The Genetic Structure of Chinese Hui Ethnic Group Revealed by Complete Mitochondrial Genome Analyses Using Massively Parallel Sequencing

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Preparation and Ethical Declaration
2.2. Library Construction and Sequencing of Mitochondrial Genome
2.3. Sequencing Data Analyses
2.4. The Worldwide Populations for a Comparison Study with Hui Group
2.5. MtDNA Haplogroup Allocation
2.6. Statistical Analyses
3. Results and Discussion
3.1. Sequencing Depth of Complete Mitogenome
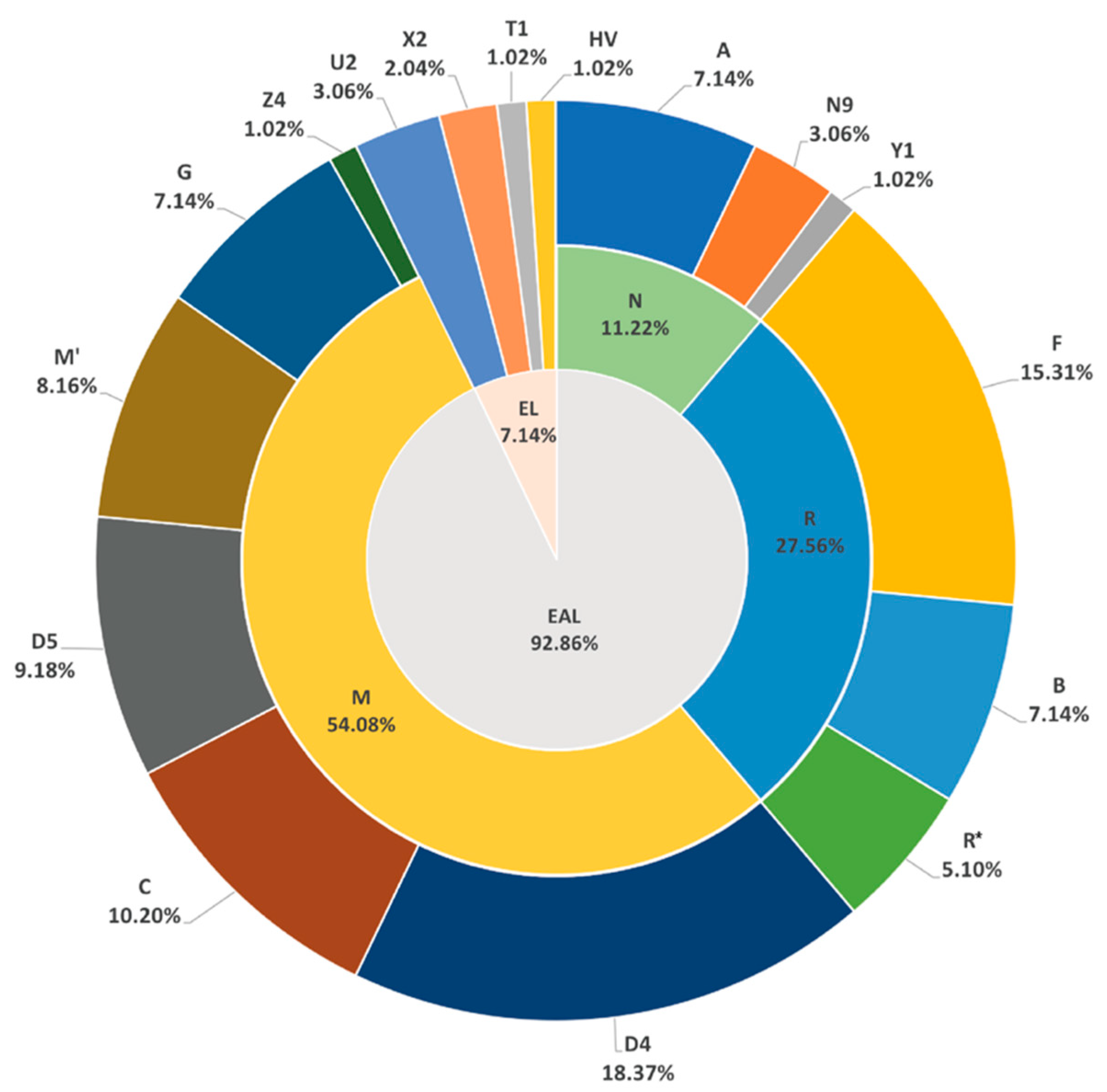
3.2. Mitochondrial Haplogroups of Hui Ethnic Group
3.3. Descriptive Statistical Indexes of Hui and Han Populations
3.4. Genetic Divergence among Populations
3.4.1. Analysis of Molecular Variance for Hui and 99 Worldwide Populations
3.4.2. Principal Component Analysis for Hui and 99 Worldwide Populations
3.4.3. Genetic Distance Analyses for Hui and 99 Worldwide Populations
3.4.4. The Population Expansion Time of Hui and Han Populations
3.4.5. Haplotypes Sharing between Hui and Worldwide Populations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Xie, X.; Shan, X. The DNA Evidence for the Origin of Chinese Hui; Research on the Hui: Gansu, China, 2002; Volume 3. [Google Scholar]
- Hong, W.; Chen, S.; Shao, H.; Fu, Y.; Hu, Z.; Xu, A. HLA class I Polymorphism in Mongolian and Hui Ethnic Groups from Northern China. Hum. Immunol. 2007, 68, 439–448. [Google Scholar] [CrossRef]
- Yao, Y.-G.; Kong, Q.-P.; Wang, C.-Y.; Zhu, C.-L.; Zhang, Y.-P. Different Matrilineal Contributions to Genetic Structure of Ethnic Groups in the Silk Road Region in China. Mol. Biol. Evol. 2004, 21, 2265–2280. [Google Scholar] [CrossRef] [PubMed]
- Zhenhua, H. The Silk Road and the Hui Population; Heilongjiang National Series: Heilongjiang, China, 2015; Volume 4. [Google Scholar] [CrossRef]
- Wang, C.-C.; Lu, Y.; Kang, L.; Ding, H.; Yan, S.; Guo, J.; Zhang, Q.; Wen, S.; Wang, L.; Zhang, M.; et al. The massive assimilation of indigenous East Asian populations in the origin of Muslim Hui people inferred from paternal Y chromosome. Am. J. Phys. Anthr. 2019, 169, 341–347. [Google Scholar] [CrossRef] [PubMed]
