DNA Associated with Circulating Exosomes as a Biomarker for Glioma


Abstract
1. Introduction
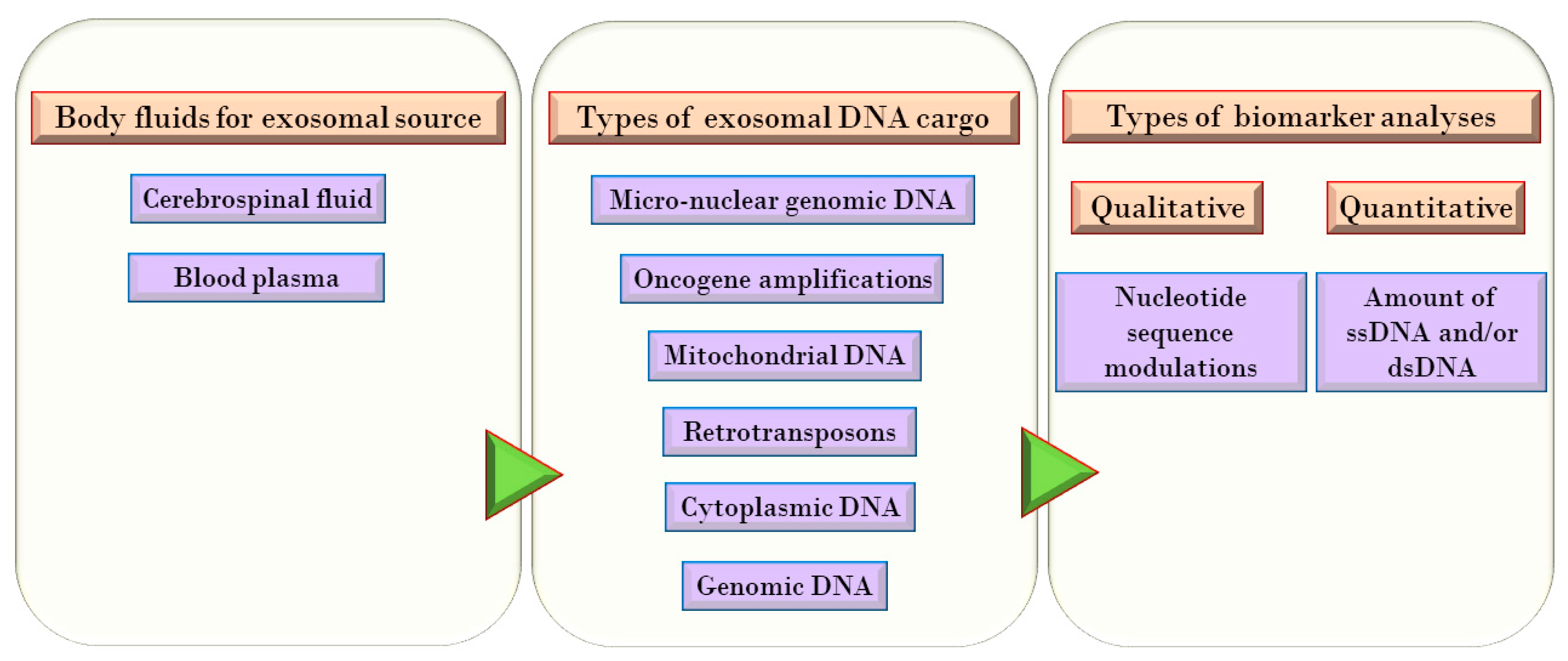
Localization, Types, and Sources of DNA in Exosomes
2. Exosomal DNA Modulations as Cancer Biomarkers
2.1. NANOG/NANOGP8
2.2. SOX2
2.3. EGFR
2.4. MGMT
2.5. IDH1/IDH2
2.6. mtDNA
2.7. CDKN2a, PTEN and TP53
2.8. ERBB2, CDK4, AKT3, MDM2, and RB1
2.9. c-Myc and POU5F1B
2.10. PD-L1
2.11. L1 and HERV
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- National Cancer Institute. Available online: https://www.cancer.gov/about-cancer/understanding/what-is-cancer (accessed on 5 May 2020).
