‘There and Back Again’—Forward Genetics and Reverse Phenotyping in Pulmonary Arterial Hypertension

 , ,
, ,
Abstract
:1. Introduction
2. Genetic and Phenotypic Heterogeneity
3. Forward Phenotyping for Genetic Studies
4. Forward Genetics
4.1. Concepts
4.2. Methodology
4.2.1. Study Design
- Selection of cases (recognition of selection bias, incident vs. prevalent cases recruitment)
- Case definition (precise definition of the phenotype that can be ascertained in a research setting)
- Selection of controls (healthy vs. disease controls, matched in respect to age, sex and ethnicity, having a comparable evaluation of presence or absence of the phenotype in question)
4.2.2. Statistical Methods
4.2.3. Molecular Genetic Techniques
4.2.4. Reference Genome
4.3. Studies
4.3.1. Rare Genetic Variation
Autosomal Dominant Mode of Inheritance
Autosomal Recessive Mode of Inheritance
4.3.2. Common Genetic Variation
4.4. Limitations, Challenges and Future Directions
5. Reverse Genetics
5.1. Concepts
5.2. Methodology
5.2.1. In Vitro
5.2.2. In Vivo
5.3. Studies
5.3.1. Rare Genetic Variation
Autosomal Dominant Mode of Inheritance
Autosomal Recessive Mode of Inheritance
5.3.2. Common Genetic Variation
5.4. Limitations, Challenges and Future Directions
6. Reverse Phenotyping
6.1. Concepts
6.2. Theoretical Basis
6.3. Methodology
6.4. Studies
6.5. Limitations, Challenges and Future Directions
7. Missing Heritability in the Postgenomic Era
7.1. Unknown Genetic Variation
7.2. Epigenetic Inheritance
7.2.1. DNA Methylation
7.2.2. Histone Acetylation
7.2.3. RNA Interference
7.3. Interactions
7.4. Population Dependent Heterogeneity
8. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Montserrat Moliner, A.; Waligóra, J. The European union policy in the field of rare diseases. Public Health Genom. 2013, 16, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Nguengang Wakap, S.; Lambert, D.M.; Olry, A.; Rodwell, C.; Gueydan, C.; Lanneau, V.; Murphy, D.; Le Cam, Y.; Rath, A. Estimating cumulative point prevalence of rare diseases: Analysis of the Orphanet database. Eur. J. Hum. Genet. 2020, 28, 165–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- OMIM Entry Statistics. Available online: https://www.omim.org/statistics/entry (accessed on 27 August 2020).
- Wright, C.F.; FitzPatrick, D.R.; Firth, H.V. Paediatric genomics: Diagnosing rare disease in children. Nat. Rev. Genet. 2018, 19, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Robbins, I.M.; Beghetti, M.; Channick, R.N.; Delcroix, M.; Denton, C.P.; Elliott, C.G.; Gaine, S.P.; Gladwin, M.T.; Jing, Z.-C.; et al. Updated clinical classification of pulmonary hypertension. J. Am. Coll. Cardiol. 2009, 54, S43–S54. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.A. XV.—The Correlation between Relatives on the Supposition of Mendelian Inheritance. Trans. R. Soc. Edinb. 1919, 52, 399–433. [Google Scholar] [CrossRef] [Green Version]
- Wright, S. The Relative Importance of Heredity and Environment in Determining the Piebald Pattern of Guinea-Pigs. Proc. Natl. Acad. Sci. USA 1920, 6, 320–332. [Google Scholar] [CrossRef] [Green Version]
- Tenesa, A.; Haley, C.S. The heritability of human disease: Estimation, uses and abuses. Nat. Rev. Genet. 2013, 14, 139–149. [Google Scholar] [CrossRef]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [Green Version]
- Maher, B. Personal genomes: The case of the missing heritability. Nature 2008, 456, 18–21. [Google Scholar] [CrossRef]
- Génin, E. Missing heritability of complex diseases: Case solved? Hum. Genet. 2020, 139, 103–113. [Google Scholar] [CrossRef]
- WSPH, Geneva. 1973. Available online: http://www.wsphassociation.org/wp-content/uploads/2019/03/Programma-WSPH-Geneva-1973.pdf (accessed on 25 November 2020).
- WSPH, Evian. 1998. Available online: http://www.wsphassociation.org/wp-content/uploads/2019/04/Primary-Pulmonary-Hypertension-Evian-1998.pdf (accessed on 25 November 2020).
- Simonneau, G.; Galiè, N.; Rubin, L.J.; Langleben, D.; Seeger, W.; Domenighetti, G.; Gibbs, S.; Lebrec, D.; Speich, R.; Beghetti, M.; et al. Clinical classification of pulmonary hypertension. J. Am. Coll. Cardiol. 2004, 43, 5S–12S. [Google Scholar] [CrossRef]
- Galiè, N.; Hoeper, M.M.; Humbert, M.; Torbicki, A.; Vachiery, J.-L.; Barbera, J.A.; Beghetti, M.; Corris, P.; Gaine, S.; Gibbs, J.S.; et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: The Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur. Heart J. 2009, 30, 2493–2537. [Google Scholar] [CrossRef] [PubMed]
- Updated Clinical Classification of Pulmonary Hypertension. J. Am. Coll. Cardiol. 2013, 62, D34–D41. [CrossRef] [PubMed] [Green Version]
- Galiè, N.; Humbert, M.; Vachiery, J.-L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Respir. J. 2015, 46, 903–975. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53. [Google Scholar] [CrossRef]
- Kato, G.J.; Piel, F.B.; Reid, C.D.; Gaston, M.H.; Ohene-Frempong, K.; Krishnamurti, L.; Smith, W.R.; Panepinto, J.A.; Weatherall, D.J.; Costa, F.F.; et al. Sickle cell disease. Nat. Rev. Dis. Primers 2018, 4, 18010. [Google Scholar] [CrossRef] [Green Version]
- Peron, A.; Au, K.S.; Northrup, H. Genetics, genomics, and genotype-phenotype correlations of TSC: Insights for clinical practice. Am. J. Med. Genet. C Semin. Med. Genet. 2018, 178, 281–290. [Google Scholar] [CrossRef]
- Bravo-Gil, N.; González-Del Pozo, M.; Martín-Sánchez, M.; Méndez-Vidal, C.; Rodríguez-de la Rúa, E.; Borrego, S.; Antiñolo, G. Unravelling the genetic basis of simplex Retinitis Pigmentosa cases. Sci. Rep. 2017, 7, 41937. [Google Scholar] [CrossRef]
- Chiurazzi, P.; Pirozzi, F. Advances in understanding - genetic basis of intellectual disability. F1000Res 2016, 5. [Google Scholar] [CrossRef]
- Southgate, L.; Machado, R.D.; Gräf, S.; Morrell, N.W. Molecular genetic framework underlying pulmonary arterial hypertension. Nat. Rev. Cardiol. 2020, 17, 85–95. [Google Scholar] [CrossRef]
- Gräf, S.; Haimel, M.; Bleda, M.; Hadinnapola, C.; Southgate, L.; Li, W.; Hodgson, J.; Liu, B.; Salmon, R.M.; Southwood, M.; et al. Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nat. Commun. 2018, 9, 1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, N.; Pauciulo, M.W.; Welch, C.L.; Lutz, K.A.; Coleman, A.W.; Gonzaga-Jauregui, C.; Wang, J.; Grimes, J.M.; Martin, L.J.; He, H.; et al. Novel risk genes and mechanisms implicated by exome sequencing of 2572 individuals with pulmonary arterial hypertension. Genome Med. 2019, 11, 69. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.-J.; Lian, T.-Y.; Jiang, X.; Liu, S.-F.; Li, S.-Q.; Jiang, R.; Wu, W.-H.; Ye, J.; Cheng, C.-Y.; Du, Y.; et al. Germline BMP9 mutation causes idiopathic pulmonary arterial hypertension. Eur. Respir. J. 2019, 53, 1801609. [Google Scholar] [CrossRef] [PubMed]
- Dweik, R.A.; Rounds, S.; Erzurum, S.C.; Archer, S.; Fagan, K.; Hassoun, P.M.; Hill, N.S.; Humbert, M.; Kawut, S.M.; Krowka, M.; et al. An official American Thoracic Society Statement: Pulmonary hypertension phenotypes. Am. J. Respir. Crit. Care Med. 2014, 189, 345–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutendra, G.; Michelakis, E.D. The metabolic basis of pulmonary arterial hypertension. Cell Metab. 2014, 19, 558–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buyske, S.; Yang, G.; Matise, T.C.; Gordon, D. When a Case Is Not a Case: Effects of Phenotype Misclassification on Power and Sample Size Requirements for the Transmission Disequilibrium Test with Affected Child Trios. Hum. Hered. 2009, 67, 287–292. [Google Scholar] [CrossRef]
- Swietlik, E.M.; Greene, D.; Zhu, N.; Megy, K.; Cogliano, M.; Rajaram, S.; Pandya, D.; Tilly, T.; Lutz, K.A.; Welch, C.C.L.; et al. Reduced transfer coefficient of carbon monoxide in pulmonary arterial hypertension implicates rare protein-truncating variants in KDR. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Pullabhatla, V.; Roberts, A.L.; Lewis, M.J.; Mauro, D.; Morris, D.L.; Odhams, C.A.; Tombleson, P.; Liljedahl, U.; Vyse, S.; Simpson, M.A.; et al. De novo mutations implicate novel genes in systemic lupus erythematosus. Hum. Mol. Genet. 2018, 27, 421–429. [Google Scholar] [CrossRef] [Green Version]
- Husson, T.; Duboc, J.-B.; Quenez, O.; Charbonnier, C.; Rotharmel, M.; Cuenca, M.; Jegouzo, X.; Richard, A.-C.; Frebourg, T.; Deleuze, J.-F.; et al. Identification of potential genetic risk factors for bipolar disorder by whole-exome sequencing. Transl. Psychiatry 2018, 8, 268. [Google Scholar] [CrossRef] [Green Version]
- Bjørnland, T.; Bye, A.; Ryeng, E.; Wisløff, U.; Langaas, M. Powerful extreme phenotype sampling designs and score tests for genetic association studies. Stat. Med. 2018, 37, 4234–4251. [Google Scholar] [CrossRef] [Green Version]