- World Medical Association. World Medical Association Declaration of Helsinki. JAMA 2013, 310, 2191–2194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, R.M.; Kubacka, I.; Chinnery, P.F.; Chrzanowska-Lightowlers, Z.; Turnbull, D.M.; Howell, N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 1999, 23, 147. [Google Scholar] [CrossRef]
- Just, R.S.; Irwin, J.A.; Parson, W. Mitochondrial DNA heteroplasmy in the emerging field of massively parallel sequencing. Forensic Sci. Int. Genet. 2015, 18, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Kong, Q.-P.; Salas, A.; Sun, C.; Fuku, N.; Tanaka, M.; Zhong, L.; Wang, C.-Y.; Yao, Y.-G.; Bandelt, H.-J. Distilling Artificial Recombinants from Large Sets of Complete mtDNA Genomes. PLoS ONE 2008, 3, e3016. [Google Scholar] [CrossRef] [Green Version]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Excoffier, L.; Laval, G.; Schneider, S. ARLEQUIN. Ver 3.0. An integrated software package for population genetic data analysis. Evol. Bioinform. 2005, 1, 47–50. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, P.; Ermini, L.; Thomson, N.; Mormina, M.; Rito, T.; Röhl, A.; Salas, A.; Oppenheimer, S.; Macaulay, V.; Richards, M.B. Correcting for Purifying Selection: An Improved Human Mitochondrial Molecular Clock. Am. J. Hum. Genet. 2009, 84, 740–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.A.; Suchard, M. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gignoux, C.R.; Henn, B.M.; Mountain, J.L. Rapid, global demographic expansions after the origins of agriculture. Proc. Natl. Acad. Sci. USA 2011, 108, 6044–6049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, J.; Yu, X. Detection of Population Expantion and Calculation of Expantion Time; Chinese Science and Technology: Beijing, China, 2017; Available online: http://www.paper.edu.cn (accessed on 13 November 2020).
