At the X-Roads of Sex and Genetics in Pulmonary Arterial Hypertension

 ,
, 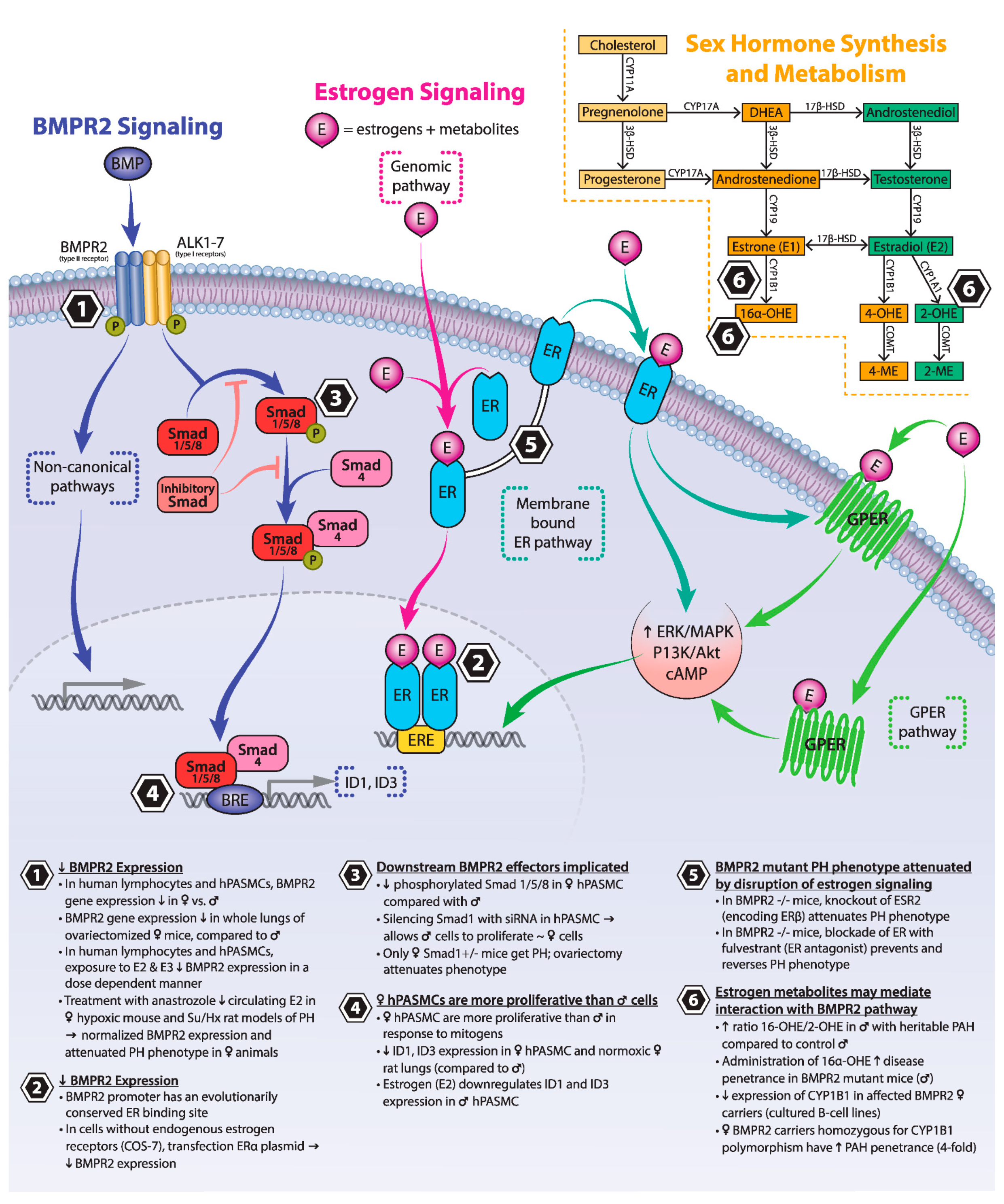{kind=link}
Abstract
:1. Introduction
2. The “Estrogen Puzzle” of PAH
3. BMPR2 Signaling
4. Estrogen Signaling
5. Estrogen and BMPR2
5.1. Estrogens and Their Receptors Reduce BMPR2 Expression and Downstream Signaling
5.2. Loss of Estrogen Signaling Attenuates Experimental PH Phenotypes Driven by Mutations in Components of the BMPR2 Signaling Pathway
5.3. Estrogen Metabolites May Mediate Interaction between BMPR2 and Estrogen Signaling
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmuller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur. Respir. J. 2019, 53, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andruska, A.; Spiekerkoetter, E. Consequences of BMPR2 Deficiency in the Pulmonary Vasculature and Beyond: Contributions to Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2018, 19, 2499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, K.B.; Machado, R.D.; Pauciulo, M.W.; Thomson, J.R.; Phillips, J.A., 3rd; Loyd, J.E.; Nichols, W.C.; Trembath, R.C. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat. Genet. 2000, 26, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Morse, J.H.; Slager, S.L.; Cuervo, N.; Moore, K.J.; Venetos, G.; Kalachikov, S.; Cayanis, E.; Fischer, S.G.; Barst, R.J.; et al. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am. J. Hum. Genet. 2000, 67, 737–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, R.D.; Aldred, M.A.; James, V.; Harrison, R.E.; Patel, B.; Schwalbe, E.C.; Gruenig, E.; Janssen, B.; Koehler, R.; Seeger, W.; et al. Mutations of the TGF-beta type II receptor BMPR2 in pulmonary arterial hypertension. Hum. Mutat. 2006, 27, 121–132. [Google Scholar] [CrossRef]
- Cogan, J.D.; Pauciulo, M.W.; Batchman, A.P.; Prince, M.A.; Robbins, I.M.; Hedges, L.K.; Stanton, K.C.; Wheeler, L.A.; Phillips, J.A., 3rd; Loyd, J.E.; et al. High frequency of BMPR2 exonic deletions/duplications in familial pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2006, 174, 590–598. [Google Scholar] [CrossRef]
- Aldred, M.A.; Vijayakrishnan, J.; James, V.; Soubrier, F.; Gomez-Sanchez, M.A.; Martensson, G.; Galie, N.; Manes, A.; Corris, P.; Simonneau, G.; et al. BMPR2 gene rearrangements account for a significant proportion of mutations in familial and idiopathic pulmonary arterial hypertension. Hum. Mutat. 2006, 27, 212–213. [Google Scholar] [CrossRef]
- Thomson, J.R.; Machado, R.D.; Pauciulo, M.W.; Morgan, N.V.; Humbert, M.; Elliott, G.C.; Ward, K.; Yacoub, M.; Mikhail, G.; Rogers, P.; et al. Sporadic primary pulmonary hypertension is associated with germline mutations of the gene encoding BMPR-II, a receptor member of the TGF-beta family. J. Med. Genet. 2000, 37, 741–745. [Google Scholar] [CrossRef]