- Chung, W.K.; Deng, L.; Carroll, J.S.; Mallory, N.; Diamond, B.; Rosenzweig, E.B.; Barst, R.J.; Morse, J.H. Polymorphism in the angiotensin II type 1 receptor (AGTR1) is associated with age at diagnosis in pulmonary arterial hypertension. J. Heart Lung Transplant. 2009, 28, 373–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Lewinger, J.P.; Gauderman, W.J.; Murcray, C.E.; Conti, D. Using extreme phenotype sampling to identify the rare causal variants of quantitative traits in association studies. Genet. Epidemiol. 2011, 35, 790–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze, T.G.; McMahon, F.J. Defining the phenotype in human genetic studies: Forward genetics and reverse phenotyping. Hum. Hered. 2004, 58, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Sweatt, A.J.; Hedlin, H.K.; Balasubramanian, V.; Hsi, A.; Blum, L.K.; Robinson, W.H.; Haddad, F.; Hickey, P.M.; Condliffe, R.; Lawrie, A.; et al. Discovery of Distinct Immune Phenotypes Using Machine Learning in Pulmonary Arterial Hypertension. Circ. Res. 2019, 124, 904–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoeper, M.M.; Pausch, C.; Grünig, E.; Klose, H.; Staehler, G.; Huscher, D.; Pittrow, D.; Olsson, K.M.; Vizza, C.D.; Gall, H.; et al. Idiopathic pulmonary arterial hypertension phenotypes determined by cluster analysis from the COMPERA registry. J. Heart Lung Transplant. 2020. [Google Scholar] [CrossRef] [PubMed]
- Köhler, S.; Carmody, L.; Vasilevsky, N.; Jacobsen, J.O.B.; Danis, D.; Gourdine, J.-P.; Gargano, M.; Harris, N.L.; Matentzoglu, N.; McMurry, J.A.; et al. Expansion of the Human Phenotype Ontology (HPO) knowledge base and resources. Nucleic Acids Res. 2019, 47, D1018–D1027. [Google Scholar] [CrossRef] [PubMed]
- Köhler, S.; Schulz, M.H.; Krawitz, P.; Bauer, S.; Dölken, S.; Ott, C.E.; Mundlos, C.; Horn, D.; Mundlos, S.; Robinson, P.N. Clinical diagnostics in human genetics with semantic similarity searches in ontologies. Am. J. Hum. Genet. 2009, 85, 457–464. [Google Scholar] [CrossRef] [Green Version]
- Greene, D.; Richardson, S.; Turro, E. ontologyX: A suite of R packages for working with ontological data. Bioinformatics 2017, 33, 1104–1106. [Google Scholar] [CrossRef] [Green Version]
- Peters, J.L.; Cnudde, F.; Gerats, T. Forward genetics and map-based cloning approaches. Trends Plant Sci. 2003, 8, 484–491. [Google Scholar] [CrossRef]
- Ionita-Laza, I.; Lee, S.; Makarov, V.; Buxbaum, J.D.; Lin, X. Family-based association tests for sequence data, and comparisons with population-based association tests. Eur. J. Hum. Genet. 2013, 21, 1158–1162. [Google Scholar] [CrossRef]
- Risch, N.; Merikangas, K. The Future of Genetic Studies of Complex Human Diseases. Science 1996, 273, 1516–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchini, J.; Donnelly, P.; Cardon, L.R. Genome-wide strategies for detecting multiple loci that influence complex diseases. Nat. Genet. 2005, 37, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Petrovski, S.; Todd, J.L.; Durheim, M.T.; Wang, Q.; Chien, J.W.; Kelly, F.L.; Frankel, C.; Mebane, C.M.; Ren, Z.; Bridgers, J.; et al. An Exome Sequencing Study to Assess the Role of Rare Genetic Variation in Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2017, 196, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef]
- Cooper, G.M.; Stone, E.A.; Asimenos, G.; NISC Comparative Sequencing Program; Green, E.D.; Batzoglou, S.; Sidow, A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005, 15, 901–913. [Google Scholar] [CrossRef] [Green Version]
- Pollard, K.S.; Hubisz, M.J.; Rosenbloom, K.R.; Siepel, A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010, 20, 110–121. [Google Scholar] [CrossRef] [Green Version]
- Siepel, A.; Bejerano, G.; Pedersen, J.S.; Hinrichs, A.S.; Hou, M.; Rosenbloom, K.; Clawson, H.; Spieth, J.; Hillier, L.W.; Richards, S.; et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005, 15, 1034–1050. [Google Scholar] [CrossRef] [Green Version]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Nasim, M.T.; Ogo, T.; Ahmed, M.; Randall, R.; Chowdhury, H.M.; Snape, K.M.; Bradshaw, T.Y.; Southgate, L.; Lee, G.J.; Jackson, I.; et al. Molecular genetic characterization of SMAD signaling molecules in pulmonary arterial hypertension. Hum. Mutat. 2011, 32, 1385–1389. [Google Scholar] [CrossRef] [Green Version]
- Potus, F.; Pauciulo, M.W.; Cook, E.K.; Zhu, N.; Hsieh, A.; Welch, C.L.; Shen, Y.; Tian, L.; Lima, P.; Mewburn, J.; et al. Novel Mutations and Decreased Expression of the Epigenetic Regulator TET2 in Pulmonary Arterial Hypertension. Circulation 2020, 141, 1986–2000. [Google Scholar] [CrossRef]
- Povysil, G.; Petrovski, S.; Hostyk, J.; Aggarwal, V.; Allen, A.S.; Goldstein, D.B. Rare-variant collapsing analyses for complex traits: Guidelines and applications. Nat. Rev. Genet. 2019, 20, 747–759. [Google Scholar] [CrossRef] [PubMed]
- Asimit, J.L.; Day-Williams, A.G.; Morris, A.P.; Zeggini, E. ARIEL and AMELIA: Testing for an accumulation of rare variants using next-generation sequencing data. Hum. Hered. 2012, 73, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.P.; Zeggini, E. An evaluation of statistical approaches to rare variant analysis in genetic association studies. Genet. Epidemiol. 2010, 34, 188–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Leal, S.M. Methods for detecting associations with rare variants for common diseases: Application to analysis of sequence data. Am. J. Hum. Genet. 2008, 83, 311–321. [Google Scholar] [CrossRef] [Green Version]
- Madsen, B.E.; Browning, S.R. A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet. 2009, 5, e1000384. [Google Scholar] [CrossRef] [Green Version]
- Ionita-Laza, I.; Buxbaum, J.D.; Laird, N.M.; Lange, C. A New Testing Strategy to Identify Rare Variants with Either Risk or Protective Effect on Disease. PLoS Genet. 2011, 7, e1001289. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.C.; Lee, S.; Cai, T.; Li, Y.; Boehnke, M.; Lin, X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet. 2011, 89, 82–93. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Emond, M.J.; Bamshad, M.J.; Barnes, K.C.; Rieder, M.J.; Nickerson, D.A.; NHLBI GO Exome Sequencing Project—ESP Lung Project Team; Christiani, D.C.; Wurfel, M.M.; Lin, X. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am. J. Hum. Genet. 2012, 91, 224–237. [Google Scholar] [CrossRef] [Green Version]
- Greene, D.; NIHR BioResource; Richardson, S.; Turro, E. A Fast Association Test for Identifying Pathogenic Variants Involved in Rare Diseases. Am. J. Hum. Genet. 2017, 101, 104–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eilbeck, K.; Quinlan, A.; Yandell, M. Settling the score: Variant prioritization and Mendelian disease. Nat. Rev. Genet. 2017, 18, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Cacheiro, P.; Muñoz-Fuentes, V.; Murray, S.A.; Dickinson, M.E.; Bucan, M.; Nutter, L.M.J.; Peterson, K.A.; Haselimashhadi, H.; Flenniken, A.M.; Morgan, H.; et al. Human and mouse essentiality screens as a resource for disease gene discovery. Nat. Commun. 2020, 11, 655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Rehm, H.L.; Berg, J.S.; Brooks, L.D.; Bustamante, C.D.; Evans, J.P.; Landrum, M.J.; Ledbetter, D.H.; Maglott, D.R.; Martin, C.L.; Nussbaum, R.L.; et al. ClinGen—The Clinical Genome Resource. N. Engl. J. Med. 2015, 372, 2235–2242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goswami, R.S.; Harada, S. An Overview of Molecular Genetic Diagnosis Techniques. Curr. Protoc. Hum. Genet. 2020, 105, e97. [Google Scholar] [CrossRef] [PubMed]
- Bean, L.J.H.; Funke, B.; Carlston, C.M.; Gannon, J.L.; Kantarci, S.; Krock, B.L.; Zhang, S.; Bayrak-Toydemir, P. Diagnostic gene sequencing panels: From design to report—A technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2020, 22, 453–461. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Eichstaedt, C.A.; Viales, R.R.; Benjamin, N.; Harutyunova, S.; Fischer, C.; Grünig, E.; Hinderhofer, K. Identification of genetic defects in pulmonary arterial hypertension by a new gene panel diagnostic tool. Clin. Sci. 2016, 130, 2043–2052. [Google Scholar] [CrossRef]
- Rabbani, B.; Tekin, M.; Mahdieh, N. The promise of whole-exome sequencing in medical genetics. J. Hum. Genet. 2014, 59, 5–15. [Google Scholar] [CrossRef]
- Retterer, K.; Juusola, J.; Cho, M.T.; Vitazka, P.; Millan, F.; Gibellini, F.; Vertino-Bell, A.; Smaoui, N.; Neidich, J.; Monaghan, K.G.; et al. Clinical application of whole-exome sequencing across clinical indications. Genet. Med. 2016, 18, 696–704. [Google Scholar] [CrossRef] [Green Version]
- Austin, E.D.; Ma, L.; LeDuc, C.; Berman Rosenzweig, E.; Borczuk, A.; Phillips, J.A., 3rd; Palomero, T.; Sumazin, P.; Kim, H.R.; Talati, M.H.; et al. Whole exome sequencing to identify a novel gene (caveolin-1) associated with human pulmonary arterial hypertension. Circ. Cardiovasc. Genet. 2012, 5, 336–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, N.; Welch, C.L.; Wang, J.; Allen, P.M.; Gonzaga-Jauregui, C.; Ma, L.; King, A.K.; Krishnan, U.; Rosenzweig, E.B.; Ivy, D.D.; et al. Rare variants in SOX17 are associated with pulmonary arterial hypertension with congenital heart disease. Genome Med. 2018, 10, 691. [Google Scholar] [CrossRef] [PubMed]
- Zare, F.; Dow, M.; Monteleone, N.; Hosny, A.; Nabavi, S. An evaluation of copy number variation detection tools for cancer using whole exome sequencing data. BMC Bioinform. 2017, 18, 286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turro, E.; Astle, W.J.; Megy, K.; Gräf, S.; Greene, D.; Shamardina, O.; Allen, H.L.; Sanchis-Juan, A.; Frontini, M.; Thys, C.; et al. Whole-genome sequencing of patients with rare diseases in a national health system. Nature 2020, 583, 96–102. [Google Scholar] [CrossRef] [PubMed]
- National Human Genome Research Institute Home. Available online: https://www.genome.gov (accessed on 17 October 2020).
