Lights and Shadows in the Genetics of Syndromic and Non-Syndromic Hearing Loss in the Italian Population

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Clinical Evaluation and Sample Collection
2.3. GJB2, GJB6, and mtDNA Analysis
2.4. Multiplex Ligation Probe Amplification (MLPA)
2.5. Whole Exome Sequencing (WES)
3. Results
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Allen, S.B.; Goldman, J. Hearing, Inner Ear, Syndromic Sensorineural Loss; StatPearls Publishing: Tampa, FL, USA, 2019. [Google Scholar]
- Likar, T.; Hasanhodžić, M.; Teran, N.; Maver, A.; Peterlin, B.; Writzl, K. Diagnostic outcomes of exome sequencing in patients with syndromic or non-syndromic hearing loss. PLoS ONE 2018, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, S.; Pandey, M. Advances in Genetic Diagnosis and Treatment of Hearing Loss—A Thirst for Revolution. In Update On Hearing Loss; InTech: London, UK, 2015. [Google Scholar]
- Smith, R.J.; Bale, J.F.; White, K.R. Sensorineural hearing loss in children. Lancet 2005, 365, 879–890. [Google Scholar] [CrossRef]
- Korver, A.M.H.; Smith, R.J.H.; Van Camp, G.; Schleiss, M.R.; Bitner-Glindzicz, M.A.K.; Lustig, L.R.; Usami, S.I.; Boudewyns, A.N. Congenital hearing loss. Nat. Rev. Dis. Prim. 2017, 3, 16094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ideura, M.; Nishio, S.Y.; Moteki, H.; Takumi, Y.; Miyagawa, M.; Sato, T.; Kobayashi, Y.; Ohyama, K.; Oda, K.; Matsui, T.; et al. Comprehensive analysis of syndromic hearing loss patients in Japan. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Wolfrum, U.; Nagel-Wolfrum, K. The Usher Syndrome, a Human Ciliopathy. Klin. Monbl. Augenheilkd. 2018, 235, 273–280. [Google Scholar] [CrossRef]
- Ahmed, J.D.N.; Mui, R.K.; Masood, S. Waardenburg Syndrome. J. Med Genet. 2020, 34, 656–665. [Google Scholar]
- Bademci, G.; Cengiz, F.B.; Foster, J.; Duman, D.; Sennaroglu, L.; Diaz-Horta, O.; Atik, T.; Kirazli, T.; Olgun, L.; Alper, H.; et al. Variations in Multiple Syndromic Deafness Genes Mimic Non-syndromic Hearing Loss. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Morgan, A.; Lenarduzzi, S.; Cappellani, S.; Pecile, V.; Morgutti, M.; Orzan, E.; Ghiselli, S.; Ambrosetti, U.; Brumat, M.; Gajendrarao, P.; et al. Genomic Studies in a Large Cohort of Hearing Impaired Italian Patients Revealed Several New Alleles, a Rare Case of Uniparental Disomy (UPD) and the Importance to Search for Copy Number Variations. Front. Genet. 2018, 9, 681. [Google Scholar] [CrossRef]
- Cabanillas, R.; Diñeiro, M.; Cifuentes, G.A.; Castillo, D.; Pruneda, P.C.; Álvarez, R.; Sánchez-Durán, N.; Capín, R.; Plasencia, A.; Viejo-Díaz, M.; et al. Comprehensive genomic diagnosis of non-syndromic and syndromic hereditary hearing loss in Spanish patients. BMC Med. Genom. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Azaiez, H.; Decker, A.R.; Booth, K.T.; Simpson, A.C.; Shearer, A.E.; Huygen, P.L.M.; Bu, F.; Hildebrand, M.S.; Ranum, P.T.; Shibata, S.B.; et al. HOMER2, a stereociliary scaffolding protein, is essential for normal hearing in humans and mice. PLoS Genet. 2015, 11, e1005137. [Google Scholar] [CrossRef] [Green Version]