- Chen, N.Y.; Collum, S.D.; Luo, F.; Weng, T.; Le, T.T.; Hernandez, A.M.; Philip, K.; Molina, J.G.; Garcia-Morales, L.J.; Cao, Y.; et al. Macrophage bone morphogenic protein receptor 2 depletion in idiopathic pulmonary fibrosis and Group III pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L238–L254. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, C.; Stewart, S.; Upton, P.D.; Machado, R.; Thomson, J.R.; Trembath, R.C.; Morrell, N.W. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation 2002, 105, 1672–1678. [Google Scholar] [CrossRef] [Green Version]
- Evans, J.D.; Girerd, B.; Montani, D.; Wang, X.J.; Galie, N.; Austin, E.D.; Elliott, G.; Asano, K.; Grunig, E.; Yan, Y.; et al. BMPR2 mutations and survival in pulmonary arterial hypertension: An individual participant data meta-analysis. Lancet Respir. Med. 2016, 4, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Sztrymf, B.; Coulet, F.; Girerd, B.; Yaici, A.; Jais, X.; Sitbon, O.; Montani, D.; Souza, R.; Simonneau, G.; Soubrier, F.; et al. Clinical outcomes of pulmonary arterial hypertension in carriers of BMPR2 mutation. Am. J. Respir. Crit. Care Med. 2008, 177, 1377–1383. [Google Scholar] [CrossRef] [PubMed]
- Elliott, C.G.; Glissmeyer, E.W.; Havlena, G.T.; Carlquist, J.; McKinney, J.T.; Rich, S.; McGoon, M.D.; Scholand, M.B.; Kim, M.; Jensen, R.L.; et al. Relationship of BMPR2 mutations to vasoreactivity in pulmonary arterial hypertension. Circulation 2006, 113, 2509–2515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenzweig, E.B.; Morse, J.H.; Knowles, J.A.; Chada, K.K.; Khan, A.M.; Roberts, K.E.; McElroy, J.J.; Juskiw, N.K.; Mallory, N.C.; Rich, S.; et al. Clinical implications of determining BMPR2 mutation status in a large cohort of children and adults with pulmonary arterial hypertension. J. Heart Lung Transpl. 2008, 27, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Austin, E.D.; Loyd, J.E.; Phillips, J.A., 3rd. Genetics of pulmonary arterial hypertension. Semin. Respir. Crit. Care Med. 2009, 30, 386–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahm, T.; Tuder, R.M.; Petrache, I. Progress in solving the sex hormone paradox in pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L7–L26. [Google Scholar] [CrossRef]
- Larkin, E.K.; Newman, J.H.; Austin, E.D.; Hemnes, A.R.; Wheeler, L.; Robbins, I.M.; West, J.D.; Phillips, J.A., 3rd; Hamid, R.; Loyd, J.E. Longitudinal analysis casts doubt on the presence of genetic anticipation in heritable pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 892–896. [Google Scholar] [CrossRef] [Green Version]
- Ge, X.; Zhu, T.; Zhang, X.; Liu, Y.; Wang, Y.; Zhang, W. Gender differences in pulmonary arterial hypertension patients with BMPR2 mutation: A meta-analysis. Respir. Res. 2020, 21, 44. [Google Scholar] [CrossRef]
- Frump, A.L.; Goss, K.N.; Vayl, A.; Albrecht, M.; Fisher, A.; Tursunova, R.; Fierst, J.; Whitson, J.; Cucci, A.R.; Brown, M.B.; et al. Estradiol improves right ventricular function in rats with severe angioproliferative pulmonary hypertension: Effects of endogenous and exogenous sex hormones. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L873–L890. [Google Scholar] [CrossRef]
- Umar, S.; Iorga, A.; Matori, H.; Nadadur, R.D.; Li, J.; Maltese, F.; Van der Laarse, A.; Eghbali, M. Estrogen rescues preexisting severe pulmonary hypertension in rats. Am. J. Respir. Crit. Care Med. 2011, 184, 715–723. [Google Scholar] [CrossRef]