- Schwarze, K.; Buchanan, J.; Fermont, J.M.; Dreau, H.; Tilley, M.W.; Taylor, J.M.; Antoniou, P.; Knight, S.J.L.; Camps, C.; Pentony, M.M.; et al. The complete costs of genome sequencing: A microcosting study in cancer and rare diseases from a single center in the United Kingdom. Genet. Med. 2020, 22, 85–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barozzi, C.; Galletti, M.; Tomasi, L.; De Fanti, S.; Palazzini, M.; Manes, A.; Sazzini, M.; Galiè, N. A Combined Targeted and Whole Exome Sequencing Approach Identified Novel Candidate Genes Involved in Heritable Pulmonary Arterial Hypertension. Sci. Rep. 2019, 9, 753. [Google Scholar] [CrossRef]
- Pulmonary Arterial Hypertension (Version 2.5). Available online: https://panelapp.genomicsengland.co.uk/panels/193/ (accessed on 14 October 2020).
- Kim, J.; Weber, J.A.; Jho, S.; Jang, J.; Jun, J.; Cho, Y.S.; Kim, H.-M.; Kim, H.; Kim, Y.; Chung, O.; et al. KoVariome: Korean National Standard Reference Variome database of whole genomes with comprehensive SNV, indel, CNV, and SV analyses. Sci. Rep. 2018, 8, 5677. [Google Scholar] [CrossRef] [Green Version]
- Finer, S.; Martin, H.C.; Khan, A.; Hunt, K.A.; MacLaughlin, B.; Ahmed, Z.; Ashcroft, R.; Durham, C.; MacArthur, D.G.; McCarthy, M.I.; et al. Cohort Profile: East London Genes & Health (ELGH), a community-based population genomics and health study in British Bangladeshi and British Pakistani people. Int. J. Epidemiol. 2020, 49, 20–21i. [Google Scholar] [CrossRef] [Green Version]
- BioBank Japan (BBJ). Available online: http://biobankjp.org (accessed on 14 October 2020).
- IBD BioResource—Translating Today’s Science into Tomorrow’s Treatments. Available online: https://www.ibdbioresource.nihr.ac.uk (accessed on 14 October 2020).
- Genetic Links to Anxiety and Depression Study—GLAD Study. Available online: https://gladstudy.org.uk (accessed on 14 October 2020).
- Eating Disorders Genetics Initiative. Available online: https://edgiuk.org/ (accessed on 14 October 2020).
- Flannick, J.; Beer, N.L.; Bick, A.G.; Agarwala, V.; Molnes, J.; Gupta, N.; Burtt, N.P.; Florez, J.C.; Meigs, J.B.; Taylor, H.; et al. Assessing the phenotypic effects in the general population of rare variants in genes for a dominant Mendelian form of diabetes. Nat. Genet. 2013, 45, 1380–1385. [Google Scholar] [CrossRef]
- Castel, S.E.; Cervera, A.; Mohammadi, P.; Aguet, F.; Reverter, F.; Wolman, A.; Guigo, R.; Iossifov, I.; Vasileva, A.; Lappalainen, T. Modified penetrance of coding variants by cis-regulatory variation contributes to disease risk. Nat. Genet. 2018, 50, 1327–1334. [Google Scholar] [CrossRef] [Green Version]
- International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar]
- International Human Genome Sequencing Consortium Finishing the euchromatic sequence of the human genome. Nature 2004, 431, 931–945. [CrossRef]
- Guo, Y.; Dai, Y.; Yu, H.; Zhao, S.; Samuels, D.C.; Shyr, Y. Improvements and impacts of GRCh38 human reference on high throughput sequencing data analysis. Genomics 2017, 109, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.; Kusko, R.; Xiao, W.; Zheng, Y.; Liu, Z.; Xiao, C.; Sakkiah, S.; Guo, W.; Gong, P.; Zhang, C.; et al. Similarities and differences between variants called with human reference genome HG19 or HG38. BMC Bioinform. 2019, 20, 17–29. [Google Scholar] [CrossRef]
- Schneider, V.A.; Graves-Lindsay, T.; Howe, K.; Bouk, N.; Chen, H.-C.; Kitts, P.A.; Murphy, T.D.; Pruitt, K.D.; Thibaud-Nissen, F.; Albracht, D.; et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 2017, 27, 849–864. [Google Scholar] [CrossRef] [Green Version]
- Church, D.M.; Schneider, V.A.; Steinberg, K.M.; Schatz, M.C.; Quinlan, A.R.; Chin, C.-S.; Kitts, P.A.; Aken, B.; Marth, G.T.; Hoffman, M.M.; et al. Extending reference assembly models. Genome Biol. 2015, 16, 13. [Google Scholar] [CrossRef] [Green Version]
- Lappalainen, T.; Scott, A.J.; Brandt, M.; Hall, I.M. Genomic Analysis in the Age of Human Genome Sequencing. Cell 2019, 177, 70–84. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Li, Y.; Zheng, H.; Luo, R.; Zhu, H.; Li, Q.; Qian, W.; Ren, Y.; Tian, G.; Li, J.; et al. Building the sequence map of the human pan-genome. Nat. Biotechnol. 2010, 28, 57–63. [Google Scholar] [CrossRef]
- Paten, B.; Novak, A.M.; Eizenga, J.M.; Garrison, E. Genome graphs and the evolution of genome inference. Genome Res. 2017, 27, 665–676. [Google Scholar] [CrossRef] [Green Version]
- Rakocevic, G.; Semenyuk, V.; Lee, W.-P.; Spencer, J.; Browning, J.; Johnson, I.J.; Arsenijevic, V.; Nadj, J.; Ghose, K.; Suciu, M.C.; et al. Fast and accurate genomic analyses using genome graphs. Nat. Genet. 2019, 51, 354–362. [Google Scholar] [CrossRef]
- Ballouz, S.; Dobin, A.; Gillis, J.A. Is it time to change the reference genome? Genome Biol. 2019, 20, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International PPH Consortium; Lane, K.B.; Machado, R.D.; Pauciulo, M.W.; Thomson, J.R.; Phillips, J.A., 3rd; Loyd, J.E.; Nichols, W.C.; Trembath, R.C. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat. Genet. 2000, 26, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.R. Sporadic primary pulmonary hypertension is associated with germline mutations of the gene encoding BMPR-II, a receptor member of the TGF-beta family. J. Med Genet. 2000, 37, 741–745. [Google Scholar] [CrossRef]
- Trembath, R.C.; Thomson, J.R.; Machado, R.D.; Morgan, N.V.; Atkinson, C.; Winship, I.; Simonneau, G.; Galie, N.; Loyd, J.E.; Humbert, M.; et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N. Engl. J. Med. 2001, 345, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Chaouat, A. Endoglin germline mutation in a patient with hereditary haemorrhagic telangiectasia and dexfenfluramine associated pulmonary arterial hypertension. Thorax 2004, 59, 446–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, R.E.; Berger, R.; Haworth, S.G.; Tulloh, R.; Mache, C.J.; Morrell, N.W.; Aldred, M.A.; Trembath, R.C. Transforming growth factor-beta receptor mutations and pulmonary arterial hypertension in childhood. Circulation 2005, 111, 435–441. [Google Scholar] [CrossRef] [Green Version]
- Shintani, M.; Yagi, H.; Nakayama, T.; Saji, T.; Matsuoka, R. A new nonsense mutation of SMAD8 associated with pulmonary arterial hypertension. J. Med. Genet. 2009, 46, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Roman-Campos, D.; Austin, E.D.; Eyries, M.; Sampson, K.S.; Soubrier, F.; Germain, M.; Trégouët, D.-A.; Borczuk, A.; Rosenzweig, E.B.; et al. A novel channelopathy in pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 351–361. [Google Scholar] [CrossRef] [Green Version]
- Kerstjens-Frederikse, W.S.; Bongers, E.M.H.F.; Roofthooft, M.T.R.; Leter, E.M.; Douwes, J.M.; Van Dijk, A.; Vonk-Noordegraaf, A.; Dijk-Bos, K.K.; Hoefsloot, L.H.; Hoendermis, E.S.; et al. TBX4 mutations (small patella syndrome) are associated with childhood-onset pulmonary arterial hypertension. J. Med. Genet. 2013, 50, 500–506. [Google Scholar] [CrossRef] [Green Version]
- Eyries, M.; Montani, D.; Girerd, B.; Perret, C.; Leroy, A.; Lonjou, C.; Chelghoum, N.; Coulet, F.; Bonnet, D.; Dorfmüller, P.; et al. EIF2AK4 mutations cause pulmonary veno-occlusive disease, a recessive form of pulmonary hypertension. Nat. Genet. 2014, 46, 65–69. [Google Scholar] [CrossRef]
- Best, D.H.; Sumner, K.L.; Smith, B.P.; Damjanovich-Colmenares, K.; Nakayama, I.; Brown, L.M.; Ha, Y.; Paul, E.; Morris, A.; Jama, M.A.; et al. EIF2AK4 Mutations in Patients Diagnosed with Pulmonary Arterial Hypertension. Chest 2017, 151, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Hadinnapola, C.; Bleda, M.; Haimel, M.; Screaton, N.; Swift, A.; Dorfmüller, P.; Preston, S.D.; Southwood, M.; Hernandez-Sanchez, J.; Martin, J.; et al. Phenotypic Characterization of Mutation Carriers in a Large Cohort of Patients Diagnosed Clinically with Pulmonary Arterial Hypertension. Circulation 2017, 136, 2022–2033. [Google Scholar] [CrossRef] [PubMed]
- Hiraide, T.; Kataoka, M.; Suzuki, H.; Aimi, Y.; Chiba, T.; Kanekura, K.; Satoh, T.; Fukuda, K.; Gamou, S.; Kosaki, K. SOX17 Mutations in Japanese Patients with Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2018, 198, 1231–1233. [Google Scholar] [CrossRef]
- Bohnen, M.S.; Ma, L.; Zhu, N.; Qi, H.; McClenaghan, C.; Gonzaga-Jauregui, C.; Dewey, F.E.; Overton, J.D.; Reid, J.G.; Shuldiner, A.R.; et al. Loss-of-Function ABCC8 Mutations in Pulmonary Arterial Hypertension. Circ. Genom. Precis. Med. 2018, 11, e002087. [Google Scholar] [CrossRef] [Green Version]