- Morgan, A.; Koboldt, D.C.; Barrie, E.S.; Crist, E.R.; García García, G.; Mezzavilla, M.; Faletra, F.; Mihalic Mosher, T.; Wilson, R.K.; Blanchet, C.; et al. Mutations in PLS1, encoding fimbrin, cause autosomal dominant nonsyndromic hearing loss. Hum. Mutat. 2019. [Google Scholar] [CrossRef] [PubMed]
- Del Castillo, I.; Moreno-Pelayo, M.A.; Del Castillo, F.J.; Brownstein, Z.; Marlin, S.; Adina, Q.; Cockburn, D.J.; Pandya, A.; Siemering, K.R.; Chamberlin, G.P.; et al. Prevalence and Evolutionary Origins of the del(GJB6-D13S1830) Mutation in the DFNB1 Locus in Hearing-Impaired Subjects: A Multicenter Study. Am. J. Hum. Genet. 2003, 73, 1452–1458. [Google Scholar] [CrossRef] [Green Version]
- Cama, E.; Melchionda, S.; Palladino, T.; Carella, M.; Santarelli, R.; Genovese, E.; Benettazzo, F.; Zelante, L.; Arslan, E. Hearing loss features in GJB2 biallelic mutations and GJB2/GJB6 digenic inheritance in a large Italian cohort. Int. J. Audiol. 2009, 48, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Morton, C.C.; Nance, W.E. Newborn Hearing Screening—A Silent Revolution. N. Engl. J. Med. 2006, 354, 2151–2164. [Google Scholar] [CrossRef]
- Yokota, Y.; Moteki, H.; Nishio, S.Y.; Yamaguchi, T.; Wakui, K.; Kobayashi, Y.; Ohyama, K.; Miyazaki, H.; Matsuoka, R.; Abe, S.; et al. Frequency and clinical features of hearing loss caused by STRC deletions. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef]
- Clark, J.G. Uses and abuses of hearing loss classification. ASHA 1981, 23, 493–500. [Google Scholar]
- Del Castillo, I.; Villamar, M.; Moreno-Pelayo, M.A.; del Castillo, F.J.; Alvarez, A.; Tellería, D.; Menéndez, I.; Moreno, F. A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N. Engl. J. Med. 2002, 346, 243–249. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 7. [Google Scholar] [CrossRef] [Green Version]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, J.M.; Rödelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef] [PubMed]
- Chun, S.; Fay, J.C. Identification of deleterious mutations within three human genomes. Genome Res. 2009, 19, 1553–1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollard, K.S.; Hubisz, M.J.; Rosenbloom, K.R.; Siepel, A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010, 20, 110–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, G.M.; Stone, E.A.; Asimenos, G.; NISC Comparative Sequencing Program; Green, E.D.; Batzoglou, S.; Sidow, A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005, 15, 901–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desmet, F.-O.; Hamroun, D.; Lalande, M.; Collod-Béroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [Green Version]