- Liu, Z.; Duan, Y.L.; Ge, S.L.; Zhang, C.X.; Gong, W.H.; Xu, J.J. Effect of estrogen on right ventricular remodeling of monocrotaline-induced pulmonary arterial hypertension in rats and its mechanism. Eur. Rev. Med. Pharm. Sci. 2019, 23, 1742–1750. [Google Scholar] [CrossRef]
- Lahm, T.; Albrecht, M.; Fisher, A.J.; Selej, M.; Patel, N.G.; Brown, J.A.; Justice, M.J.; Brown, M.B.; Van Demark, M.; Trulock, K.M.; et al. 17beta-Estradiol attenuates hypoxic pulmonary hypertension via estrogen receptor-mediated effects. Am. J. Respir. Crit. Care Med. 2012, 185, 965–980. [Google Scholar] [CrossRef] [PubMed]
- Nadadur, R.D.; Umar, S.; Wong, G.; Eghbali, M.; Iorga, A.; Matori, H.; Partow-Navid, R.; Eghbali, M. Reverse right ventricular structural and extracellular matrix remodeling by estrogen in severe pulmonary hypertension. J. Appl. Physiol. 2012, 113, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Lahm, T.; Frump, A.L.; Albrecht, M.E.; Fisher, A.J.; Cook, T.G.; Jones, T.J.; Yakubov, B.; Whitson, J.; Fuchs, R.K.; Liu, A.; et al. 17beta-Estradiol mediates superior adaptation of right ventricular function to acute strenuous exercise in female rats with severe pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L375–L388. [Google Scholar] [CrossRef] [PubMed]
- Ventetuolo, C.E.; Ouyang, P.; Bluemke, D.A.; Tandri, H.; Barr, R.G.; Bagiella, E.; Cappola, A.R.; Bristow, M.R.; Johnson, C.; Kronmal, R.A.; et al. Sex hormones are associated with right ventricular structure and function: The MESA-right ventricle study. Am. J. Respir. Crit. Care Med. 2011, 183, 659–667. [Google Scholar] [CrossRef]
- Swift, A.J.; Capener, D.; Hammerton, C.; Thomas, S.M.; Elliot, C.; Condliffe, R.; Wild, J.M.; Kiely, D.G. Right ventricular sex differences in patients with idiopathic pulmonary arterial hypertension characterised by magnetic resonance imaging: Pair-matched case controlled study. PLoS ONE 2015, 10, e0127415. [Google Scholar] [CrossRef]
- Jacobs, W.; Van de Veerdonk, M.C.; Trip, P.; De Man, F.; Heymans, M.W.; Marcus, J.T.; Kawut, S.M.; Bogaard, H.J.; Boonstra, A.; Vonk Noordegraaf, A. The right ventricle explains sex differences in survival in idiopathic pulmonary arterial hypertension. Chest 2014, 145, 1230–1236. [Google Scholar] [CrossRef] [Green Version]
- Kozu, K.; Sugimura, K.; Aoki, T.; Tatebe, S.; Yamamoto, S.; Yaoita, N.; Shimizu, T.; Nochioka, K.; Sato, H.; Konno, R.; et al. Sex differences in hemodynamic responses and long-term survival to optimal medical therapy in patients with pulmonary arterial hypertension. Heart Vessel. 2018, 33, 939–947. [Google Scholar] [CrossRef] [Green Version]
- Umar, S.; Cunningham, C.M.; Itoh, Y.; Moazeni, S.; Vaillancourt, M.; Sarji, S.; Centala, A.; Arnold, A.P.; Eghbali, M. The Y Chromosome Plays a Protective Role in Experimental Hypoxic Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2018, 197, 952–955. [Google Scholar] [CrossRef]