- Toschi, L.; Finocchiaro, G.; Nguyen, T.T.; Skokan, M.C.; Giordano, L.; Gianoncelli, L.; Perrino, M.; Siracusano, L.; Di Tommaso, L.; Infante, M.; et al. Increased SOX2 Gene Copy Number Is Associated with FGFR1 and PIK3CA Gene Gain in Non-Small Cell Lung Cancer and Predicts Improved Survival in Early Stage Disease. PLoS ONE 2014, 9, e95303. [Google Scholar] [CrossRef] [PubMed]
- García-Romero, N.; Carrión-Navarro, J.; Esteban-Rubio, S.; Lázaro-Ibáñez, E.; Peris-Celda, M.; Alonso, M.M.; Guzmán-De-Villoria, J.; Fernández-Carballal, C.; De Mendivil, A.O.; García-Duque, S.; et al. DNA sequences within glioma-derived extracellular vesicles can cross the intact blood-brain barrier and be detected in peripheral blood of patients. Oncotarget 2016, 8, 1416–1428. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Wang, G.; Hu, W.; Yao, Y.; Yu, X.-F. Emerging roles and therapeutic value of exosomes in cancer metastasis. Mol. Cancer 2019, 18, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ciregia, F.; Urbani, A.; Palmisano, G. Extracellular Vesicles in Brain Tumors and Neurodegenerative Diseases. Front. Mol. Neurosci. 2017, 10, 276. [Google Scholar] [CrossRef]
- Rodriguez-Dorantes, M.; Romero-Cordoba, S.; Peralta-Zaragoza, O.; Salido-Guadarrama, I.; Hidalgo-Miranda, A. MicroRNAs transported by exosomes in body fluids as mediators of intercellular communication in cancer. OncoTargets Ther. 2014, 7, 1327–1338. [Google Scholar] [CrossRef]
- Fischer, S.; Cornils, K.; Speiseder, T.; Badbaran, A.; Reimer, R.; Indenbirken, D.; Grundhoff, A.; Brunswig-Spickenheier, B.; Alawi, M.; Lange, C. Indication of Horizontal DNA Gene Transfer by Extracellular Vesicles. PLoS ONE 2016, 11, e0163665. [Google Scholar] [CrossRef]
- Balaj, L.; Lessard, R.T.; Dai, L.; Cho, Y.-J.; Pomeroy, S.L.; Breakefield, X.O.; Skog, J. Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat. Commun. 2011, 2, 1–9. [Google Scholar] [CrossRef]
- Takahashi, A.; Okada, R.; Nagao, K.; Kawamata, Y.; Hanyu, A.; Yoshimoto, S.; Takasugi, M.; Watanabe, S.; Kanemaki, M.T.; Obuse, C.; et al. Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat. Commun. 2017, 8, 15287. [Google Scholar] [CrossRef]
- Vanpouille-Box, C.; DeMaria, S.; Formenti, S.C.; Galluzzi, L. Cytosolic DNA Sensing in Organismal Tumor Control. Cancer Cell 2018, 34, 361–378. [Google Scholar] [CrossRef]
- Thakur, B.K.; Zhang, H.; Becker, A.; Matei, I.; Huang, Y.; Costa-Silva, B.; Zheng, Y.; Hoshino, A.; Brazier, H.; Xiang, J.; et al. Double-stranded DNA in exosomes: A novel biomarker in cancer detection. Cell Res. 2014, 24, 766–769. [Google Scholar] [CrossRef]
- Rajagopal, C.; Harikumar, K.B. The Origin and Functions of Exosomes in Cancer. Front. Oncol. 2018, 8, 66. [Google Scholar] [CrossRef]
- Sancho-Albero, M.; Navascués, N.; Mendoza, G.; Sebastián, V.; Arruebo, M.; Martín-Duque, P.; Santamaría, J. Exosome origin determines cell targeting and the transfer of therapeutic nanoparticles towards target cells. J. Nanobiotechnol. 2019, 17, 1–13. [Google Scholar] [CrossRef]
- Smyth, T.J.; Redzic, J.S.; Graner, M.W.; Anchordoquy, T.J. Examination of the specificity of tumor cell derived exosomes with tumor cells in vitro. Biochim. Biophys. Acta Biomembr. 2014, 1838, 2954–2965. [Google Scholar] [CrossRef] [PubMed]
- Fernando, M.R.; Jiang, C.; Krzyzanowski, G.D.; Ryan, W.L. New evidence that a large proportion of human blood plasma cell-free DNA is localized in exosomes. PLoS ONE 2017, 12, e0183915. [Google Scholar] [CrossRef]
- Santiago-Dieppa, D.R.; Steinberg, J.; Gonda, D.; Cheung, V.J.; Carter, B.S.; Chen, C.C. Extracellular vesicles as a platform for ‘liquid biopsy’ in glioblastoma patients. Expert Rev. Mol. Diagn. 2014, 14, 819–825. [Google Scholar] [CrossRef]
- Available online: https://cbtrus.org/cbtrus-fact-sheet/ (accessed on 28 October 2020).