- Shearer, A.E.; Kolbe, D.L.; Azaiez, H.; Sloan, C.M.; Frees, K.L.; Weaver, A.E.; Clark, E.T.; Nishimura, C.J.; Black-Ziegelbein, E.A.; Smith, R.J.H. Copy number variants are a common cause of non-syndromic hearing loss. Genome Med. 2014, 6, 37. [Google Scholar] [CrossRef] [Green Version]
- McGuirt, W.T.; Prasad, S.D.; Griffith, A.J.; Kunst, H.P.M.; Green, G.E.; Shpargel, K.B.; Runge, C.; Huybrechts, C.; Mueller, R.F.; Lynch, E.; et al. Mutations in COL11A2 cause non-syndromic hearing loss (DFNA13). Nat. Genet. 1999, 23, 413–419. [Google Scholar] [CrossRef]
- Vona, B.; Hofrichter, M.A.H.; Neuner, C.; Schröder, J.; Gehrig, A.; Hennermann, J.B.; Kraus, F.; Shehata-Dieler, W.; Klopocki, E.; Nanda, I.; et al. DFNB16 is a frequent cause of congenital hearing impairment: Implementation of STRC mutation analysis in routine diagnostics. Clin. Genet. 2015, 87, 49–55. [Google Scholar] [CrossRef] [Green Version]
- Numakura, C.; Lin, C.; Ikegami, T.; Guldberg, P.; Hayasaka, K. Molecular analysis in Japanese patients with Charcot-Marie-Tooth disease: DGGE analysis for PMP22, MPZ, and Cx32/GJB1 mutations. Hum. Mutat. 2002, 20, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Wattenhofer, M.; Reymond, A.; Falciola, V.; Charollais, A.; Caille, D.; Borel, C.; Lyle, R.; Estivill, X.; Petersen, M.B.; Meda, P.; et al. Different mechanisms preclude mutant CLDN14 proteins from forming tight junctions in vitro. Hum. Mutat. 2005, 25, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Ruf, R.G.; Xu, P.X.; Silvius, D.; Otto, E.A.; Beekmann, F.; Muerb, U.T.; Kumar, S.; Neuhaus, T.J.; Kemper, M.J.; Raymond, R.M.; et al. SIX1 mutations cause branchio-oto-renal syndrome by disruption of EYA1-SIX1-DNA complexes. Proc. Natl. Acad. Sci. USA 2004, 101, 8090–8095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheppard, S.; Biswas, S.; Li, M.H.; Jayaraman, V.; Slack, I.; Romasko, E.J.; Sasson, A.; Brunton, J.; Rajagopalan, R.; Sarmady, M.; et al. Utility and limitations of exome sequencing as a genetic diagnostic tool for children with hearing loss. Genet. Med. 2018, 20, 1663–1676. [Google Scholar] [CrossRef] [Green Version]
- Lenarduzzi, S.; Morgan, A.; Faletra, F.; Cappellani, S.; Morgutti, M.; Mezzavilla, M.; Peruzzi, A.; Ghiselli, S.; Ambrosetti, U.; Graziano, C.; et al. Next generation sequencing study in a cohort of Italian patients with syndromic hearing loss. Hear. Res. 2019, 381. [Google Scholar] [CrossRef]
- Rodríguez-Ballesteros, M.; Reynoso, R.; Olarte, M.; Villamar, M.; Morera, C.; Santarelli, R.; Arslan, E.; Medá, C.; Curet, C.; Völter, C.; et al. A multicenter study on the prevalence and spectrum of mutations in the otoferlin gene ( OTOF ) in subjects with nonsyndromic hearing impairment and auditory neuropathy. Hum. Mutat. 2008, 29, 823–831. [Google Scholar] [CrossRef]