- Palanichamy, M.G.; Mitra, B.; Zhang, C.-L.; Debnath, M.; Li, G.-M.; Wang, H.-W.; Agrawal, S.; Chaudhuri, T.K.; Zhang, Y.-P. West Eurasian mtDNA lineages in India: An insight into the spread of the Dravidian language and the origins of the caste system. Hum. Genet. 2015, 134, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Derenko, M.; Malyarchuk, B.; Denisova, G.; Perkova, M.; Litvinov, A.; Grzybowski, T.; Dambueva, I.; Skonieczna, K.; Rogalla, U.; Tsybovsky, I.; et al. Western Eurasian ancestry in modern Siberians based on mitogenomic data. BMC Evol. Biol. 2014, 14, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kivisild, T.; Tolk, H.V.; Parik, J.; Wang, Y.; Papiha, S.S.; Bandelt, H.J.; Villems, R. The emerging limbs and twigs of the East Asian mtDNA tree. Mol. Biol. Evol. 2002, 19, 1737–1751. [Google Scholar] [CrossRef] [Green Version]
- Derenko, M.; Malyarchuk, B.; Denisova, G.; Perkova, M.; Rogalla, U.; Grzybowski, T.; Khusnutdinova, E.; Dambueva, I.; Zakharov, I. Complete Mitochondrial DNA Analysis of Eastern Eurasian Haplogroups Rarely Found in Populations of Northern Asia and Eastern Europe. PLoS ONE 2012, 7, e32179. [Google Scholar] [CrossRef] [Green Version]
- Umetsu, K.; Tanaka, M.; Yuasa, I.; Adachi, N.; Miyoshi, A.; Kashimura, S.; Park, K.S.; Wei, Y.-H.; Watanabe, G.; Osawa, M. Multiplex amplified product-length polymorphism analysis of 36 mitochondrial single-nucleotide polymorphisms for haplogrouping of East Asian populations. Electrophoresis 2005, 26, 91–98. [Google Scholar] [CrossRef]
- Li, Y.-C.; Ye, W.-J.; Jiang, C.-G.; Zeng, Z.; Tian, J.-Y.; Yang, L.-Q.; Liu, K.-J.; Kong, Q.-P. River Valleys Shaped the Maternal Genetic Landscape of Han Chinese. Mol. Biol. Evol. 2019, 36, 1643–1652. [Google Scholar] [CrossRef]
- Peng, M.-S.; He, J.-D.; Liu, H.-X.; Zhang, Y.-P. Tracing the legacy of the early Hainan Islanders—A perspective from mitochondrial DNA. BMC Evol. Biol. 2011, 11, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabbada, K.A.; Trejaut, J.A.; Loo, J.-H.; Chen, Y.-M.; Lin, M.; Lahr, M.M.; Kivisild, T.; De Ungria, M.C.A. Philippine Mitochondrial DNA Diversity: A Populated Viaduct between Taiwan and Indonesia? Mol. Biol. Evol. 2009, 27, 21–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutanan, W.; Kampuansai, J.; Srikummool, M.; Kangwanpong, D.; Ghirotto, S.; Brunelli, A.; Stoneking, M. Complete mitochondrial genomes of Thai and Lao populations indicate an ancient origin of Austroasiatic groups and demic diffusion in the spread of Tai–Kadai languages. Qual. Life Res. 2017, 136, 85–98. [Google Scholar] [CrossRef] [Green Version]
- Derenko, M.; Malyarchuk, B.; Grzybowski, T.; Denisova, G.; Rogalla, U.; Perkova, M.; Dambueva, I.; Zakharov, I. Origin and Post-Glacial Dispersal of Mitochondrial DNA Haplogroups C and D in Northern Asia. PLoS ONE 2010, 5, e15214. [Google Scholar] [CrossRef] [Green Version]
