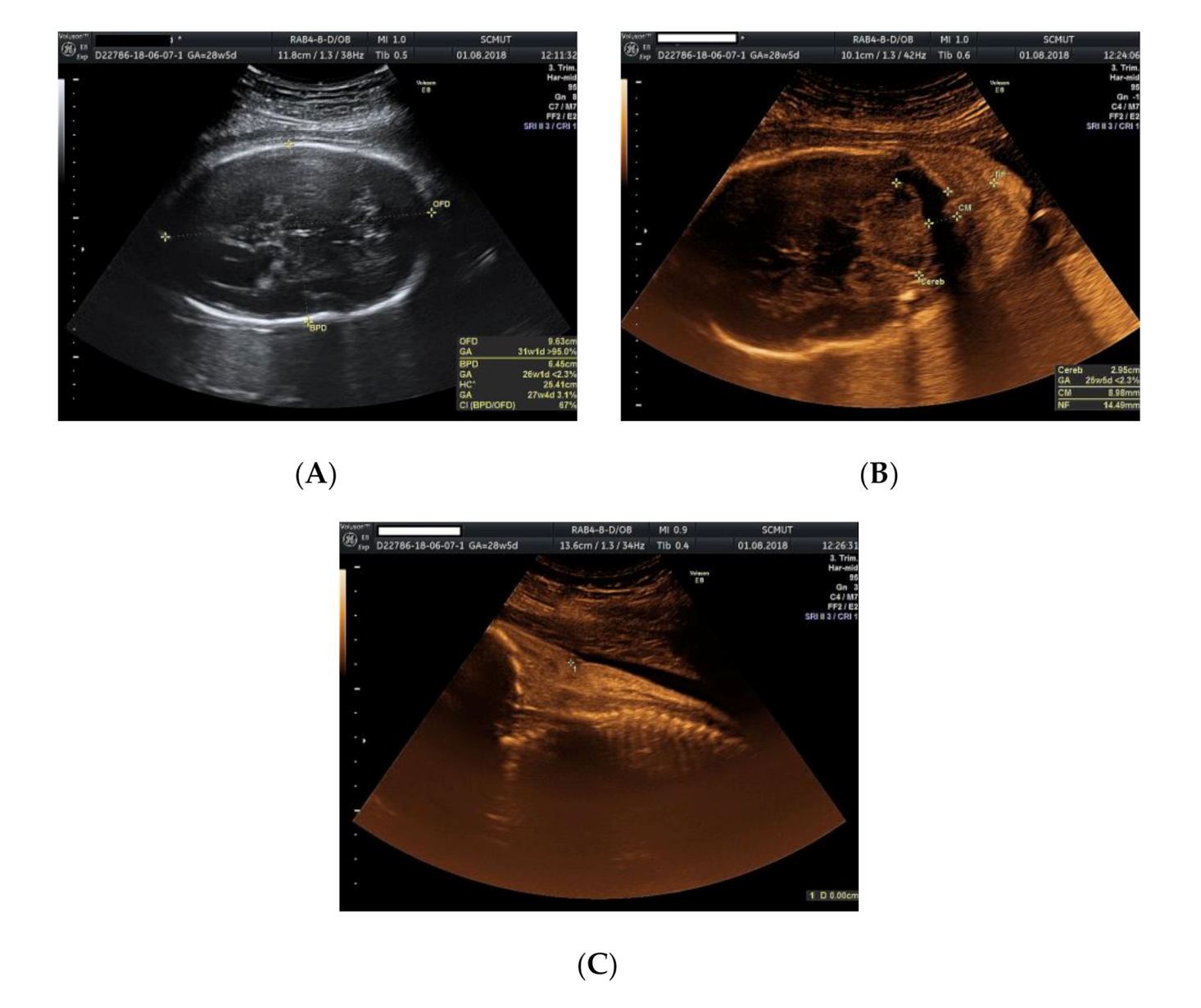
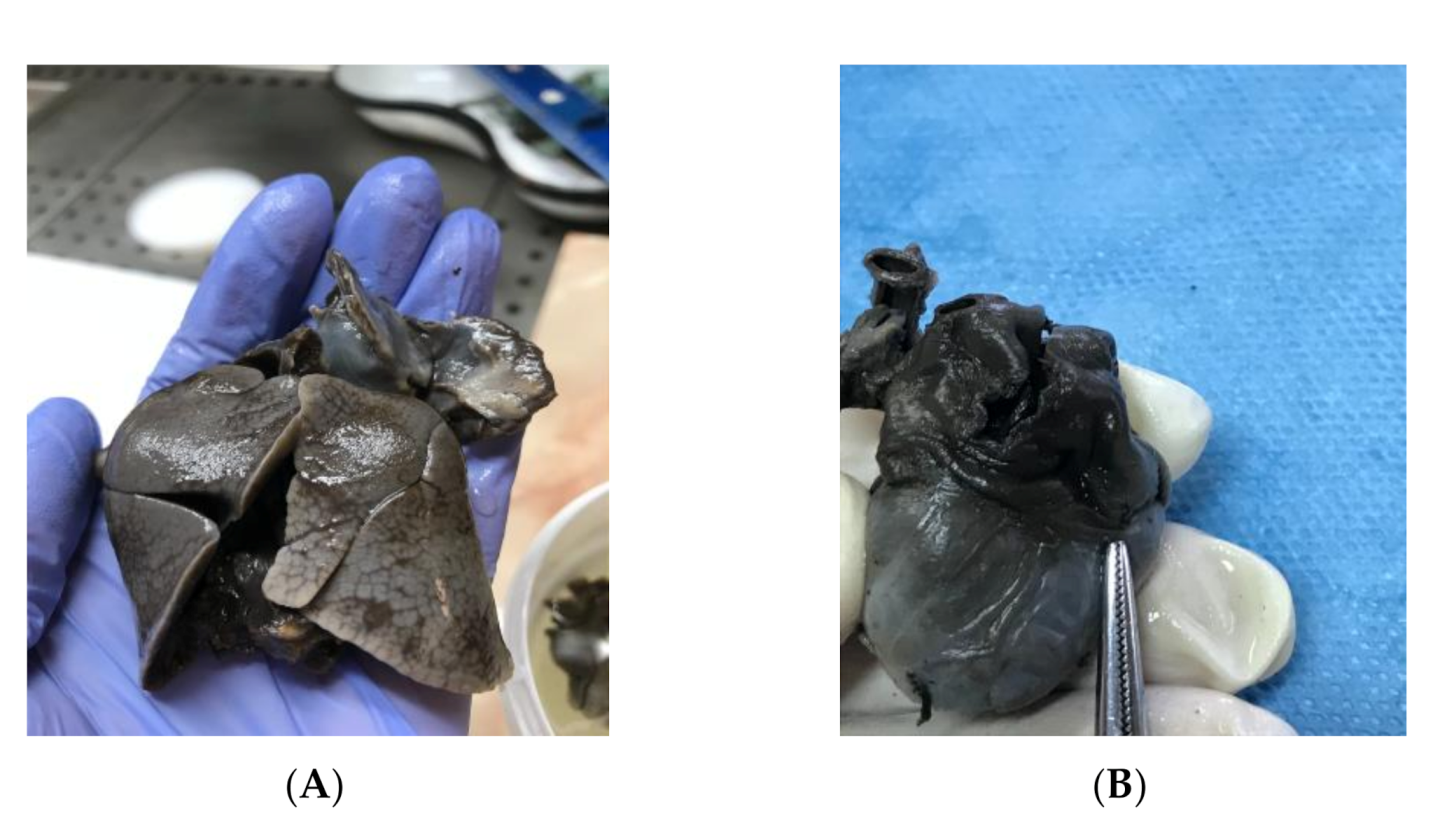
2. Case Presentation
3. Discussion and Conclusions
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Abuhamad, A.; Chaoui, R. A Practical Guide to Fetal Echocardiography, 3rd ed.; Wolters Kluwer: London, UK, 2016; pp. 485–503. [Google Scholar]
- Chaoui, R. Cardiac malposition and syndromes with right or left isomerism. In Fetal Cardiology: Embriology, Genetics, Physiology, Echocardiographic Evaluation, Diagnosis and Perinatal Management of Cardiac Diseases; Yagel, S., Gembrugh, U., Silverman, N., Eds.; Informa Healthcare: New York, NY, USA, 2008; pp. 239–250. [Google Scholar]
- O’Leary, P.M.; Hagler, D.J. Cardiac malpositions and abnormalities of atrial and visceral situs. In Moss and Adam’s Heart Disease in Infants, Children and Adolescents, 8th ed.; Allen, H.D., Driscoll, D.J., Shaddy, R.E., Feltes, T.F., Eds.; Williams & Wilkins: Baltimore, MD, USA, 2012; pp. 1195–1216. [Google Scholar]
- Yoo, S.J.; Friedberg, M.K.; Jaeggi, E. Abnormal visceral and atrial situs and congenital heart disease’. In Fetal Cardiology: Embriology, Genetics, Physiology, Echocardiographic Evaluation, Diagnosis and Perinatal Management of Cardiac Diseases; Yagel, S., Gembrugh, U., Silverman, N., Eds.; Informa Healthcare: New York, NY, USA, 2008; pp. 347–362. [Google Scholar]
- Yagel, S.; Silverman, N.H.; Gembruch, U. Fetal Cardiology: Embriology, Genetics, Physiology, Echocardiographic Evaluation, Diagnosis and Perinatal Management of Cardiac Diseases, 3rd ed; CRC Press: Boca Raton, FL, USA, 2019; pp. 683–689. [Google Scholar]