- Magliulo, G.; Iannella, G.; Gagliardi, S.; Iozzo, N.; Plateroti, R.; Mariottini, A.; Torricelli, F. Usher’s Syndrome Type II: A Comparative Study of Genetic Mutations and Vestibular System Evaluation. Otolaryngol. Head Neck Surg. 2017, 157, 853–860. [Google Scholar] [CrossRef]
- Rivolta, C.; Sweklo, E.A.; Berson, E.L.; Dryja, T.P. Missense mutation in the USH2A gene: Association with recessive retinitis pigmentosa without hearing loss. Am. J. Hum. Genet. 2000, 66, 1975–1978. [Google Scholar] [CrossRef] [Green Version]
- Van Wijk, E.; Pennings, R.J.E.; Te Brinke, H.; Claassen, A.; Yntema, H.G.; Hoefsloot, L.H.; Cremers, F.P.M.; Cremers, W.R.J.; Kremer, H. Identification of 51 Novel Exons of the Usher Syndrome Type 2A (USH2A) Gene That Encode Multiple Conserved Functional Domains and That Are Mutated in Patients with Usher Syndrome Type II. Am. J. Hum. Genet. 2004, 74, 738–744. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.Y.; Park, G.; Gim, J.; Kim, A.R.; Kim, B.J.; Kim, H.S.; Park, J.H.; Park, T.; Oh, S.H.; Han, K.H.; et al. Diagnostic Application of Targeted Resequencing for Familial Nonsyndromic Hearing Loss. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Ebermann, I.; Phillips, J.B.; Liebau, M.C.; Koenekoop, R.K.; Schermer, B.; Lopez, I.; Schäfer, E.; Roux, A.F.; Dafinger, C.; Bernd, A.; et al. PDZD7 is a modifier of retinal disease and a contributor to digenic Usher syndrome. J. Clin. Invest. 2010, 120, 1812–1823. [Google Scholar] [CrossRef] [Green Version]
- Shearer, A.E.; DeLuca, A.P.; Hildebrand, M.S.; Taylor, K.R.; Gurrola, J.; Scherer, S.; Scheetz, T.E.; Smith, R.J.H. Comprehensive genetic testing for hereditary hearing loss using massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2010, 107, 21104–21109. [Google Scholar] [CrossRef] [Green Version]
- Sloan-Heggen, C.M.; Bierer, A.O.; Shearer, A.E.; Kolbe, D.L.; Nishimura, C.J.; Frees, K.L.; Ephraim, S.S.; Shibata, S.B.; Booth, K.T.; Campbell, C.A.; et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum. Genet. 2016, 135, 441–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atik, T.; Onay, H.; Aykut, A.; Bademci, G.; Kirazli, T.; Tekin, M.; Ozkinay, F. Comprehensive analysis of deafness genes in families with autosomal recessive nonsyndromic hearing loss. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [Green Version]
- Tassabehji, M.; Read, A.P.; Newton, V.E.; Patton, M.; Gruss, P.; Harris, R.; Strachan, T. Mutations in the PAX3 gene causing Waardenburg syndrome type 1 and type 2. Nat. Genet. 