- Yan, L.; Cogan, J.D.; Hedges, L.K.; Nunley, B.; Hamid, R.; Austin, E.D. The Y Chromosome Regulates BMPR2 Expression via SRY: A Possible Reason “Why” Fewer Males Develop Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2018, 198, 1581–1583. [Google Scholar] [CrossRef]
- Hester, J.; Ventetuolo, C.; Lahm, T. Sex, Gender, and Sex Hormones in Pulmonary Hypertension and Right Ventricular Failure. Compr. Physiol. 2019, 10, 125–170. [Google Scholar] [CrossRef] [PubMed]
- Tofovic, S.P.; Jackson, E.K. Estradiol Metabolism: Crossroads in Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2019, 21, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Li, X.; Morrell, N.W. Id proteins in the vasculature: From molecular biology to cardiopulmonary medicine. Cardiovasc Res. 2014, 104, 388–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vinuesa, A.G.; Abdelilah-Seyfried, S.; Knaus, P.; Zwijsen, A.; Bailly, S. BMP signaling in vascular biology and dysfunction. Cytokine Growth Factor Rev. 2016, 27, 65–79. [Google Scholar] [CrossRef] [Green Version]
- Girerd, B.; Montani, D.; Coulet, F.; Sztrymf, B.; Yaici, A.; Jais, X.; Tregouet, D.; Reis, A.; Drouin-Garraud, V.; Fraisse, A.; et al. Clinical outcomes of pulmonary arterial hypertension in patients carrying an ACVRL1 (ALK1) mutation. Am. J. Respir. Crit. Care Med. 2010, 181, 851–861. [Google Scholar] [CrossRef] [Green Version]
- Shintani, M.; Yagi, H.; Nakayama, T.; Saji, T.; Matsuoka, R. A new nonsense mutation of SMAD8 associated with pulmonary arterial hypertension. J. Med. Genet. 2009, 46, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Fan, R.; Ji, R.; Zou, W.; Penny, D.J.; Varghese, N.P.; Fan, Y. Novel homozygous BMP9 nonsense mutation causes pulmonary arterial hypertension: A case report. BMC Pulm. Med. 2016, 16, 17. [Google Scholar] [CrossRef] [Green Version]
- Austin, E.D.; Ma, L.; LeDuc, C.; Berman Rosenzweig, E.; Borczuk, A.; Phillips, J.A., 3rd; Palomero, T.; Sumazin, P.; Kim, H.R.; Talati, M.H.; et al. Whole exome sequencing to identify a novel gene (caveolin-1) associated with human pulmonary arterial hypertension. Circ. Cardiovasc. Genet. 2012, 5, 336–343. [Google Scholar] [CrossRef] [Green Version]
- Hamidi, S.A.; Dickman, K.G.; Berisha, H.; Said, S.I. 17beta-estradiol protects the lung against acute injury: Possible mediation by vasoactive intestinal polypeptide. Endocrinology 2011, 152, 4729–4737. [Google Scholar] [CrossRef]
- Dougherty, S.M.; Mazhawidza, W.; Bohn, A.R.; Robinson, K.A.; Mattingly, K.A.; Blankenship, K.A.; Huff, M.O.; McGregor, W.G.; Klinge, C.M. Gender difference in the activity but not expression of estrogen receptors alpha and beta in human lung adenocarcinoma cells. Endocr. Relat. Cancer 2006, 13, 113–134. [Google Scholar] [CrossRef] [Green Version]
- Carmeci, C.; Thompson, D.A.; Ring, H.Z.; Francke, U.; Weigel, R.J. Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics 1997, 45, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Simoncini, T.; Hafezi-Moghadam, A.; Brazil, D.P.; Ley, K.; Chin, W.W.; Liao, J.K. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature 2000, 407, 538–541. [Google Scholar] [CrossRef] [PubMed]