- Eyries, M.; Montani, D.; Nadaud, S.; Girerd, B.; Levy, M.; Bourdin, A.; Trésorier, R.; Chaouat, A.; Cottin, V.; Sanfiorenzo, C.; et al. Widening the landscape of heritable pulmonary hypertension mutations in paediatric and adult cases. Eur. Respir. J. 2019, 53. [Google Scholar] [CrossRef] [Green Version]
- Hodgson, J.; Swietlik, E.M.; Salmon, R.M.; Hadinnapola, C.; Nikolic, I.; Wharton, J.; Guo, J.; Liley, J.; Haimel, M.; Bleda, M.; et al. Characterization of Mutations and Levels of BMP9 and BMP10 in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2020, 201, 575–585. [Google Scholar] [CrossRef]
- Rhodes, C.J.; Batai, K.; Bleda, M.; Haimel, M.; Southgate, L.; Germain, M.; Pauciulo, M.W.; Hadinnapola, C.; Aman, J.; Girerd, B.; et al. Genetic determinants of risk in pulmonary arterial hypertension: International genome-wide association studies and meta-analysis. Lancet Respir Med. 2019, 7, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Eyries, M.; Montani, D.; Girerd, B.; Favrolt, N.; Riou, M.; Faivre, L.; Manaud, G.; Perros, F.; Gräf, S.; Morrell, N.W.; et al. Familial pulmonary arterial hypertension by KDR heterozygous loss of function. Eur. Respir. J. 2020, 55, 1902165. [Google Scholar] [CrossRef]
- Zhu, N.; Swietlik, E.M.; Welch, C.L.; Pauciulo, M.W.; Hagen, J.J.; Zhou, X.; Guo, Y.; Karten, J.; Pandya, D.; Tilly, T.; et al. Rare variant analysis of 4,241 pulmonary arterial hypertension cases from an international consortium implicate FBLN2, PDGFD and rare de novo variants in PAH. bioRxiv 2020. [Google Scholar] [CrossRef]
- Deng, Z.; Morse, J.H.; Slager, S.L.; Cuervo, N.; Moore, K.J.; Venetos, G.; Kalachikov, S.; Cayanis, E.; Fischer, S.G.; Barst, R.J.; et al. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am. J. Hum. Genet. 2000, 67, 737–744. [Google Scholar] [CrossRef] [Green Version]
- Navas Tejedor, P.; Tenorio Castaño, J.; Palomino Doza, J.; Arias Lajara, P.; Gordo Trujillo, G.; López Meseguer, M.; Román Broto, A.; Lapunzina Abadía, P.; Escribano Subía, P. An homozygous mutation in KCNK3 is associated with an aggressive form of hereditary pulmonary arterial hypertension. Clin. Genet. 2017, 91, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Fan, R.; Ji, R.; Zou, W.; Penny, D.J.; Varghese, N.P.; Fan, Y. Novel homozygous BMP9 nonsense mutation causes pulmonary arterial hypertension: A case report. BMC Pulm. Med. 2016, 16, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, E.K.; Newman, J.H.; Austin, E.D.; Hemnes, A.R.; Wheeler, L.; Robbins, I.M.; West, J.D.; Phillips, J.A., 3rd; Hamid, R.; Loyd, J.E. Longitudinal analysis casts doubt on the presence of genetic anticipation in heritable pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 892–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beppu, H.; Ichinose, F.; Kawai, N.; Jones, R.C.; Yu, P.B.; Zapol, W.M.; Miyazono, K.; Li, E.; Bloch, K.D. BMPR-II heterozygous mice have mild pulmonary hypertension and an impaired pulmonary vascular remodeling response to prolonged hypoxia. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2004, 287, L1241–L1247. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Long, L.; Southwood, M.; Rudarakanchana, N.; Upton, P.D.; Jeffery, T.K.; Atkinson, C.; Chen, H.; Trembath, R.C.; Morrell, N.W. Dysfunctional Smad Signaling Contributes to Abnormal Smooth Muscle Cell Proliferation in Familial Pulmonary Arterial Hypertension. Circ. Res. 2005, 96, 1053–1063. [Google Scholar] [CrossRef] [Green Version]
- Hong, K.-H.; Lee, Y.J.; Lee, E.; Park, S.O.; Han, C.; Beppu, H.; Li, E.; Raizada, M.K.; Bloch, K.D.; Oh, S.P. Genetic ablation of the BMPR2 gene in pulmonary endothelium is sufficient to predispose to pulmonary arterial hypertension. Circulation 2008, 118, 722–730. [Google Scholar] [CrossRef] [Green Version]
- West, J.; Harral, J.; Lane, K.; Deng, Y.; Ickes, B.; Crona, D.; Albu, S.; Stewart, D.; Fagan, K. Mice expressing BMPR2R899X transgene in smooth muscle develop pulmonary vascular lesions. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L744–L755. [Google Scholar] [CrossRef] [Green Version]
- Gangopahyay, A.; Oran, M.; Bauer, E.M.; Wertz, J.W.; Comhair, S.A.; Erzurum, S.C.; Bauer, P.M. Bone morphogenetic protein receptor II is a novel mediator of endothelial nitric-oxide synthase activation. J. Biol. Chem. 2011, 286, 33134–33140. [Google Scholar] [CrossRef] [Green Version]
- Hemnes, A.R.; Brittain, E.L.; Trammell, A.W.; Fessel, J.P.; Austin, E.D.; Penner, N.; Maynard, K.B.; Gleaves, L.; Talati, M.; Absi, T.; et al. Evidence for right ventricular lipotoxicity in heritable pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2014, 189, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Ranchoux, B.; Antigny, F.; Rucker-Martin, C.; Hautefort, A.; Péchoux, C.; Bogaard, H.J.; Dorfmüller, P.; Remy, S.; Lecerf, F.; Planté, S.; et al. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation 2015, 131, 1006–1018. [Google Scholar] [CrossRef] [Green Version]
- Caruso, P.; Dunmore, B.J.; Schlosser, K.; Schoors, S.; Dos Santos, C.; Perez-Iratxeta, C.; Lavoie, J.R.; Zhang, H.; Long, L.; Flockton, A.R.; et al. Identification of MicroRNA-124 as a Major Regulator of Enhanced Endothelial Cell Glycolysis in Pulmonary Arterial Hypertension via PTBP1 (Polypyrimidine Tract Binding Protein) and Pyruvate Kinase M2. Circulation 2017, 136, 2451–2467. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Jiang, X.; Sung, Y.K.; Shuffle, E.; Wu, T.-H.; Kao, P.N.; Tu, A.B.; Dorfmüller, P.; Cao, A.; Wang, L.; et al. Phenotypically Silent Bone Morphogenetic Protein Receptor 2 Mutations Predispose Rats to Inflammation-Induced Pulmonary Arterial Hypertension by Enhancing the Risk for Neointimal Transformation. Circulation 2019, 140, 1409–1425. [Google Scholar] [CrossRef] [PubMed]
- Gore, B.; Izikki, M.; Mercier, O.; Dewachter, L.; Fadel, E.; Humbert, M.; Dartevelle, P.; Simonneau, G.; Naeije, R.; Lebrin, F.; et al. Key role of the endothelial TGF-β/ALK1/endoglin signaling pathway in humans and rodents pulmonary hypertension. PLoS ONE 2014, 9, e100310. [Google Scholar] [CrossRef] [PubMed]
- Drake, K.M.; Zygmunt, D.; Mavrakis, L.; Harbor, P.; Wang, L.; Comhair, S.A.; Erzurum, S.C.; Aldred, M.A. Altered MicroRNA Processing in Heritable Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2011, 184, 1400–1408. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Wang, D.; Ihida-Stansbury, K.; Jones, P.L.; Martin, J.F. Defective pulmonary vascular remodeling in Smad8 mutant mice. Hum. Mol. Genet. 2009, 18, 2791–2801. [Google Scholar] [CrossRef] [Green Version]
- Drake, K.M.; Dunmore, B.J.; McNelly, L.N.; Morrell, N.W.; Aldred, M.A. Correction of nonsense BMPR2 and SMAD9 mutations by ataluren in pulmonary arterial hypertension. Am. J. Respir. Cell Mol. Biol. 2013, 49, 403–409. [Google Scholar] [CrossRef] [Green Version]
- Han, C.; Hong, K.-H.; Kim, Y.H.; Kim, M.-J.; Song, C.; Kim, M.J.; Kim, S.-J.; Raizada, M.K.; Oh, S.P. SMAD1 deficiency in either endothelial or smooth muscle cells can predispose mice to pulmonary hypertension. Hypertension 2013, 61, 1044–1052. [Google Scholar] [CrossRef] [Green Version]
- Copeland, C.A.; Han, B.; Tiwari, A.; Austin, E.D.; Loyd, J.E.; West, J.D.; Kenworthy, A.K. A disease-associated frameshift mutation in caveolin-1 disrupts caveolae formation and function through introduction of a de novo ER retention signal. Mol. Biol. Cell 2017, 28, 3095–3111. [Google Scholar] [CrossRef]
- Zhao, Y.-Y.; Liu, Y.; Stan, R.-V.; Fan, L.; Gu, Y.; Dalton, N.; Chu, P.-H.; Peterson, K.; Ross, J., Jr.; Chien, K.R. Defects in caveolin-1 cause dilated cardiomyopathy and pulmonary hypertension in knockout mice. Proc. Natl. Acad. Sci. USA 2002, 99, 11375–11380. [Google Scholar] [CrossRef] [Green Version]
- Drab, M.; Verkade, P.; Elger, M.; Kasper, M.; Lohn, M.; Lauterbach, B.; Menne, J.; Lindschau, C.; Mende, F.; Luft, F.C.; et al. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science 2001, 293, 2449–2452. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Gonzaga-Jauregui, C.; Welch, C.L.; Ma, L.; Qi, H.; King, A.K.; Krishnan, U.; Rosenzweig, E.B.; Ivy, D.D.; Austin, E.D.; et al. Exome Sequencing in Children with Pulmonary Arterial Hypertension Demonstrates Differences Compared with Adults. Circ Genom Precis Med 2018, 11, e001887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, M.; Eyries, M.; Szezepanski, I.; Ladouceur, M.; Nadaud, S.; Bonnet, D.; Soubrier, F. Genetic analyses in a cohort of children with pulmonary hypertension. Eur. Respir. J. 2016, 48, 1118–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thoré, P.; Girerd, B.; Jaïs, X.; Savale, L.; Ghigna, M.-R.; Eyries, M.; Levy, M.; Ovaert, C.; Servettaz, A.; Guillaumot, A.; et al. Phenotype and outcome of pulmonary arterial hypertension patients carrying a TBX4 mutation. Eur. Respir. J. 2020, 55, 1902340. [Google Scholar] [CrossRef] [PubMed]