1993, 3, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.H.; Walsh, C.A. FLNA-Related Periventricular Nodular Heterotopia; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Schultz, J.M.; Yang, Y.; Caride, A.J.; Filoteo, A.G.; Penheiter, A.R.; Lagziel, A.; Morell, R.J.; Mohiddin, S.A.; Fananapazir, L.; Madeo, A.C.; et al. Modification of human hearing loss by plasma-membrane calcium pump PMCA2. N. Engl. J. Med. 2005, 352, 1557–1564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smits, J.J.; Oostrik, J.; Beynon, A.J.; Kant, S.G.; de Koning Gans, P.A.M.; Rotteveel, L.J.C.; Klein Wassink-Ruiter, J.S.; Free, R.H.; Maas, S.M.; van de Kamp, J.; et al. De novo and inherited loss-of-function variants of ATP2B2 are associated with rapidly progressive hearing impairment. Hum. Genet. 2019, 138, 61–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Wang, Q.; Gu, H.; Zhang, X.; Qi, Y.; Liu, Y. Whole exome sequencing identified a second pathogenic variant in HOMER2 for autosomal dominant non-syndromic deafness. Clin. Genet. 2018, 94, 419–428. [Google Scholar] [CrossRef]
- Vozzi, D.; Morgan, A.; Vuckovic, D.; D’Eustacchio, A.; Abdulhadi, K.; Rubinato, E.; Badii, R.; Gasparini, P.; Girotto, G. Hereditary hearing loss: A 96 gene targeted sequencing protocol reveals novel alleles in a series of Italian and Qatari patients. Gene 2014, 542, 209–216. [Google Scholar] [CrossRef]
- Shearer, A.E.; Eppsteiner, R.W.; Frees, K.; Tejani, V.; Sloan-Heggen, C.M.; Brown, C.; Abbas, P.; Dunn, C.; Hansen, M.R.; Gantz, B.J.; et al. Genetic variants in the peripheral auditory system significantly affect adult cochlear implant performance. Hear. Res. 2017, 348, 138–142. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | Gene | Variant 1 | Variant 2 | Status |
|---|---|---|---|---|
| Patient 1 | GJB2 | c.35delG, p.(Gly12Valfs * 2) | c.-27C > T | compound heterozygous |
| Patient 2 | c.35delG, p.(Gly12Valfs * 2) | c.229T > C, p.(Trp77Arg) | compound heterozygous | |
| Patient 3 | c.35delG, p.(Gly12Valfs * 2) | c.35delG, p.(Gly12Valfs * 2) | homozygous | |
| Patient 4 | c.35delG, p.(Gly12Valfs * 2) | c.139G > T p.(Glu47 *) | compound heterozygous | |
| Patient 5 | c.35delG, p.(Gly12Valfs * 2) | c.358_360delGAG, p.(Glu120del) | compound heterozygous | |
| Patient 6 | c.35delG, p.(Gly12Valfs * 2) | c.-23 + 1G > A | compound heterozygous | |
| Patient 7 | c.35delG, p.(Gly12Valfs * 2) | c.71G > A, p.(Glu24 *) | compound heterozygous | |
| Patient 8 | c.35delG, p.(Gly12Valfs * 2) | c.71G > A, p.(Glu24 *) | compound heterozygous | |
| Patient 9 | c.35delG, p.(Gly12Valfs * 2) | c.35delG, p.(Gly12Valfs * 2) | homozygous | |
| Patient 10 | c.35delG, p.(Gly12Valfs * 2) | c.35delG, p.(Gly12Valfs * 2) | homozygous | |
| Patient 11 | c.35delG, p.(Gly12Valfs * 2) | c.314_327del14, p.(Lys105Argfs * 5) | compound heterozygous | |
| Patient 12 | c.35delG, p.(Gly12Valfs * 2) | c.35delG, p.(Gly12Valfs * 2) | homozygous | |
| Patient 13 | c.35delG, p.(Gly12Valfs * 2) | c.95G > A, p.(Arg32His) | compound heterozygous | |
| Patient 14 | c.35delG, p.(Gly12Valfs * 2) | c.35delG, p.(Gly12Valfs * 2) | homozygous | |