- Rode, A.; Maass, K.K.; Willmund, K.V.; Ernst, A.; Lichter, P. Chromothripsis in cancer cells: An update. Int. J. Cancer 2015, 138, 2322–2333. [Google Scholar] [CrossRef] [PubMed]
- Koltsova, A.S.; Pendina, A.A.; Efimova, O.A.; Chiryaeva, O.G.; Kuznetzova, T.V.; Baranov, V.S. On the Complexity of Mechanisms and Consequences of Chromothripsis: An Update. Front. Genet. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Luijten, M.N.H.; Lee, J.X.T.; Crasta, K.C. Mutational game changer: Chromothripsis and its emerging relevance to cancer. Mutat. Res. 2018, 777, 29–51. [Google Scholar] [CrossRef] [PubMed]
- Ratnaparkhe, M.; Wong, J.K.L.; Wei, P.-C.; Hlevnjak, M.; Kolb, T.; Simovic, M.; Haag, D.; Paul, Y.; Devens, F.; Northcott, P.; et al. Defective DNA damage repair leads to frequent catastrophic genomic events in murine and human tumors. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Maciejowski, J.; Li, Y.; Bosco, N.; Campbell, P.J.; De Lange, T. Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 2015, 163, 1641–1654. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nat. Cell Biol. 2015, 522, 179–184. [Google Scholar] [CrossRef]
- Yokoi, A.; Villar-Prados, A.; Oliphint, P.A.; Zhang, J.; Song, X.; De Hoff, P.; Morey, R.; Liu, J.; Roszik, J.; Clise-Dwyer, K.; et al. Mechanisms of nuclear content loading to exosomes. Sci. Adv. 2019, 5, eaax8849. [Google Scholar] [CrossRef] [PubMed]
- Bolukbasi, M.F.; Mizrak, A.; Ozdener, G.B.; Madlener, S.; Ströbel, T.; Erkan, E.P.; Fan, J.-B.; Breakefield, X.O.; Saydam, O. miR-1289 and “Zipcode”-like Sequence Enrich mRNAs in Microvesicles. Mol. Ther. Nucleic Acids 2012, 1, e10. [Google Scholar] [CrossRef]
- Kawamura, Y.; Yamamoto, Y.; Sato, T.-A.; Ochiya, T. Extracellular vesicles as trans-genomic agents: Emerging roles in disease and evolution. Cancer Sci. 2017, 108, 824–830. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, M.; Bacchus, M.; Sugaya, K. Differential sequences of exosomal NANOG DNA as a potential diagnostic cancer marker. PLoS ONE 2018, 13, e0197782. [Google Scholar] [CrossRef]
- Buzás, E.I.; Tóth, E.Á.; Sódar, B.W.; Szabó-Taylor, K.É. Molecular interactions at the surface of extracellular vesicles. Semin. Immunopathol. 2018, 40, 453–464. [Google Scholar] [CrossRef]
- Paludan, S.R.; Bowie, A.G. Immune Sensing of DNA. Immunology 2013, 38, 870–880. [Google Scholar] [CrossRef]
- Li, T.; Chen, Z.J. The cGAS–cGAMP–STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 2018, 215, 1287–1299. [Google Scholar] [CrossRef]
- Higgins, D.M.; Wang, R.; Milligan, B.; Schroeder, M.; Carlson, B.; Pokorny, J.; Cheshier, S.H.; Meyer, F.B.; Weissman, I.L.; Sarkaria, J.N.; et al. Brain Tumor Stem Cell Multipotency Correlates with Nanog Expression and Extent of Passaging in Human Glioblastoma Xenografts. Oncotarget 2013, 4, 792–801. [Google Scholar] [CrossRef]
- Gong, S.; Li, Q.; Jeter, C.R.; Fan, Q.; Tang, D.G.; Liu, B. Regulation of NANOG in cancer cells. Mol. Carcinog. 2015, 54, 679–687. [Google Scholar] [CrossRef]