- Fontana, G.P.; Burke, R.P. Straddling and overriding atrioventricularvalves. In Glenn’s Thoracic and Cardiovascular Surgery, 6th ed.; Baue, A.E., Geha, A.S., Hammond, G.L., Lacks, H., Naunheim, K.S., Glen, W.W.L., Eds.; Appleton & Lange: Stamford, CT, USA, 1996. [Google Scholar]
- Van Praagh, R. Cardiac anatomy. In Pediatric Cardiac Intensive Care; Chang, A.C., Hanley, F.L., Wernovsky, G., Wessel, D.L., Eds.; Williams and Wilkins: Baltimore, MD, USA, 1998; pp. 3–15. [Google Scholar]
- Nakhleh, N.; Francis, R.; Giese, R.A.; Tian, X.; Li, Y.; Zariwala, M.A.; Yagi, H.; Khalifa, O.; Kureshi, S.; Chatterjee, B.; et al. High prevalence of respiratory ciliary dysfunction in congenital heart disease patients with heterotaxy. Circulation 2012, 125, 2232–2242. [Google Scholar] [CrossRef] [PubMed]
- Bruckner, M. Impact of genetic diagnosis on clinical management of patients with congenital heart disease: Cilia point the way. Circulation 2012, 125, 2178–2180. [Google Scholar] [CrossRef]
- Kennedy, M.P.; Omran, H.; Leigh, M.W.; Dell, S.; Morgan, L.; Zariwala, M.A.; Molina, P.L.; Minnix, S.L.; Severin, T.; Ahrens, P.; et al. Congenital heart disease and other heterotaxic defects in a large cohort of patients with primary ciliary dyskinesia. Circulation 2007, 115, 2814–2821. [Google Scholar] [CrossRef] [PubMed]
- Leigh, M.W.; Pittman, J.E.; Carson, J.L.; Ferkol, T.W.; Dell, S.D.; Davis, S.D.; Knowles, M.R.; Zariwala, M.A. Clinical and genetic aspects of primary ciliary dyskinesia/Kartagener syndrome. Genet. Med. 2009, 11, 473–487. [Google Scholar] [CrossRef]