| Patient 15 | c.59T > C, p.(Ile20Thr) | c.314_327del14, p.(Lys105Argfs * 5) | compound heterozygous | |
| Patient 16 | c.35delG, p.(Gly12Valfs * 2) | c.71G > A, p.(Glu24 * ) | compound heterozygous | |
| Patient 17 | c.35delG, p.(Gly12Valfs * 2) | c.35delG, p.(Gly12Valfs * 2) | homozygous | |
| Patient 18 | c.35delG, p.(Gly12Valfs * 2) | c.139G > T p.(Glu47 *) | compound heterozygous | |
| Patient 19 | c.35delG, p.(Gly12Valfs * 2) | c.35delG, p.(Gly12Valfs * 2) | homozygous | |
| Patient 20 | c.35delG, p.(Gly12Valfs * 2) | c.35delG, p.(Gly12Valfs * 2) | homozygous | |
| Patient 21 | c.101T > C, p.(Met34Thr) | c.358_360delGAG, p.(Glu120del) | compound heterozygous | |
| Patient 22 | c.35delG, p.(Gly12Valfs * 2) | c.35delG, p.(Gly12Valfs * 2) | homozygous | |
| Patient 23 | c.35delG, p.(Gly12Valfs * 2) | c.35delG, p.(Gly12Valfs * 2) | homozygous | |
| Patient 24 | c.35delG, p.(Gly12Valfs * 2) | c.358_360delGAG, p.(Glu120del) | compound heterozygous | |
| Patient 25 | c.35delG, p.(Gly12Valfs * 2) | c.35delG, p.(Gly12Valfs * 2) | homozygous |
| Family ID | Gene | cDNA Change | Protein Change | dbSNP | gnomAD_ALL | CADD PHRED | GERP ++_RS | Polyphen-2 | SIFT | Mutation Taster | References |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Family 3 | OTOA (NM_144672.3) | entire gene deletion (hom) | NA | NA | NA | NA | NA | NA | NA | NA | Shearer et al., (2014) Genome Med [30] |
| Family 4 | COL11A2 (NM_080680.2) | c.3100C > T (het) | p.(Arg1034Cys) | rs121912947 | NA | 32 | 2.95 | D | D | D | McGuirt et al., (1999) Nat Genet [31] |
| Patient 41 | MYO7A (NM_000260.3) | c.6236G > A (hom) | p.(Arg2079Gln) | rs765083332 | 0.00004188 | 25.5 | 4.24 | P | T | D | NA |
| Patient 42 | ADGRV1 (NM_032119.3) | c.10084C > T (het) | p.(Gln3362 *) | NA | NA | 42 | 3.02 | NA | NA | A | NA |
| c.13655dupT (het) | p.(Asn4553Glufs * 18) | rs765376986 | NA | NA | NA | NA | NA | NA | NA | ||
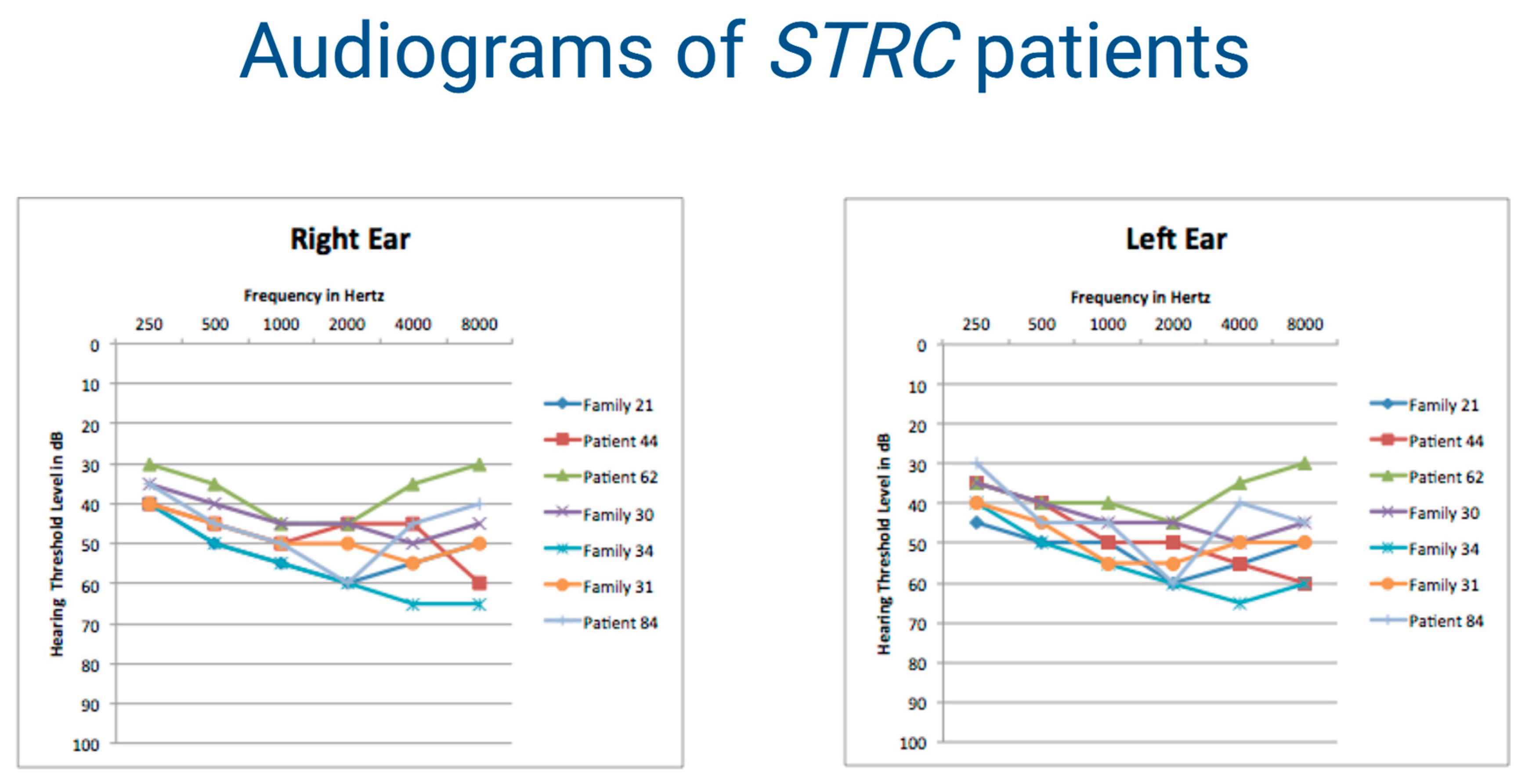
| Patient 44 | STRC (NM_153700.2) | entire gene deletion (hom) | NA | NA | NA | NA | NA | NA | NA | NA | Vona et al.,(2015) Clin Genet [32] |
| Patient 45 | GJB1 (NM_000166.5) | c.790C > T (het) | p.(Arg264Cys) | rs587777879 | 0.00005714 | 25.8 | 4.9 | D | D | D | Numakura et al., (2002) Hum Mutat [33] |
| Patient 51 | CLDN14 (NM_144492.2) | c.301G > A (hom) | p.(Gly101Arg) | rs74315438 | 0.00004302 | 26.5 | 5.42 | D | D | D | Wattenhofer et al.,(2005) Hum Mutat [34] |
| Family 6 | SMPX (NM_014332.2) | c.45+1G > A (hemizygous) | NA | NA | NA | 24.4 | 4.9 | NA | NA | D | NA |
| Family 7 | SIX1 (NM_005982.3) | c.397_399delGAG (het) | p.(Glu133del) | rs80356460 | NA | NA | NA | NA | NA | NA | Ruf et al., (2004) Proc Natl Acad Sci U S A [35] |
| Family 8 | KARS (NM_001130089.1) | c.1423C > T (het) | p.(Leu475Phe) | NA | NA | 28.2 | 5.91 | P | T | D | NA |
| c.1570T > C (het) | p.(Cys524Arg) | NA | NA | 26.9 | 5.81 | D | D | D | NA | ||