- Oliveto, S.; Mancino, M.; Manfrini, N.; Biffo, S. Role of microRNAs in translation regulation and cancer. World J. Biol. Chem. 2017, 8, 45–56. [Google Scholar] [CrossRef]
- Soni, P.; Qayoom, S.; Husain, N.; Kumar, P.; Chandra, A.; Ojha, B.K.; Gupta, R.K. CD24 and Nanog expression in Stem Cells in Glioblastoma: Correlation with Response to Chemoradiation and Overall Survival. Asian Pac. J. Cancer Prev. 2017, 18, 2215–2219. [Google Scholar] [PubMed]
- Wuebben, E.L.; Rizzino, A. The dark side of SOX2: Cancer—A comprehensive overview. Oncotarget 2017, 8, 44917–44943. [Google Scholar] [CrossRef]
- De La Rocha, A.M.A.; Sampron, N.; Alonso, M.M.; Matheu, A. Role of SOX family of transcription factors in central nervous system tumors. Am. J. Cancer Res. 2014, 4, 312–324. [Google Scholar] [PubMed]
- Annovazzi, L.; Mellai, M.; Caldera, V.; Valente, G.; Schiffer, M.M.A.D. SOX2 expression and amplification in gliomas and glioma cell lines. Cancer Genom. Proteom. 2011, 8, 139–147. [Google Scholar]
- Lasky, J.L.; Choe, M.; Nakano, I. Cancer stem cells in pediatric brain tumors. Curr. Stem Cell Res. Ther. 2009, 4, 298–305. [Google Scholar]
- Bahmad, H.F.; Elajami, M.K.; El Zarif, T.; Bou-Gharios, J.; Abou-Antoun, T.J.; Abou-Kheir, W. Drug repurposing towards targeting cancer stem cells in pediatric brain tumors. Cancer Metastasis Rev. 2020, 39, 127–148. [Google Scholar] [CrossRef]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef]
- Vaidya, M.; Sugaya, K. Differential sequences and single nucleotide polymorphism of exosomal SOX2 DNA in cancer. PLoS ONE 2020, 15, e0229309. [Google Scholar] [CrossRef]
- Luo, W.; Yan, D.; Song, Z.; Zhu, X.; Liu, X.; Li, X.; Zhao, S. miR-126-3p sensitizes glioblastoma cells to temozolomide by inactivating Wnt/β-catenin signaling via targeting SOX2. Life Sci. 2019, 226, 98–106. [Google Scholar] [CrossRef]
- Lui, W.-O.; Pourmand, N.; Patterson, B.K.; Fire, A. Patterns of Known and Novel Small RNAs in Human Cervical Cancer. Cancer Res. 2007, 67, 6031–6043. [Google Scholar] [CrossRef]
- An, Z.; Aksoy, O.; Zheng, T.; Fan, Q.-W.; Weiss, W.A. Epidermal growth factor receptor and EGFRvIII in glioblastoma: Signaling pathways and targeted therapies. Oncogene 2018, 37, 1561–1575. [Google Scholar] [CrossRef]
- Salkeni, M.A.; Zarzour, A.; Ansay, T.Y.; McPherson, C.M.; Warnick, R.E.; Rixe, O.; Bahassi, E.M. Detection of EGFRvIII mutant DNA in the peripheral blood of brain tumor patients. J. Neuro Oncol. 2013, 115, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Saenz-Antoñanzas, A.; Auzmendi-Iriarte, J.; Carrasco-Garcia, E.; Moreno-Cugnon, L.; Ruiz, I.; Villanua, J.; Egaña, L.; Otaegui, D.; Samprón, N.; Matheu, A. Liquid Biopsy in Glioblastoma: Opportunities, Applications and Challenges. Cancers 2019, 11, 950. [Google Scholar] [CrossRef] [PubMed]
- Oldrini, B.; Vaquero-Siguero, N.; Mu, Q.; Kroon, P.; Zhang, Y.; Galán-Ganga, M.; Bao, Z.; Wang, Z.; Liu, H.; Sa, J.K.; et al. MGMT genomic rearrangements contribute to chemotherapy resistance in gliomas. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef]