- Jansen, S.M.A.; van den Heuvel, L.; Meijboom, L.J.; Alsters, S.I.M.; Vonk Noordegraaf, A.; Houweling, A.; Bogaard, H.J. Correspondence regarding “T-box protein 4 mutation causing pulmonary arterial hypertension and lung disease”: A single-centre case series. Eur. Respir. J. 2020, 55, 1902272. [Google Scholar] [CrossRef]
- Hernandez-Gonzalez, I.; Tenorio, J.; Palomino-Doza, J.; Martinez Meñaca, A.; Morales Ruiz, R.; Lago-Docampo, M.; Valverde Gomez, M.; Gomez Roman, J.; Enguita Valls, A.B.; Perez-Olivares, C.; et al. Clinical heterogeneity of Pulmonary Arterial Hypertension associated with variants in TBX4. PLoS ONE 2020, 15, e0232216. [Google Scholar] [CrossRef]
- Bohnen, M.S.; Roman-Campos, D.; Terrenoire, C.; Jnani, J.; Sampson, K.J.; Chung, W.K.; Kass, R.S. The Impact of Heterozygous KCNK3 Mutations Associated with Pulmonary Arterial Hypertension on Channel Function and Pharmacological Recovery. J. Am. Heart Assoc. 2017, 6, e006465. [Google Scholar] [CrossRef]
- Lambert, M.; Capuano, V.; Boet, A.; Tesson, L.; Bertero, T.; Nakhleh, M.K.; Remy, S.; Anegon, I.; Pechoux, C.; Hautefort, A.; et al. Characterization of Kcnk3 -Mutated Rat, a Novel Model of Pulmonary Hypertension. Circ. Res. 2019, 125, 678–695. [Google Scholar] [CrossRef]
- Legchenko, E.; Liu, B.; West, J.; Vangheluwe, P.; Upton, P.; Morrell, N. Protein truncating mutations in ATP13A3 promote pulmonary arterial hypertension in mice. ERJ Open Res. 2020, 6, 83. [Google Scholar]
- Liu, Y.; Sun, Z.; Zhu, J.; Xiao, B.; Dong, J.; Li, X. LncRNA-TCONS_00034812 in cell proliferation and apoptosis of pulmonary artery smooth muscle cells and its mechanism. J. Cell. Physiol. 2018, 233, 4801–4814. [Google Scholar] [CrossRef]
- Yun, X.; Jiang, H.; Lai, N.; Wang, J.; Shimoda, L.A. Aquaporin 1-mediated changes in pulmonary arterial smooth muscle cell migration and proliferation involve β-catenin. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L889–L898. [Google Scholar] [CrossRef] [Green Version]
- Saadoun, S.; Papadopoulos, M.C.; Hara-Chikuma, M.; Verkman, A.S. Impairment of angiogenesis and cell migration by targeted aquaporin-1 gene disruption. Nature 2005, 434, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Yang, B.; Gillespie, A.; Carlson, E.J.; Epstein, C.J.; Verkman, A.S. Severely impaired urinary concentrating ability in transgenic mice lacking aquaporin-1 water channels. J. Biol. Chem. 1998, 273, 4296–4299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, L.; Desroches-Castan, A.; Mallet, C.; Guyon, L.; Cumont, A.; Phan, C.; Robert, F.; Thuillet, R.; Bordenave, J.; Sekine, A.; et al. Selective BMP-9 Inhibition Partially Protects Against Experimental Pulmonary Hypertension. Circ. Res. 2019, 124, 846–855. [Google Scholar] [CrossRef] [PubMed]
- Lange, A.W.; Haitchi, H.M.; LeCras, T.D.; Sridharan, A.; Xu, Y.; Wert, S.E.; James, J.; Udell, N.; Thurner, P.J.; Whitsett, J.A. Sox17 is required for normal pulmonary vascular morphogenesis. Dev. Biol. 2014, 387, 109–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, J.; Liu, B.; Huang, C.; Haimel, M.; Bleda, M.; Gräf, S.; Rana, A.; Upton, P.D.; Morrell, N.W. SOX17 Deficiency Impairs Tube Network Formation Through the Reduction of Arterial Identity in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2020, 201, A2366. [Google Scholar]
- Winter, M.-P.; Sharma, S.; Altmann, J.; Seidl, V.; Panzenböck, A.; Alimohammadi, A.; Zelniker, T.; Redwan, B.; Nagel, F.; Santer, D.; et al. Interruption of vascular endothelial growth factor receptor 2 signaling induces a proliferative pulmonary vasculopathy and pulmonary hypertension. Basic Res. Cardiol. 2020, 115, 58. [Google Scholar] [CrossRef]
- Lee, H.-W.; Adachi, T.; Park, S.; Kowalski, P.; Anderson, D.; Simons, M.; Jin, S.W.; Chun, H.J. Bmpr1a and Its Role in Endomt in Pulmonary Arterial Hypertension. Circulation 2018, 138, A17070. [Google Scholar]
- Lee, H.W.; Adachi, T.; Park, S.; Pak, B.; Kowalski, P.; Choi, W.; Clapham, K.; Lee, A.; Anderson, D.; Simons, M.; et al. Abstract 14616: Loss of Endothelial BMPR1A Results in Pulmonary Hypertension Through Endothelial to Mesenchymal Transition. Circulation 2019, 140, A14616. [Google Scholar] [CrossRef]
- Du, L.; Sullivan, C.C.; Chu, D.; Cho, A.J.; Kido, M.; Wolf, P.L.; Yuan, J.X.-J.; Deutsch, R.; Jamieson, S.W.; Thistlethwaite, P.A. Signaling molecules in nonfamilial pulmonary hypertension. N. Engl. J. Med. 2003, 348, 500–509. [Google Scholar] [CrossRef]
- El-Bizri, N.; Wang, L.; Merklinger, S.L.; Guignabert, C.; Desai, T.; Urashima, T.; Sheikh, A.Y.; Knutsen, R.H.; Mecham, R.P.; Mishina, Y.; et al. Smooth muscle protein 22alpha-mediated patchy deletion of Bmpr1a impairs cardiac contractility but protects against pulmonary vascular remodeling. Circ. Res. 2008, 102, 380–388. [Google Scholar] [CrossRef] [Green Version]
- Chida, A.; Shintani, M.; Nakayama, T.; Furutani, Y.; Hayama, E.; Inai, K.; Saji, T.; Nonoyama, S.; Nakanishi, T. Missense mutations of the BMPR1B (ALK6) gene in childhood idiopathic pulmonary arterial hypertension. Circ. J. 2012, 76, 1501–1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Keller, S.H.; Remillard, C.V.; Safrina, O.; Nicholson, A.; Zhang, S.L.; Jiang, W.; Vangala, N.; Landsberg, J.W.; Wang, J.-Y.; et al. A functional single-nucleotide polymorphism in the TRPC6 gene promoter associated with idiopathic pulmonary arterial hypertension. Circulation 2009, 119, 2313–2322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Knight, L.; Ji, R.; Lawrence, P.; Kanaan, U.; Li, L.; Das, A.; Cui, B.; Zou, W.; Penny, D.J.; et al. Early onset severe pulmonary arterial hypertension with “two-hit” digenic mutations in both BMPR2 and KCNA5 genes. Int. J. Cardiol. 2014, 177, e167–e169. [Google Scholar] [CrossRef] [PubMed]
- Remillard, C.V.; Tigno, D.D.; Platoshyn, O.; Burg, E.D.; Brevnova, E.E.; Conger, D.; Nicholson, A.; Rana, B.K.; Channick, R.N.; Rubin, L.J.; et al. Function of Kv1.5 channels and genetic variations of KCNA5 in patients with idiopathic pulmonary arterial hypertension. Am. J. Physiol. Cell Physiol. 2007, 292, C1837–C1853. [Google Scholar] [CrossRef]
- Zhang, P.; McGrath, B.C.; Reinert, J.; Olsen, D.S.; Lei, L.; Gill, S.; Wek, S.A.; Vattem, K.M.; Wek, R.C.; Kimball, S.R.; et al. The GCN2 eIF2alpha kinase is required for adaptation to amino acid deprivation in mice. Mol. Cell. Biol. 2002, 22, 6681–6688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germain, M.; Eyries, M.; Montani, D.; Poirier, O.; Girerd, B.; Dorfmüller, P.; Coulet, F.; Nadaud, S.; Maugenre, S.; Guignabert, C.; et al. Genome-wide association analysis identifies a susceptibility locus for pulmonary arterial hypertension. Nat. Genet. 2013, 45, 518–521. [Google Scholar] [CrossRef]
- Paulin, R.; Dromparis, P.; Sutendra, G.; Gurtu, V.; Zervopoulos, S.; Bowers, L.; Haromy, A.; Webster, L.; Provencher, S.; Bonnet, S.; et al. Sirtuin 3 deficiency is associated with inhibited mitochondrial function and pulmonary arterial hypertension in rodents and humans. Cell Metab. 2014, 20, 827–839. [Google Scholar] [CrossRef] [Green Version]
- Michelakis, E.D.; Gurtu, V.; Webster, L.; Barnes, G.; Watson, G.; Howard, L.; Cupitt, J.; Paterson, I.; Thompson, R.B.; Chow, K.; et al. Inhibition of pyruvate dehydrogenase kinase improves pulmonary arterial hypertension in genetically susceptible patients. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Dromparis, P.; Paulin, R.; Sutendra, G.; Qi, A.C.; Bonnet, S.; Michelakis, E.D. Uncoupling protein 2 deficiency mimics the effects of hypoxia and endoplasmic reticulum stress on mitochondria and triggers pseudohypoxic pulmonary vascular remodeling and pulmonary hypertension. Circ. Res. 2013, 113, 126–136. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.Y.; Baffy, G.; Perret, P.; Krauss, S.; Peroni, O.; Grujic, D.; Hagen, T.; Vidal-Puig, A.J.; Boss, O.; Kim, Y.B.; et al. Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, beta cell dysfunction, and type 2 diabetes. Cell 2001, 105, 745–755. [Google Scholar] [CrossRef] [Green Version]