| Patient 57 | OTOG (NM_001277269.1) | c.2500C > T (hom) | p.(Gln834 *) | rs554847663 | 0.0004274 | 37 | 2.01 | NA | NA | D | Sheppard et al.,(2018) Genet Med [36] |
| Family 10 | PAX3 (NM_181457.3) | c.220C > T (het) | p.(Arg74Cys) | NA | NA | 35 | 5.24 | D | D | D | Lenarduzzi et al., (2019) Hear Res [37] |
| Patient 62 | STRC (NM_153700.2) | entire gene deletion (hom) | NA | NA | NA | NA | NA | NA | NA | NA | Vona et al.,(2015) Clin Genet [32] |
| Family 13 | ESPN (NM_031475.2) | entire gene deletion (hom) | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| Family 14 | MYO6 (NM_004999.3) | c.1525delG (het) | p.(Val509Trpfs * 7) | NA | NA | NA | NA | NA | NA | D | NA |
| Family 15 | OTOF (NM_194248.2) | c.2891C > A (hom) | p.(Ala964Glu) | rs201329629 | NA | 32 | 5.41 | D | D | D | Rodriguez-Ballesteros et al. (2008) Hum Mutat [38] |
| Patient 65 | ADGRV1 (NM_032119.3) | c.4378G>A (het) | p.(Gly1460Ser) | rs1303930496 | 0.00001248 | 32 | 5.53 | D | D | D | Magliulo et al.,(2017) Otolaryngol Head Neck Surg [39] |
| c.13655dupT (het) | p.(Asn4553Glufs * 18) | rs765376986 | NA | NA | NA | NA | NA | NA | NA | ||
| Family 18 | DIAPH1 (NM_005219.4) | c.3556delC (het) | p.(Leu1186Serfs * 2) | NA | NA | NA | NA | NA | NA | D | NA |
| Patient 70 | GATA3 (NM_002051.2) | c.925-1G > T (het, de novo) | NA | NA | NA | 25.9 | 5.3 | NA | NA | NA | NA |
| Family 20 | TECTA (NM_005422.2) | c.3841T > C (het) | p.(Cys1281Arg) | NA | NA | 17.03 | 5.76 | T | D | D | NA |
| Family 21 | STRC (NM_153700.2) | entire gene deletion (hom) | NA | NA | NA | NA | NA | NA | NA | NA | Vona et al.,(2015) Clin Genet [32] |
| Family 23 | USH2A (NM_206933.2) | c.2276G > T (het) | p.(Cys759Phe) | rs80338902 | 0.000947 | 33 | 5.79 | D | D | D | Rivolta et al.,(2000) Am J Hum Genet [40] |
| c.11864G > A (het) | p.(Trp3955 *) | rs111033364 | 0.000119 | 51 | 5.53 | NA | D | D | van Wijk et al., (2004) Am J Hum Genet [41] | ||
| EYA4 (NM_004100.4) | c.714C > A (het) | p.(Tyr238 *) | rs1264401894 | 0.000004 | 37 | 4.77 | NA | D | D | NA | |
| Family 24 | WFS1 (NM_006005.3) | c.2567C > A (het) | p.(Pro856His) | NA | NA | 23.7 | 4.82 | D | D | D | NA |
| Family 26 | MYO6 (NM_004999.3) | c.613C > T (het) | p.(Arg205 *) | rs557441143 | 0.00000398 | 37 | 3.31 | NA | NA | A | Choi ET AL.,(2013) PLoS One 8 [42] |
| Family 27 | COL2A1 (NM_001844.5) | c.4201G > C (het) | p.(Asp1401His) | NA | NA | 19.9 | 5.06 | D | D | D | NA |
| Family 28 | ATP2B2 (NM_001001331.4) | c.962C > G (het) | p.(Ser321 *) | NA | NA | 42 | 5.34 | NA | T | A | NA |
| Patient 79 | LARS2 | 4 Kb gene deletion (hom) | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| Family 29 | PDZD7 (NM_024895.4) | c.166dupC (het) | p.(Arg56Profs * 24) | rs587776894 | NA | NA | NA | NA | NA | NA | Ebermann et al.,(2010) J Clin Invest [43] |
| c.305G > A (het) | p.(Arg102His) | rs760825921 | 0.00001061 | 34 | 5.07 | D | D | D | NA | ||