- Berg, C.; Geipel, A.; Kamil, D.; Knüppel, M.; Breuer, J.; Krapp, M.; Baschat, A.; Germer, U.; Hansmann, M.; Gembruch, U. The syndrome of left isomerism: Sonographic findings and outcome in prenatally diagnosed cases. J. Ultrasound Med. 2005, 24, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Salehi Karlslätt, K.; Pettersson, M.; Jäntti, N.; Szafranski, P.; Wester, T.; Husberg, B.; Ullberg, U.; Stankiewicz, P.; Nordgren, A.; Lundin, J.; et al. Rare copy number variants contribute pathogenic alleles in patients with intestinal malrotation. Mol. Genet. Genomic Med. 2019, 7, e549. [Google Scholar] [CrossRef] [PubMed]
- Bartolini, L.; Sartori, S.; Lenzini, E.; Rigon, C.; Cainelli, E.; Agrati, C.; Toldo, I.; Donà, M.; Trevisson, E. De novo trisomy 20p characterized by array comparative genomic hybridization: Report of a novel case and review of the literature. Gene 2013, 524, 368–372. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ilian, A.; Motoc, A.; Balulescu, L.; Secosan, C.; Grigoras, D.; Pirtea, L. A Case Report of Left Atrial Isomerism in a Syndromic Context. Genes 2020, 11, 1211. https://doi.org/10.3390/genes11101211
Ilian A, Motoc A, Balulescu L, Secosan C, Grigoras D, Pirtea L. A Case Report of Left Atrial Isomerism in a Syndromic Context. Genes. 2020; 11(10):1211. https://doi.org/10.3390/genes11101211
Chicago/Turabian StyleIlian, Aurora, Andrei Motoc, Ligia Balulescu, Cristina Secosan, Dorin Grigoras, and Laurentiu Pirtea. 2020. "A Case Report of Left Atrial Isomerism in a Syndromic Context" Genes 11, no. 10: 1211. https://doi.org/10.3390/genes11101211
APA StyleIlian, A., Motoc, A., Balulescu, L., Secosan, C., Grigoras, D., & Pirtea, L. (2020). A Case Report of Left Atrial Isomerism in a Syndromic Context. Genes, 11(10), 1211. https://doi.org/10.3390/genes11101211