- Derenko, M.; Malyarchuk, B.; Grzybowski, T.; Denisova, G.; Dambueva, I.; Perkova, M.; Dorzhu, C.; Luzina, F.; Lee, H.K.; Vanecek, T.; et al. Phylogeographic Analysis of Mitochondrial DNA in Northern Asian Populations. Am. J. Hum. Genet. 2007, 81, 1025–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, R.-J.; The Genographic Consortium; Pan, S.-L.; Mustavich, L.F.; Qin, Z.-D.; Cai, X.-Y.; Qian, J.; Liu, C.-W.; Peng, J.-H.; Li, S.-L.; et al. Pinghua population as an exception of Han Chinese’s coherent genetic structure. J. Hum. Genet. 2008, 53, 303–313. [Google Scholar] [CrossRef] [Green Version]
- Lan, Q.; Xie, T.; Jin, X.; Fang, Y.; Mei, S.; Yang, G.; Zhu, B. MtDNA polymorphism analyses in the Chinese Mongolian group: Efficiency evaluation and further matrilineal genetic structure exploration. Mol. Genet. Genom. Med. 2019, 7, e00934. [Google Scholar] [CrossRef] [Green Version]
- Derenko, M.; Malyarchuk, B.; Bahmanimehr, A.; Denisova, G.; Perkova, M.; Farjadian, S.; Yepiskoposyan, L. Complete Mitochondrial DNA Diversity in Iranians. PLoS ONE 2013, 8, e80673. [Google Scholar] [CrossRef] [Green Version]
- Maji, S.; Krithika, S.; Vasulu, T.S. Distribution of Mitochondrial DNA Macrohaplogroup N in India with Special Reference to Haplogroup R and its Sub-Haplogroup, U. Int. J. Hum. Genet. 2008, 8, 85–96. [Google Scholar] [CrossRef]
- Palanichamy, M.G.; Sun, C.; Agrawal, S.; Bandelt, H.-J.; Kong, Q.-P.; Khan, F.; Wang, C.-Y.; Chaudhuri, T.K.; Palla, V.; Zhang, Y.-P. Phylogeny of Mitochondrial DNA Macrohaplogroup N in India, Based on Complete Sequencing: Implications for the Peopling of South Asia. Am. J. Hum. Genet. 2004, 75, 966–978. [Google Scholar] [CrossRef] [Green Version]
- Mittal, B.; Tripathy, V.; Aruna, M.; Reddy, A.G.; Thanseem, I.; Thangaraj, K.; Singh, L.; Reddy, B.M. Mitochondrial DNA variation and substructure among the tribal populations of Andhra Pradesh, India. Am. J. Hum. Biol. 2008, 20, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Metspalu, M.; Kivisild, T.; Metspalu, E.; Parik, J.; Hudjashov, G.; Kaldma, K.; Serk, P.; Karmin, M.; Behar, D.M.; Gilbert, M.T.P.; et al. Most of the extant mtDNA boundaries in South and Southwest Asia were likely shaped during the initial settlement of Eurasia by anatomically modern humans. BMC Genet. 2004, 5, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of Molecular Variance Inferred from Metric Distances among DNA Haplotypes: Application to Human Mitochondrial DNA Restriction Data. Genetics 1992, 131, 479–491. [Google Scholar]
- Amaral, M.R.X.; Albrecht, M.; McKinley, A.S.; De Carvalho, A.M.F.; De Sousa, J.S.C.; Diniz, F.M.; Junior, S.C.D.S. Mitochondrial DNA Variation Reveals a Sharp Genetic Break within the Distribution of the Blue Land Crab Cardisoma guanhumi in the Western Central Atlantic. Molecules 2015, 20, 15158–15174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auton, A.; Abecasis, G.R.; Altshuler, D.M.; Durbin, R.M.; Abecasis, G.R.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar]