| Family 30 | STRC (NM_153700.2) | entire gene deletion (hom) | NA | NA | NA | NA | NA | NA | NA | NA | Vona et al.,(2015) Clin Genet [32] |
| Family 31 | STRC (NM_153700.2) | entire gene deletion (hom) | NA | NA | NA | NA | NA | NA | NA | NA | Vona et al.,(2015) Clin Genet [32] |
| Family 32 | HOMER2 (NM_199330.2) | c.592_597delACCACA (het) | p.(Thr198_Thr199del) | NA | NA | NA | NA | NA | NA | P | NA |
| Patient 84 | STRC (NM_153700.2) | entire gene deletion (hom) | NA | NA | NA | NA | NA | NA | NA | NA | Vona et al.,(2015) Clin Genet [32] |
| Family 34 | STRC (NM_153700.2) | c.4057C > T (hom) | p.(Gln1353 *) | rs774312182 | 0.00006374 | 37 | 4.16 | NA | NA | D | Shearer et al., (2010) Proc Natl Acad Sci U S A [44] |
| Patient 86 | TCOF1 (NM_001135243.1) | c.4362_4366del (het) | p.(Lys1457Glufs * 11) | NA | NA | NA | NA | NA | NA | NA | NA |
| Family 35 | COL4A3 (NM_000091.4) | c.3943C > T (het) | p.(Pro1315Ser) | rs760703010 | 0.00002793 | 25 | 5.57 | D | D | D | NA |
| Patient 87 | USH2A (NM_206933.2) | c.11864G > A (hom) | p.(Trp3955 *) | rs111033364 | 0.000119 | 51 | 5.53 | NA | D | A | van Wijk et al., (2004) Am J Hum Genet [41] |
| Patient 88 | USH2A (NM_206933.2) | c.4933G > T (hom) | p.(Gly1645 *) | NA | NA | 38 | 3.86 | NA | NA | A | Sloan-Heggen et al.,(2016) Hum Genet [45] |
| Patient 89 | USH2A (NM_206933.2) | c.2035G > T (het) | p.(Gly679 *) | NA | NA | 38 | 5.26 | NA | NA | A | NA |
| c.11864G > A (het) | p.(Trp3955 *) | rs111033364 | 0.000119 | 51 | 5.53 | NA | D | A | van Wijk et al., (2004) Am J Hum Genet [41] | ||
| Patient 90 | MYO7A (NM_000260.3) | c.735G > A (het) | (p.Gln245Gln) | NA | NA | NA | NA | NA | NA | A | Atik et al.,(2015) PLoS One 10 [46] |
| c.1834_1836delAGC (het) | p.(Ser612del) | NA | NA | NA | NA | NA | NA | D | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morgan, A.; Lenarduzzi, S.; Spedicati, B.; Cattaruzzi, E.; Murru, F.M.; Pelliccione, G.; Mazzà, D.; Zollino, M.; Graziano, C.; Ambrosetti, U.; et al. Lights and Shadows in the Genetics of Syndromic and Non-Syndromic Hearing Loss in the Italian Population. Genes 2020, 11, 1237. https://doi.org/10.3390/genes11111237
Morgan A, Lenarduzzi S, Spedicati B, Cattaruzzi E, Murru FM, Pelliccione G, Mazzà D, Zollino M, Graziano C, Ambrosetti U, et al. Lights and Shadows in the Genetics of Syndromic and Non-Syndromic Hearing Loss in the Italian Population. Genes. 2020; 11(11):1237. https://doi.org/10.3390/genes11111237
Chicago/Turabian StyleMorgan, Anna, Stefania Lenarduzzi, Beatrice Spedicati, Elisabetta Cattaruzzi, Flora Maria Murru, Giulia Pelliccione, Daniela Mazzà, Marcella Zollino, Claudio Graziano, Umberto Ambrosetti, and et al. 2020. "Lights and Shadows in the Genetics of Syndromic and Non-Syndromic Hearing Loss in the Italian Population" Genes 11, no. 11: 1237. https://doi.org/10.3390/genes11111237