- Baloira Villar, A.; Valverde, D.; Lago, M. Functional study of polymorphisms in the promoter region of the endothelin-1 gene in Pulmonary Arterial Hypertension. Eur. Respir. J. 2019, 54, PA5043. [Google Scholar]
- Giaid, A.; Yanagisawa, M.; Langleben, D.; Michel, R.P.; Levy, R.; Shennib, H.; Kimura, S.; Masaki, T.; Duguid, W.P.; Stewart, D.J. Expression of Endothelin-1 in the Lungs of Patients with Pulmonary Hypertension. N. Engl. J. Med. 1993, 328, 1732–1739. [Google Scholar] [CrossRef] [PubMed]
- De Jesus Perez, V.A.; Yuan, K.; Lyuksyutova, M.A.; Dewey, F.; Orcholski, M.E.; Shuffle, E.M.; Mathur, M.; Yancy, L., Jr.; Rojas, V.; Li, C.G.; et al. Whole-exome sequencing reveals TopBP1 as a novel gene in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2014, 189, 1260–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damico, R.; Kolb, T.M.; Valera, L.; Wang, L.; Housten, T.; Tedford, R.J.; Kass, D.A.; Rafaels, N.; Gao, L.; Barnes, K.C.; et al. Serum endostatin is a genetically determined predictor of survival in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2015, 191, 208–218. [Google Scholar] [CrossRef] [Green Version]
- Maloney, J.P.; Stearman, R.S.; Bull, T.M.; Calabrese, D.W.; Tripp-Addison, M.L.; Wick, M.J.; Broeckel, U.; Robbins, I.M.; Wheeler, L.A.; Cogan, J.D.; et al. Loss-of-function thrombospondin-1 mutations in familial pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L541–L554. [Google Scholar] [CrossRef] [Green Version]
- Pfarr, N.; Fischer, C.; Ehlken, N.; Becker-Grünig, T.; López-González, V.; Gorenflo, M.; Hager, A.; Hinderhofer, K.; Miera, O.; Nagel, C.; et al. Hemodynamic and genetic analysis in children with idiopathic, heritable, and congenital heart disease associated pulmonary arterial hypertension. Respir. Res. 2013, 14, 3. [Google Scholar] [CrossRef] [Green Version]
- Runo, J.R.; Vnencak-Jones, C.L.; Prince, M.; Loyd, J.E.; Wheeler, L.; Robbins, I.M.; Lane, K.B.; Newman, J.H.; Johnson, J.; Nichols, W.C.; et al. Pulmonary veno-occlusive disease caused by an inherited mutation in bone morphogenetic protein receptor II. Am. J. Respir. Crit. Care Med. 2003, 167, 889–894. [Google Scholar] [CrossRef]
- Machado, R.D.; Southgate, L.; Eichstaedt, C.A.; Aldred, M.A.; Austin, E.D.; Best, D.H.; Chung, W.K.; Benjamin, N.; Elliott, C.G.; Eyries, M.; et al. Pulmonary Arterial Hypertension: A Current Perspective on Established and Emerging Molecular Genetic Defects. Hum. Mutat. 2015, 36, 1113–1127. [Google Scholar] [CrossRef] [Green Version]
- Machado, R.D.; Eickelberg, O.; Elliott, C.G.; Geraci, M.W.; Hanaoka, M.; Loyd, J.E.; Newman, J.H.; Phillips, J.A., 3rd; Soubrier, F.; Trembath, R.C.; et al. Genetics and genomics of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2009, 54, S32–S42. [Google Scholar] [CrossRef] [Green Version]
- Machado, R.D.; Aldred, M.A.; James, V.; Harrison, R.E.; Patel, B.; Schwalbe, E.C.; Gruenig, E.; Janssen, B.; Koehler, R.; Seeger, W.; et al. Mutations of the TGF-beta type II receptor BMPR2 in pulmonary arterial hypertension. Hum. Mutat. 2006, 27, 121–132. [Google Scholar] [CrossRef]
- Evans, J.D.W.; Girerd, B.; Montani, D.; Wang, X.-J.; Galiè, N.; Austin, E.D.; Elliott, G.; Asano, K.; Grünig, E.; Yan, Y.; et al. BMPR2 mutations and survival in pulmonary arterial hypertension: An individual participant data meta-analysis. Lancet Respir. Med. 2016, 4, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, C.; Stewart, S.; Upton, P.D.; Machado, R.; Thomson, J.R.; Trembath, R.C.; Morrell, N.W. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation 2002, 105, 1672–1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavoie, J.R.; Ormiston, M.L.; Perez-Iratxeta, C.; Courtman, D.W.; Jiang, B.; Ferrer, E.; Caruso, P.; Southwood, M.; Foster, W.S.; Morrell, N.W.; et al. Proteomic analysis implicates translationally controlled tumor protein as a novel mediator of occlusive vascular remodeling in pulmonary arterial hypertension. Circulation 2014, 129, 2125–2135. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.-Y.; D Collum, S.; Luo, F.; Weng, T.; Le, T.-T.; M Hernandez, A.; Philip, K.; Molina, J.G.; Garcia-Morales, L.J.; Cao, Y.; et al. Macrophage bone morphogenic protein receptor 2 depletion in idiopathic pulmonary fibrosis and Group III pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L238–L254. [Google Scholar] [CrossRef] [Green Version]
- Donaldson, J.W.; McKeever, T.M.; Hall, I.P.; Hubbard, R.B.; Fogarty, A.W. The UK prevalence of hereditary haemorrhagic telangiectasia and its association with sex, socioeconomic status and region of residence: A population-based study. Thorax 2014, 69, 161–167. [Google Scholar] [CrossRef] [Green Version]
- Harrison, R.E.; Flanagan, J.A.; Sankelo, M.; Abdalla, S.A.; Rowell, J.; Machado, R.D.; Elliott, C.G.; Robbins, I.M.; Olschewski, H.; McLaughlin, V.; et al. Molecular and functional analysis identifies ALK-1 as the predominant cause of pulmonary hypertension related to hereditary haemorrhagic telangiectasia. J. Med. Genet. 2003, 40, 865–871. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, M.; Yagi, H.; Matsuoka, R.; Akimoto, K.; Furutani, M.; Imamura, S.-I.; Uehara, R.; Nakayama, T.; Takao, A.; Nakazawa, M.; et al. Implications of mutations of activin receptor-like kinase 1 gene (ALK1) in addition to bone morphogenetic protein receptor II gene (BMPR2) in children with pulmonary arterial hypertension. Circ. J. 2008, 72, 127–133. [Google Scholar] [CrossRef] [Green Version]
- Kwasnickacrawford, D.; Carson, A.; Roberts, W.; Summers, A.; Rehnstrom, K.; Jarvela, I.; Scherer, S. Characterization of a novel cation transporter ATPase gene (ATP13A4) interrupted by 3q25–q29 inversion in an individual with language delay. Genomics 2005, 86, 182–194. [Google Scholar] [CrossRef]
- Madan, M.; Patel, A.; Skruber, K.; Geerts, D.; Altomare, D.A.; Iv, O.P. ATP13A3 and caveolin-1 as potential biomarkers for difluoromethylornithine-based therapies in pancreatic cancers. Am. J. Cancer Res. 2016, 6, 1231–1252. [Google Scholar]
- Sui, H.; Han, B.G.; Lee, J.K.; Walian, P.; Jap, B.K. Structural basis of water-specific transport through the AQP1 water channel. Nature 2001, 414, 872–878. [Google Scholar] [CrossRef] [Green Version]
- Montani, D.; Girerd, B.; Günther, S.; Riant, F.; Tournier-Lasserve, E.; Magy, L.; Maazi, N.; Guignabert, C.; Savale, L.; Sitbon, O.; et al. Pulmonary arterial hypertension in familial hemiplegic migraine with ATP1A2 channelopathy. Eur. Respir. J. 2014, 43, 641–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firth, A.L.; Remillard, C.V.; Platoshyn, O.; Fantozzi, I.; Ko, E.A.; Yuan, J.X.-J. Functional ion channels in human pulmonary artery smooth muscle cells: Voltage-dependent cation channels. Pulm. Circ. 2011, 1, 48–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makino, A.; Firth, A.L.; Yuan, J.X.-J. Endothelial and smooth muscle cell ion channels in pulmonary vasoconstriction and vascular remodeling. Compr. Physiol. 2011, 1, 1555–1602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burg, E.D.; Remillard, C.V.; Yuan, J.X.-J. Potassium channels in the regulation of pulmonary artery smooth muscle cell proliferation and apoptosis: Pharmacotherapeutic implications. Br. J. Pharmacol. 2008, 153 (Suppl. 1), S99–S111. [Google Scholar] [CrossRef] [Green Version]
- Lambert, M.; Capuano, V.; Olschewski, A.; Sabourin, J.; Nagaraj, C.; Girerd, B.; Weatherald, J.; Humbert, M.; Antigny, F. Ion Channels in Pulmonary Hypertension: A Therapeutic Interest? Int. J. Mol. Sci. 2018, 19, 3162. [Google Scholar] [CrossRef] [Green Version]
- Babenko, A.P.; Polak, M.; Cavé, H.; Busiah, K.; Czernichow, P.; Scharfmann, R.; Bryan, J.; Aguilar-Bryan, L.; Vaxillaire, M.; Froguel, P. Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N. Engl. J. Med. 2006, 355, 456–466. [Google Scholar] [CrossRef]
- Flanagan, S.E.; Clauin, S.; Bellanné-Chantelot, C.; de Lonlay, P.; Harries, L.W.; Gloyn, A.L.; Ellard, S. Update of mutations in the genes encoding the pancreatic beta-cell KATPchannel subunits Kir6.2 (KCNJ11) and sulfonylurea receptor 1 (ABCC8) in diabetes mellitus and hyperinsulinism. Hum. Mutat. 2009, 30, 170–180. [Google Scholar] [CrossRef]