- De Botton, S.; Mondesir, J.; Willekens, C.; Touat, M. IDH1 and IDH2 mutations as novel therapeutic targets: Current perspectives. J. Blood Med. 2016, 7, 171–180. [Google Scholar] [CrossRef]
- Stewart, J.B.; Chinnery, P.F. The dynamics of mitochondrial DNA heteroplasmy: Implications for human health and disease. Nat. Rev. Genet. 2015, 16, 530–542. [Google Scholar] [CrossRef]
- Kirches, E. MtDNA As a Cancer Marker: A Finally Closed Chapter? Curr. Genom. 2017, 18, 255–267. [Google Scholar] [CrossRef][Green Version]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [CrossRef]
- Wallace, D.C.; Chalkia, D. Mitochondrial DNA Genetics and the Heteroplasmy Conundrum in Evolution and Disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a021220. [Google Scholar] [CrossRef]
- Kirches, E.; Michael, M.; Woy, C.; Schneider, T.; Warich-Kirches, M.; Schneider-Stock, R.; Winkler, K.; Wittig, H.; Dietzmann, K. Loss of heteroplasmy in the displacement loop of brain mitochondrial DNA in astrocytic tumors. Genes Chromosom. Cancer 1999, 26, 80–83. [Google Scholar] [CrossRef]
- Dickinson, A.M.; Yeung, K.Y.; Donoghue, J.P.; Baker, M.J.; Kelly, R.D.; McKenzie, M.; Johns, T.G.; John, J.C.S. The regulation of mitochondrial DNA copy number in glioblastoma cells. Cell Death Differ. 2013, 20, 1644–1653. [Google Scholar] [CrossRef]
- Guerra, F.; Arbini, A.A.; Moro, L. Mitochondria and cancer chemoresistance. Biochim. Biophys. Acta Bioenergy 2017, 1858, 686–699. [Google Scholar] [CrossRef]
- Guescini, M.; Genedani, S.; Stocchi, V.; Agnati, L.F. Astrocytes and Glioblastoma cells release exosomes carrying mtDNA. J. Neural Transm. 2009, 117, 1–4. [Google Scholar] [CrossRef]
- Kahlert, C.; Melo, S.A.; Protopopov, A.; Tang, J.; Seth, S.; Koch, M.; Zhang, J.; Weitz, J.; Chin, L.; Futreal, A.; et al. Identification of Double-stranded Genomic DNA Spanning All Chromosomes with MutatedKRASandp53DNA in the Serum Exosomes of Patients with Pancreatic Cancer. J. Biol. Chem. 2014, 289, 3869–3875. [Google Scholar] [CrossRef]
- Ariyoshi, K.; Miura, T.; Kasai, K.; Fujishima, Y.; Nakata, A.; Yoshida, M. Radiation-Induced Bystander Effect is Mediated by Mitochondrial DNA in Exosome-Like Vesicles. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef]
- Rausch, T.; Jones, D.T.; Zapatka, M.; Stütz, A.M.; Zichner, T.; Weischenfeldt, J.; Jäger, N.; Remke, M.; Shih, D.; Northcott, P.A.; et al. Genome Sequencing of Pediatric Medulloblastoma Links Catastrophic DNA Rearrangements with TP53 Mutations. Cell 2012, 148, 59–71. [Google Scholar] [CrossRef]
- Collins, V.P. Brain tumours: Classification and genes. J. Neurol. Neurosurg. Psychiatry 2004, 75, ii2–ii11. [Google Scholar] [CrossRef]
- Yang, J.-K.; Song, J.; Huo, H.-R.; Zhao, Y.-L.; Zhang, G.-Y.; Zhao, Z.-M.; Sun, G.-Z.; Jiao, B.-H. DNM3, p65 and p53 from exosomes represent potential clinical diagnosis markers for glioblastoma multiforme. Ther. Adv. Med Oncol. 2017, 9, 741–754. [Google Scholar] [CrossRef]