- Wright, S. Evolution and Genetics of Populations; University of Chicago: Chicago, IL, USA, 1978. [Google Scholar]
- Govindajuru, R. Variation in gene flow levels among predominantly self-pollinated plants. J. Evol. Biol. 1989, 2, 173–181. [Google Scholar] [CrossRef]
- Lipman, J.N. Familiar Strangers: A History of Muslims in Northwest China; University of Washington Press: Seattle, WA, USA, 1997. [Google Scholar] [CrossRef]
- Xie, M.; Song, F.; Li, J.; Lang, M.; Luo, H.; Wang, Z.; Wu, J.; Li, C.; Tian, C.; Wang, W.; et al. Genetic substructure and forensic characteristics of Chinese Hui populations using 157 Y-SNPs and 27 Y-STRs. Forensic Sci. Int. Genet. 2019, 41, 11–18. [Google Scholar] [CrossRef]
- He, G.; Wang, Z.; Wang, M.; Luo, T.; Liu, J.; Zhou, Y.; Gao, B.; Hou, Y. Forensic ancestry analysis in two Chinese minority populations using massively parallel sequencing of 165 ancestry-informative SNPs. Electrophoresis 2018, 39, 2732–2742. [Google Scholar] [CrossRef]
- Zhou, B.; Wen, S.; Sun, H.; Zhang, H.; Shi, R. Genetic affinity between Ningxia Hui and eastern Asian populations revealed by a set of InDel loci. R. Soc. Open Sci. 2020, 7, 190358. [Google Scholar] [CrossRef] [Green Version]
- Yao, H.-B.; Wang, C.-C.; Tao, X.; Shang, L.; Wen, S.-Q.; Zhu, B.; Kang, L.; Jin, L.; Li, H. Genetic evidence for an East Asian origin of Chinese Muslim populations Dongxiang and Hui. Sci. Rep. 2016, 6, 38656. [Google Scholar] [CrossRef] [Green Version]
- Xie, T.; Guo, Y.-X.; Chen, L.; Fang, Y.; Tai, Y.; Zhu, B.-F.; Qiu, P.; Zhu, B. A set of autosomal multiple InDel markers for forensic application and population genetic analysis in the Chinese Xinjiang Hui group. Forensic Sci. Int. Genet. 2018, 35, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.-T.; Han, J.-T.; Zhang, Y.-D.; Liu, W.-J.; Wang, T.-J.; Yan, J.-W.; Huang, J.-F.; Du, W.-A.; Guo, J.-X.; Wang, H.-D.; et al. Diversity study of 12 X-chromosomal STR loci in Hui ethnic from China. Electrophoresis 2014, 35, 2001–2007. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Guo, Y.-X.; Xie, T.; Jin, X.; Lan, Q.; Zhu, B.-F.; Zhu, B. Forensic molecular genetic diversity analysis of Chinese Hui ethnic group based on a novel STR panel. Int. J. Leg. Med. 2018, 132, 1297–1299. [Google Scholar] [CrossRef] [PubMed]
- Lan, Q.; Chen, J.; Guo, Y.; Xie, T.; Fang, Y.; Jin, X.; Cui, W.; Zhou, Y.; Zhu, B. Genetic structure and polymorphism analysis of Xinjiang Hui ethnic minority based on 21 STRs. Mol. Biol. Rep. 2018, 45, 99–108. [Google Scholar] [CrossRef]
- Lu, D.; Lou, H.; Yuan, K.; Wang, X.; Wang, Y.; Zhang, C.; Lu, Y.; Yang, X.; Deng, L.; Zhou, Y.; et al. Ancestral Origins and Genetic History of Tibetan Highlanders. Am. J. Hum. Genet. 2016, 99, 580–594. [Google Scholar] [CrossRef] [Green Version]
- Berzin, A. History of the Muslims of Tibet. 2019. Available online: Studybuddhism.com (accessed on 22 October 2019).