- Lago-Docampo, M.; Tenorio, J.; Hernández-González, I.; Pérez-Olivares, C.; Escribano-Subías, P.; Pousada, G.; Baloira, A.; Arenas, M.; Lapunzina, P.; Valverde, D. Characterization of rare ABCC8 variants identified in Spanish pulmonary arterial hypertension patients. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef]
- Arora, R.; Metzger, R.J.; Papaioannou, V.E. Multiple roles and interactions of Tbx4 and Tbx5 in development of the respiratory system. PLoS Genet. 2012, 8, e1002866. [Google Scholar] [CrossRef] [Green Version]
- Karolak, J.A.; Vincent, M.; Deutsch, G.; Gambin, T.; Cogné, B.; Pichon, O.; Vetrini, F.; Mefford, H.C.; Dines, J.N.; Golden-Grant, K.; et al. Complex Compound Inheritance of Lethal Lung Developmental Disorders Due to Disruption of the TBX-FGF Pathway. Am. J. Hum. Genet. 2019, 104, 213–228. [Google Scholar] [CrossRef] [Green Version]
- Bongers, E.M.H.F.; Duijf, P.H.G.; van Beersum, S.E.M.; Schoots, J.; Van Kampen, A.; Burckhardt, A.; Hamel, B.C.J.; Losan, F.; Hoefsloot, L.H.; Yntema, H.G.; et al. Mutations in the human TBX4 gene cause small patella syndrome. Am. J. Hum. Genet. 2004, 74, 1239–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galambos, C.; Mullen, M.P.; Shieh, J.T.; Schwerk, N.; Kielt, M.J.; Ullmann, N.; Boldrini, R.; Stucin-Gantar, I.; Haass, C.; Bansal, M.; et al. Phenotype characterisation of TBX4 mutation and deletion carriers with neonatal and paediatric pulmonary hypertension. Eur. Respir. J. 2019, 54, 1801965. [Google Scholar] [CrossRef] [PubMed]
- Francois, M.; Koopman, P.; Beltrame, M. SoxF genes: Key players in the development of the cardio-vascular system. Int. J. Biochem. Cell Biol. 2010, 42, 445–448. [Google Scholar] [CrossRef] [PubMed]
- De Vilder, E.Y.G.; Debacker, J.; Vanakker, O.M. GGCX-Associated Phenotypes: An Overview in Search of Genotype-Phenotype Correlations. Int. J. Mol. Sci. 2017, 18, 240. [Google Scholar] [CrossRef] [PubMed]
- Lantuéjoul, S.; Sheppard, M.N.; Corrin, B.; Burke, M.M.; Nicholson, A.G. Pulmonary veno-occlusive disease and pulmonary capillary hemangiomatosis: A clinicopathologic study of 35 cases. Am. J. Surg. Pathol. 2006, 30, 850–857. [Google Scholar] [CrossRef]
- Davies, P.; Reid, L. Pulmonary veno-occlusive disease in siblings: Case reports and morphometric study. Hum. Pathol. 1982, 13, 911–915. [Google Scholar] [CrossRef]
- Voordes, C.G.; Kuipers, J.R.; Elema, J.D. Familial pulmonary veno-occlusive disease: A case report. Thorax 1977, 32, 763–766. [Google Scholar] [CrossRef] [Green Version]
- Austin, E.D.; Cogan, J.D.; West, J.D.; Hedges, L.K.; Hamid, R.; Dawson, E.P.; Wheeler, L.A.; Parl, F.F.; Loyd, J.E.; Phillips, J.A., 3rd. Alterations in oestrogen metabolism: Implications for higher penetrance of familial pulmonary arterial hypertension in females. Eur. Respir. J. 2009, 34, 1093–1099. [Google Scholar] [CrossRef] [Green Version]
- Austin, E.D.; Hamid, R.; Hemnes, A.R.; Loyd, J.E.; Blackwell, T.; Yu, C.; Phillips, J.A., III; Gaddipati, R.; Gladson, S.; Gu, E.; et al. BMPR2 expression is suppressed by signaling through the estrogen receptor. Biol. Sex Differ. 2012, 3, 6. [Google Scholar] [CrossRef] [Green Version]
- Hamid, R.; Cogan, J.D.; Hedges, L.K.; Austin, E.; Phillips, J.A., 3rd; Newman, J.H.; Loyd, J.E. Penetrance of pulmonary arterial hypertension is modulated by the expression of normal BMPR2 allele. Hum. Mutat. 2009, 30, 649–654. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.A., 3rd; Poling, J.S.; Phillips, C.A.; Stanton, K.C.; Austin, E.D.; Cogan, J.D.; Wheeler, L.; Yu, C.; Newman, J.H.; Dietz, H.C.; et al. Synergistic heterozygosity for TGFbeta1 SNPs and BMPR2 mutations modulates the age at diagnosis and penetrance of familial pulmonary arterial hypertension. Genet. Med. 2008, 10, 359–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirschey, M.D.; Shimazu, T.; Jing, E.; Grueter, C.A.; Collins, A.M.; Aouizerat, B.; Stančáková, A.; Goetzman, E.; Lam, M.M.; Schwer, B.; et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol. Cell 2011, 44, 177–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, W.T.; Raynolds, M.V.; Badesch, D.B.; Wynne, K.M.; Groves, B.M.; Roden, R.L.; Robertson, A.D.; Lowes, B.D.; Zisman, L.S.; Voelkel, N.F.; et al. Angiotensin-converting enzyme DD genotype in patients with primary pulmonary hypertension: Increased frequency and association with preserved haemodynamics. J. Renin-Angiotensin-Aldosterone Syst. 2003, 4, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Solari, V.; Puri, P. Genetic polymorphisms of angiotensin system genes in congenital diaphragmatic hernia associated with persistent pulmonary hypertension. J. Pediatr. Surg. 2004, 39, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Sutliff, R.L.; Kang, B.-Y.; Michael Hart, C. PPARγ as a potential therapeutic target in pulmonary hypertension. Ther. Adv. Respir. Dis. 2010, 4, 143–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, D.E.; Sutliff, R.L.; Michael Hart, C. Is Peroxisome Proliferator-Activated Receptor Gamma (PPARγ) a Therapeutic Target for the Treatment of Pulmonary Hypertension? Pulm. Circ. 2011, 1, 33–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malenfant, S.; Neyron, A.-S.; Paulin, R.; Potus, F.; Meloche, J.; Provencher, S.; Bonnet, S. Signal Transduction in the Development of Pulmonary Arterial Hypertension. Pulm. Circ. 2013, 3, 278–293. [Google Scholar] [CrossRef] [Green Version]
- Shatat, M.A.; Tian, H.; Zhang, R.; Tandon, G.; Hale, A.; Fritz, J.S.; Zhou, G.; Martínez-González, J.; Rodríguez, C.; Champion, H.C.; et al. Endothelial Krüppel-Like Factor 4 Modulates Pulmonary Arterial Hypertension. Am. J. Respir. Cell Mol. Biol. 2014, 50, 647–653. [Google Scholar] [CrossRef] [Green Version]
- Crosby, A.; Toshner, M.R.; Southwood, M.R.; Soon, E.; Dunmore, B.J.; Groves, E.; Moore, S.; Wright, P.; Ottersbach, K.; Bennett, C.; et al. Hematopoietic stem cell transplantation alters susceptibility to pulmonary hypertension in Bmpr2-deficient mice. Pulm. Circ. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Kallunki, T.; Barisic, M.; Jäättelä, M.; Liu, B. How to Choose the Right Inducible Gene Expression System for Mammalian Studies? Cells 2019, 8, 796. [Google Scholar] [CrossRef] [Green Version]
- Urnov, F.D.; Rebar, E.J.; Holmes, M.C.; Steve Zhang, H.; Gregory, P.D. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 2010, 11, 636–646. [Google Scholar] [CrossRef]
- Joung, J.K.; Keith Joung, J.; Sander, J.D. TALENs: A widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 2013, 14, 49–55. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Rossi, J.J. RNAi mechanisms and applications. Biotech. 2008, 44, 613–616. [Google Scholar] [CrossRef]
- Lewandoski, M. Conditional control of gene expression in the mouse. Nat. Rev. Genet. 2001, 2, 743–755. [Google Scholar] [CrossRef]
- Van der Weyden, L.; White, J.K.; Adams, D.J.; Logan, D.W. The mouse genetics toolkit: Revealing function and mechanism. Genome Biol. 2011, 12, 224. [Google Scholar] [CrossRef] [Green Version]
- Van Boxtel, R.; Cuppen, E. Rat traps: Filling the toolbox for manipulating the rat genome. Genome Biol. 2010, 11, 217. [Google Scholar] [CrossRef]
- Toshner, M.; Voswinckel, R.; Southwood, M.; Al-Lamki, R.; Howard, L.S.G.; Marchesan, D.; Yang, J.; Suntharalingam, J.; Soon, E.; Exley, A.; et al. Evidence of dysfunction of endothelial progenitors in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2009, 180, 780–787. [Google Scholar] [CrossRef] [Green Version]
- Morrell, N.W.; Adnot, S.; Archer, S.L.; Dupuis, J.; Jones, P.L.; MacLean, M.R.; McMurtry, I.F.; Stenmark, K.R.; Thistlethwaite, P.A.; Weissmann, N.; et al. Cellular and molecular basis of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2009, 54, S20–S31. [Google Scholar] [CrossRef] [Green Version]
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmüller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur. Respir. J. 2019, 53. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Haimel, M.; Bleda, M.; Li, W.; Gräf, S.; Upton, P.D.; Morrell, N.W. S42 Characterizing ATP13A3 loss of function in pulmonary arterial hypertension (PAH). Fundam. Mech. Pulm. Arter. Hypertens. 2018. [Google Scholar]
- Tian, L.; Gao, J.; Garcia, I.M.; Chen, H.J.; Castaldi, A.; Chen, Y.-W. Human pluripotent stem cell-derived lung organoids: Potential applications in development and disease modeling. Wiley Interdiscip. Rev. Dev. Biol. 2020, e399. [Google Scholar] [CrossRef]