- Shaul, P.W. Rapid activation of endothelial nitric oxide synthase by estrogen. Steroids 1999, 64, 28–34. [Google Scholar] [CrossRef]
- Heldring, N.; Pike, A.; Andersson, S.; Matthews, J.; Cheng, G.; Hartman, J.; Tujague, M.; Strom, A.; Treuter, E.; Warner, M.; et al. Estrogen receptors: How do they signal and what are their targets. Physiol. Rev. 2007, 87, 905–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubey, R.K.; Gillespie, D.G.; Zacharia, L.C.; Rosselli, M.; Korzekwa, K.R.; Fingerle, J.; Jackson, E.K. Methoxyestradiols mediate the antimitogenic effects of estradiol on vascular smooth muscle cells via estrogen receptor-independent mechanisms. Biochem. Biophys. Res. Commun. 2000, 278, 27–33. [Google Scholar] [CrossRef]
- Fenoy, F.J.; Hernandez, M.E.; Hernandez, M.; Quesada, T.; Salom, M.G.; Hernandez, I. Acute effects of 2-methoxyestradiol on endothelial aortic No release in male and ovariectomized female rats. Nitric Oxide 2010, 23, 12–19. [Google Scholar] [CrossRef]
- Tofovic, S.P. Estrogens and development of pulmonary hypertension: Interaction of estradiol metabolism and pulmonary vascular disease. J. Cardiovasc. Pharm. 2010, 56, 696–708. [Google Scholar] [CrossRef] [Green Version]
- Fishman, J.; Martucci, C. Biological properties of 16 alpha-hydroxyestrone: Implications in estrogen physiology and pathophysiology. J. Clin. Endocrinol. Metab. 1980, 51, 611–615. [Google Scholar] [CrossRef]
- Austin, E.D.; Hamid, R.; Hemnes, A.R.; Loyd, J.E.; Blackwell, T.; Yu, C.; Phillips Iii, J.A.; Gaddipati, R.; Gladson, S.; Gu, E.; et al. BMPR2 expression is suppressed by signaling through the estrogen receptor. Biol. Sex Differ. 2012, 3, 6. [Google Scholar] [CrossRef] [Green Version]
- Mair, K.M.; Yang, X.D.; Long, L.; White, K.; Wallace, E.; Ewart, M.A.; Docherty, C.K.; Morrell, N.W.; MacLean, M.R. Sex affects bone morphogenetic protein type II receptor signaling in pulmonary artery smooth muscle cells. Am. J. Respir. Crit. Care Med. 2015, 191, 693–703. [Google Scholar] [CrossRef] [Green Version]
- Mair, K.M.; Wright, A.F.; Duggan, N.; Rowlands, D.J.; Hussey, M.J.; Roberts, S.; Fullerton, J.; Nilsen, M.; Loughlin, L.; Thomas, M.; et al. Sex-dependent influence of endogenous estrogen in pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2014, 190, 456–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Arroyo, J.; Saleem, S.J.; Mizuno, S.; Syed, A.A.; Bogaard, H.J.; Abbate, A.; Taraseviciene-Stewart, L.; Sung, Y.; Kraskauskas, D.; Farkas, D.; et al. A brief overview of mouse models of pulmonary arterial hypertension: Problems and prospects. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L977–L991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Austin, E.D.; Talati, M.; Fessel, J.P.; Farber-Eger, E.H.; Brittain, E.L.; Hemnes, A.R.; Loyd, J.E.; West, J. Oestrogen inhibition reverses pulmonary arterial hypertension and associated metabolic defects. Eur. Respir. J. 2017, 50, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, C.; Hong, K.H.; Kim, Y.H.; Kim, M.J.; Song, C.; Kim, M.J.; Kim, S.J.; Raizada, M.K.; Oh, S.P. SMAD1 deficiency in either endothelial or smooth muscle cells can predispose mice to pulmonary hypertension. Hypertension 2013, 61, 1044–1052. [Google Scholar] [CrossRef] [Green Version]