- Minshu, Y.; Xinfang, Q.; Jinglun, X.; Zudong, L.; Jiazhen, T.; Houjun, L.; Dexiang, L.; Li, L.; Wuzhong, Y.; Xianzhen, T.; et al. Study on mitochondrial DNA polymorphism in Chinese Han, Uygur, Kazak and Hui populations. Chin. Sci. 1988, 1, 60–70. [Google Scholar]
- Daftary, F. A Modern History of the Ismailis: Continuity and Change in a Muslim Community; Bloomsbury Publishing: London, UK, 2010. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genetic Diversities | Neutrality Tests | Mismatch Distributions | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Population | Abbreviation | n | h | Hd (SD) | S (Eta) | k | Pi (SD) | Tajima’s D (p) | Fu’s Fs (p) | SSD (p) | HRI (p) |
| Hui in Northwest China | HU | 98 | 97 | 1.000(0.002) | 640(650) | 37.06 | 0.0022(0.00006) | −2.39 (0.000) | −24.09 (0.001) | 0.0015 (0.520) | 0.0005 (0.989) |
| Beijing Han Chinese | CHB | 85 | 85 | 1.000(0.002) | 614(619) | 38.37 | 0.0023(0.00005) | −2.37 (0.000) | −24.15 (0.001) | 0.0029 (0.429) | 0.0007 (0.999) |
| Southern Han Chinese | CHS | 50 | 50 | 1.000(0.004) | 442(445) | 38.36 | 0.0023(0.00007) | −2.23 (0.001) | −21.91 (0.000) | 0.0021 (0.652) | 0.0015 (0.981) |
| Minnan Han | MIN | 50 | 48 | 0.998(0.004) | 397(400) | 36.09 | 0.0022(0.00006) | −2.16 (0.003) | −17.98 (0.000) | 0.0013 (0.744) | 0.0014 (0.983) |
| Hakka Han | HAK | 45 | 39 | 0.994(0.006) | 369(372) | 34.73 | 0.0021(0.00007) | −2.16 (0.002) | −7.38 (0.035) | 0.0022 (0.510) | 0.0046 (0.171) |
| Percentage of Variation | |||||
|---|---|---|---|---|---|
| Groupings | Number of Populations | Number of Groups | Within Populations | Among Populations with in Groups | Among Groups |
| Linguistic families of worldwide populations | 100 | 13 | 92.01 | 5.57 | 2.42 |
| Geographic distributions of worldwide populations | 100 | 9 | 92.08 | 5.95 | 1.97 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.; Li, Y.; Tao, R.; Jin, X.; Guo, Y.; Cui, W.; Chen, A.; Yang, Y.; Zhang, X.; Zhang, J.; et al. The Genetic Structure of Chinese Hui Ethnic Group Revealed by Complete Mitochondrial Genome Analyses Using Massively Parallel Sequencing. Genes 2020, 11, 1352. https://doi.org/10.3390/genes11111352
Chen C, Li Y, Tao R, Jin X, Guo Y, Cui W, Chen A, Yang Y, Zhang X, Zhang J, et al. The Genetic Structure of Chinese Hui Ethnic Group Revealed by Complete Mitochondrial Genome Analyses Using Massively Parallel Sequencing. Genes. 2020; 11(11):1352. https://doi.org/10.3390/genes11111352
Chicago/Turabian StyleChen, Chong, Yuchun Li, Ruiyang Tao, Xiaoye Jin, Yuxin Guo, Wei Cui, Anqi Chen, Yue Yang, Xingru Zhang, Jingyi Zhang, and et al. 2020. "The Genetic Structure of Chinese Hui Ethnic Group Revealed by Complete Mitochondrial Genome Analyses Using Massively Parallel Sequencing" Genes 11, no. 11: 1352. https://doi.org/10.3390/genes11111352
APA StyleChen, C., Li, Y., Tao, R., Jin, X., Guo, Y., Cui, W., Chen, A., Yang, Y., Zhang, X., Zhang, J., Li, C., & Zhu, B. (2020). The Genetic Structure of Chinese Hui Ethnic Group Revealed by Complete Mitochondrial Genome Analyses Using Massively Parallel Sequencing. Genes, 11(11), 1352. https://doi.org/10.3390/genes11111352