- Andersson, U.; Guo, D.; Malmer, B.; Bergenheim, A.T.; Henriksson, R. Epidermal growth factor receptor family (EGFR, ErbB2?4) in gliomas and meningiomas. Acta Neuropathol. 2004, 108, 135–142. [Google Scholar] [CrossRef]
- He, J.; Allen, J.R.; Collins, V.P.; Allalunis-Turner, M.J.; Godbout, R.; Day, R.S.; James, C.D. CDK4 amplification is an alternative mechanism to p16 gene homozygous deletion in glioma cell lines. Cancer Res. 1994, 54, 5804–5807. [Google Scholar]
- Mure, H.; Matsuzaki, K.; Kitazato, K.T.; Mizobuchi, Y.; Kuwayama, K.; Kageji, T.; Nagahiro, S. Akt2 and Akt3 play a pivotal role in malignant gliomas. Neuro Oncol. 2009, 12, 221–232. [Google Scholar] [CrossRef]
- Reifenberger, G.; Liu, L.; Ichimura, K.; Schmidt, E.E.; Collins, V.P. Amplification and overexpression of the MDM2 gene in a subset of human malignant gliomas without p53 mutations. Cancer Res. 1993, 53, 2736–2739. [Google Scholar]
- Witkiewicz, A.K.; Cox, D.W.; Rivadeneira, D.B.; Ertel, A.E.; Fortina, P.; Schwartz, G.F.; Knudsen, E.S. The retinoblastoma tumor suppressor pathway modulates the invasiveness of ErbB2-positive breast cancer. Oncogene 2013, 33, 3980–3991. [Google Scholar] [CrossRef]
- Goldhoff, P.; Clarke, J.; Smirnov, I.; Berger, M.S.; Prados, M.D.; James, C.D.; Perry, A.; Phillips, J.J. Clinical Stratification of Glioblastoma Based on Alterations in Retinoblastoma Tumor Suppressor Protein (RB1) and Association With the Proneural Subtype. J. Neuropathol. Exp. Neurol. 2012, 71, 83–89. [Google Scholar] [CrossRef]
- Zhao, S.; Yuan, Q.; Hao, H.; Guo, Y.; Liu, S.; Zhang, Y.; Wang, J.; Liu, H.; Wang, F.; Liu, K.; et al. Expression of OCT4 pseudogenes in human tumours: Lessons from glioma and breast carcinoma. J. Pathol. 2011, 223, 672–682. [Google Scholar] [CrossRef]
- Hayashi, H.; Arao, T.; Matsumoto, K.; Nagai, T.; Kimura, H.; De Velasco, M.; Fujita, Y.; Yamada, Y.; Nakagawa, K.; Nishio, K. Amplification of OCT4-Pseudogene POU5F1B is a Poor Prognostic Factor in Gastric Cancer. Ann. Oncol. 2012, 23, xi112. [Google Scholar] [CrossRef]
- Sharma, A.; Johnson, A. Exosome DNA: Critical regulator of tumor immunity and a diagnostic biomarker. J. Cell. Physiol. 2019, 235, 1921–1932. [Google Scholar] [CrossRef]
- Ricklefs, F.L.; Alayo, Q.; Krenzlin, H.; Mahmoud, A.B.; Speranza, M.C.; Nakashima, H.; Hayes, J.L.; Lee, K.; Balaj, L.; Passaro, C.; et al. Immune evasion mediated by PD-L1 on glioblastoma-derived extracellular vesicles. Sci. Adv. 2018, 4, eaar2766. [Google Scholar] [CrossRef]
- Achanta, P.; Steranka, J.P.; Tang, Z.; Rodić, N.; Sharma, R.; Yang, W.R.; Ma, S.; Grivainis, M.; Huang, C.R.L.; Schneider, A.M.; et al. Somatic retrotransposition is infrequent in glioblastomas. Mob. DNA 2016, 7, 1–9. [Google Scholar] [CrossRef]
- Scott, E.C.; Devine, S.E. The Role of Somatic L1 Retrotransposition in Human Cancers. Viruses 2017, 9, 131. [Google Scholar] [CrossRef]
- Carreira, P.; Ewing, A.D.; Li, G.; Schauer, S.N.; Upton, K.R.; Fagg, A.C.; Morell, S.; Kindlova, M.; Gerdes, P.; Richardson, S.R.; et al. Evidence for L1-associated DNA rearrangements and negligible L1 retrotransposition in glioblastoma multiforme. Mob. DNA 2016, 7, 1–14. [Google Scholar] [CrossRef]