- Muti, P.; Bradlow, H.L.; Micheli, A.; Krogh, V.; Freudenheim, J.L.; Schunemann, H.J.; Stanulla, M.; Yang, J.; Sepkovic, D.W.; Trevisan, M.; et al. Estrogen metabolism and risk of breast cancer: A prospective study of the 2:16alpha-hydroxyestrone ratio in premenopausal and postmenopausal women. Epidemiology 2000, 11, 635–640. [Google Scholar] [CrossRef] [Green Version]
- West, J.; Cogan, J.; Geraci, M.; Robinson, L.; Newman, J.; Phillips, J.A.; Lane, K.; Meyrick, B.; Loyd, J. Gene expression in BMPR2 mutation carriers with and without evidence of pulmonary arterial hypertension suggests pathways relevant to disease penetrance. BMC Med. Genom. 2008, 1, 45. [Google Scholar] [CrossRef] [Green Version]
- Gajjar, K.; Martin-Hirsch, P.L.; Martin, F.L. CYP1B1 and hormone-induced cancer. Cancer Lett. 2012, 324, 13–30. [Google Scholar] [CrossRef]
- Austin, E.D.; Cogan, J.D.; West, J.D.; Hedges, L.K.; Hamid, R.; Dawson, E.P.; Wheeler, L.A.; Parl, F.F.; Loyd, J.E.; Phillips, J.A., 3rd. Alterations in oestrogen metabolism: Implications for higher penetrance of familial pulmonary arterial hypertension in females. Eur. Respir. J. 2009, 34, 1093–1099. [Google Scholar] [CrossRef] [Green Version]
- Fessel, J.P.; Chen, X.; Frump, A.; Gladson, S.; Blackwell, T.; Kang, C.; Johnson, J.; Loyd, J.E.; Hemnes, A.; Austin, E.; et al. Interaction between bone morphogenetic protein receptor type 2 and estrogenic compounds in pulmonary arterial hypertension. Pulm. Circ. 2013, 3, 564–577. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Talati, M.; Fessel, J.P.; Hemnes, A.R.; Gladson, S.; French, J.; Shay, S.; Trammell, A.; Phillips, J.A.; Hamid, R.; et al. Estrogen Metabolite 16alpha-Hydroxyestrone Exacerbates Bone Morphogenetic Protein Receptor Type II-Associated Pulmonary Arterial Hypertension Through MicroRNA-29-Mediated Modulation of Cellular Metabolism. Circulation 2016, 133, 82–97. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cirulis, M.M.; Dodson, M.W.; Brown, L.M.; Brown, S.M.; Lahm, T.; Elliott, G. At the X-Roads of Sex and Genetics in Pulmonary Arterial Hypertension. Genes 2020, 11, 1371. https://doi.org/10.3390/genes11111371
Cirulis MM, Dodson MW, Brown LM, Brown SM, Lahm T, Elliott G. At the X-Roads of Sex and Genetics in Pulmonary Arterial Hypertension. Genes. 2020; 11(11):1371. https://doi.org/10.3390/genes11111371
Chicago/Turabian StyleCirulis, Meghan M., Mark W. Dodson, Lynn M. Brown, Samuel M. Brown, Tim Lahm, and Greg Elliott. 2020. "At the X-Roads of Sex and Genetics in Pulmonary Arterial Hypertension" Genes 11, no. 11: 1371. https://doi.org/10.3390/genes11111371
APA StyleCirulis, M. M., Dodson, M. W., Brown, L. M., Brown, S. M., Lahm, T., & Elliott, G. (2020). At the X-Roads of Sex and Genetics in Pulmonary Arterial Hypertension. Genes, 11(11), 1371. https://doi.org/10.3390/genes11111371