- Kim, Y.-J.; Lee, J.; Han, K. Transposable Elements: No More ‘Junk DNA’. Genom. Inform. 2012, 10, 226–233. [Google Scholar] [CrossRef]
- Kong, Y.; Rose, C.M.; Cass, A.A.; Williams, A.G.; Darwish, M.; Lianoglou, S.; Haverty, P.M.; Tong, A.-J.; Blanchette, C.; Albert, M.L.; et al. Transposable element expression in tumors is associated with immune infiltration and increased antigenicity. Nat. Commun. 2019, 10, 5228. [Google Scholar] [CrossRef]
- Enciso-Mora, V.; Hosking, F.J.; Kinnersley, B.; Wang, Y.; Shete, S.; Zelenika, D.; Broderick, P.; Idbaih, A.; Delattre, J.-Y.; Hoang-Xuan, K.; et al. Deciphering the 8q24.21 association for glioma. Hum. Mol. Genet. 2013, 22, 2293–2302. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Selleck, M.J.; Senthil, M.; Wall, N.R. Making Meaningful Clinical Use of Biomarkers. Biomark. Insights 2017, 12. [Google Scholar] [CrossRef]


{kind=link}
| Gene/DNA | Type of Modulation in Exosomal DNA | Reference |
|---|---|---|
| NANOG/NANOGP8 | SNP, insertions | [27,31,32,33,34] |
| SOX2 | SNP, breast cancer specific SNP | [35,36,37,38,39,40,41,42,43] |
| EGFR | amplification, mutation | [44,45] |
| MGMT | methylated promoter, gene alterations, genomic rearrangements | [3,46,47] |
| IDH1/IDH2 | mutations | [3,48,49] |
| mtDNA | SNP, elevated quantities | [50,51,52,53,54,55,56,57,58,59] |
| TP53 | gDNA absent (loss of heterozygosity of chromosome 17 at position p13.1) | [3,21,60,61,62] |
| CDKN2a | gDNA absent (loss of heterozygosity of chromosome 9 at p21.3) | [3] |
| PTEN | gDNA absent (loss of heterozygosity of chromosome 10 at q23.1) | [3] |
| ERBB2 | GBM-specific gDNA | [3,63,64,65,66,67] |
| CDK4 | GBM-specific gDNA | [3,63,64,65,66] |
| AKT3 | GBM-specific gDNA | [3,63,64,65,66] |
| MDM2 | GBM-specific gDNA | [3,63,64,65,66] |
| RB1 | GBM-specific gDNA | [3,67,68] |
| c-Myc | amplified sequences of gDNA (including full introns) | [8,69,70] |
| POU5F1B | elevated levels of gDNA, sits side by side to c-Myc on chromosome 8q24. | [8,69,70] |
| PD-L1 | elevated levels of DNA | [71,72] |
| L1 | enriched levels of transposable element DNA | [8,73,74] |
| HERV | enriched levels of transposable element DNA | [8,75] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vaidya, M.; Sugaya, K. DNA Associated with Circulating Exosomes as a Biomarker for Glioma. Genes 2020, 11, 1276. https://doi.org/10.3390/genes11111276
Vaidya M, Sugaya K. DNA Associated with Circulating Exosomes as a Biomarker for Glioma. Genes. 2020; 11(11):1276. https://doi.org/10.3390/genes11111276
Chicago/Turabian StyleVaidya, Manjusha, and Kiminobu Sugaya. 2020. "DNA Associated with Circulating Exosomes as a Biomarker for Glioma" Genes 11, no. 11: 1276. https://doi.org/10.3390/genes11111276
APA StyleVaidya, M., & Sugaya, K. (2020). DNA Associated with Circulating Exosomes as a Biomarker for Glioma. Genes, 11(11), 1276. https://doi.org/10.3390/genes11111276