- D’Amico, R.W.; Faley, S.; Shim, H.-N.; Prosser, J.R.; Agrawal, V.; Bellan, L.M.; West, J.D. Pulmonary Vascular Platform Models the Effects of Flow and Pressure on Endothelial Dysfunction in BMPR2 Associated Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2018, 19, 2561. [Google Scholar] [CrossRef] [Green Version]
- Nogueira-Ferreira, R.; Faria-Costa, G.; Ferreira, R.; Henriques-Coelho, T. Animal models for the study of pulmonary hypertension: Potential and limitations. Cardiol. Cardiovasc. Med. 2016, 1, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Mao, L.; Rajagopal, S. Hemodynamic Characterization of Rodent Models of Pulmonary Arterial Hypertension. J. Vis. Exp. 2016. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Arroyo, J.G.; Farkas, L.; Alhussaini, A.A.; Farkas, D.; Kraskauskas, D.; Voelkel, N.F.; Bogaard, H.J. The monocrotaline model of pulmonary hypertension in perspective. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L363–L369. [Google Scholar] [CrossRef]
- Akhavein, F.; St-Michel, E.J.; Seifert, E.; Rohlicek, C.V. Decreased left ventricular function, myocarditis, and coronary arteriolar medial thickening following monocrotaline administration in adult rats. J. Appl. Physiol. 2007, 103, 287–295. [Google Scholar] [CrossRef] [Green Version]
- Taraseviciene-stewart, L.; Kasahara, Y.; Alger, L.; Hirth, P.; Mc Mahon, G.; Waltenberger, J.; Voelkel, N.F.; Tuder, R.M. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J. 2001, 15, 427–438. [Google Scholar] [CrossRef]
- Kojonazarov, B.; Hadzic, S.; Ghofrani, H.A.; Grimminger, F.; Seeger, W.; Weissmann, N.; Schermuly, R.T. Severe Emphysema in the SU5416/Hypoxia Rat Model of Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2019, 200, 515–518. [Google Scholar] [CrossRef]
- Mancuso, M.R.; Davis, R.; Norberg, S.M.; O’Brien, S.; Sennino, B.; Nakahara, T.; Yao, V.J.; Inai, T.; Brooks, P.; Freimark, B.; et al. Rapid vascular regrowth in tumors after reversal of VEGF inhibition. J. Clin. Investig. 2006, 116, 2610–2621. [Google Scholar] [CrossRef] [Green Version]
- Tabima, D.M.; Hacker, T.A.; Chesler, N.C. Measuring right ventricular function in the normal and hypertensive mouse hearts using admittance-derived pressure-volume loops. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H2069–H2075. [Google Scholar] [CrossRef] [Green Version]
- Luo, F.; Wang, X.; Luo, X.; Li, B.; Zhu, D.; Sun, H.; Tang, Y. Invasive Hemodynamic Assessment for the Right Ventricular System and Hypoxia-Induced Pulmonary Arterial Hypertension in Mice. J. Vis. Exp. 2019. [Google Scholar] [CrossRef]
- Stenmark, K.R.; Meyrick, B.; Galie, N.; Mooi, W.J.; McMurtry, I.F. Animal models of pulmonary arterial hypertension: The hope for etiological discovery and pharmacological cure. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L1013–L1032. [Google Scholar] [CrossRef]
- Akazawa, Y.; Okumura, K.; Ishii, R.; Slorach, C.; Hui, W.; Ide, H.; Honjo, O.; Sun, M.; Kabir, G.; Connelly, K.; et al. Pulmonary artery banding is a relevant model to study the right ventricular remodeling and dysfunction that occurs in pulmonary arterial hypertension. J. Appl. Physiol. 2020, 129, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Lawrie, A. A report on the use of animal models and phenotyping methods in pulmonary hypertension research. Pulm. Circ. 2014, 4, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Jones, J.E.; Beppu, H.; Keaney, J.F., Jr.; Loscalzo, J.; Zhang, Y.-Y. Increased susceptibility to pulmonary hypertension in heterozygous BMPR2-mutant mice. Circulation 2005, 112, 553–562. [Google Scholar] [CrossRef] [Green Version]
- Long, L.; MacLean, M.R.; Jeffery, T.K.; Morecroft, I.; Yang, X.; Rudarakanchana, N.; Southwood, M.; James, V.; Trembath, R.C.; Morrell, N.W. Serotonin increases susceptibility to pulmonary hypertension in BMPR2-deficient mice. Circ. Res. 2006, 98, 818–827. [Google Scholar] [CrossRef] [Green Version]
- Peacock, A.J.; Vonk Noordegraaf, A. Cardiac magnetic resonance imaging in pulmonary arterial hypertension. Eur. Respir. Rev. 2013, 22, 526–534. [Google Scholar] [CrossRef]
- Provencher, S.; Archer, S.L.; Ramirez, F.D.; Hibbert, B.; Paulin, R.; Boucherat, O.; Lacasse, Y.; Bonnet, S. Standards and Methodological Rigor in Pulmonary Arterial Hypertension Preclinical and Translational Research. Circ. Res. 2018, 122, 1021–1032. [Google Scholar] [CrossRef]
- Rol, N.; Kurakula, K.B.; Happé, C.; Bogaard, H.J.; Goumans, M.-J. TGF-β and BMPR2 Signaling in PAH: Two Black Sheep in One Family. Int. J. Mol. Sci. 2018, 19, 2585. [Google Scholar] [CrossRef] [Green Version]
- Ten Dijke, P.; Goumans, M.-J.; Pardali, E. Endoglin in angiogenesis and vascular diseases. Angiogenesis 2008, 11, 79–89. [Google Scholar] [CrossRef]
- Upton, P.D.; Davies, R.J.; Trembath, R.C.; Morrell, N.W. Bone morphogenetic protein (BMP) and activin type II receptors balance BMP9 signals mediated by activin receptor-like kinase-1 in human pulmonary artery endothelial cells. J. Biol. Chem. 2009, 284, 15794–15804. [Google Scholar] [CrossRef] [Green Version]
- Barnes, J.W.; Kucera, E.T.; Tian, L.; Mellor, N.E.; Dvorina, N.; Baldwin, W.W.; Aldred, M.A.; Farver, C.F.; Comhair, S.A.A.; Aytekin, M.; et al. Bone Morphogenic Protein Type 2 Receptor Mutation-Independent Mechanisms of Disrupted Bone Morphogenetic Protein Signaling in Idiopathic Pulmonary Arterial Hypertension. Am. J. Respir. Cell Mol. Biol. 2016, 55, 564–575. [Google Scholar] [CrossRef] [Green Version]
- Good, R.B.; Gilbane, A.J.; Trinder, S.L.; Denton, C.P.; Coghlan, G.; Abraham, D.J.; Holmes, A.M. Endothelial to Mesenchymal Transition Contributes to Endothelial Dysfunction in Pulmonary Arterial Hypertension. Am. J. Pathol. 2015, 185, 1850–1858. [Google Scholar] [CrossRef]
- Liu, T.; Zou, X.-Z.; Huang, N.; Ge, X.-Y.; Yao, M.-Z.; Liu, H.; Zhang, Z.; Hu, C.-P. miR-27a promotes endothelial-mesenchymal transition in hypoxia-induced pulmonary arterial hypertension by suppressing BMP signaling. Life Sci. 2019, 227, 64–73. [Google Scholar] [CrossRef]
- Vanderpool, R.R.; El-Bizri, N.; Rabinovitch, M.; Chesler, N.C. Patchy deletion of Bmpr1a potentiates proximal pulmonary artery remodeling in mice exposed to chronic hypoxia. Biomech. Model. Mechanobiol. 2013, 12, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Tie, L.; Wang, D.; Shi, Y.; Li, X. Aquaporins in Cardiovascular System. Adv. Exp. Med. Biol. 2017, 969, 105–113. [Google Scholar] [CrossRef]
- Clapp, C.; Martínez de la Escalera, G. Aquaporin-1: A novel promoter of tumor angiogenesis. Trends Endocrinol. Metab. 2006, 17, 1–2. [Google Scholar] [CrossRef]
- Schuoler, C.; Haider, T.J.; Leuenberger, C.; Vogel, J.; Ostergaard, L.; Kwapiszewska, G.; Kohler, M.; Gassmann, M.; Huber, L.C.; Brock, M. Aquaporin 1 controls the functional phenotype of pulmonary smooth muscle cells in hypoxia-induced pulmonary hypertension. Basic Res. Cardiol. 2017, 112, 30. [Google Scholar] [CrossRef] [Green Version]
- Kunichika, N.; Landsberg, J.W.; Yu, Y.; Kunichika, H.; Thistlethwaite, P.A.; Rubin, L.J.; Yuan, J.X.-J. Bosentan inhibits transient receptor potential channel expression in pulmonary vascular myocytes. Am. J. Respir. Crit. Care Med. 2004, 170, 1101–1107. [Google Scholar] [CrossRef] [Green Version]
- Pozeg, Z.I.; Michelakis, E.D.; McMurtry, M.S.; Thébaud, B.; Wu, X.-C.; Dyck, J.R.B.; Hashimoto, K.; Wang, S.; Moudgil, R.; Harry, G.; et al. In vivo gene transfer of the O2-sensitive potassium channel Kv1.5 reduces pulmonary hypertension and restores hypoxic pulmonary vasoconstriction in chronically hypoxic rats. Circulation 2003, 107, 2037–2044. [Google Scholar] [CrossRef] [Green Version]
- Sheeba, C.J.; Logan, M.P.O. The Roles of T-Box Genes in Vertebrate Limb Development. Curr. Top. Dev. Biol. 2017, 122, 355–381. [Google Scholar] [CrossRef]
- Zhang, W.; Menke, D.B.; Jiang, M.; Chen, H.; Warburton, D.; Turcatel, G.; Lu, C.-H.; Xu, W.; Luo, Y.; Shi, W. Spatial-temporal targeting of lung-specific mesenchyme by a Tbx4 enhancer. BMC Biol. 2013, 11, 111. [Google Scholar] [CrossRef] [Green Version]
- Rieder, M.J.; Reiner, A.P.; Rettie, A.E. Gamma-glutamyl carboxylase (GGCX) tagSNPs have limited utility for predicting warfarin maintenance dose. J. Thromb. Haemost. 2007, 5, 2227–2234. [Google Scholar] [CrossRef]
- Napolitano, M.; Mariani, G.; Lapecorella, M. Hereditary combined deficiency of the vitamin K-dependent clotting factors. Orphanet J. Rare Dis. 2010, 5, 21. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Schurgers, L.J.; Smith, A.C.M.; Tsokos, M.; Uitto, J.; Cowen, E.W. Co-existent pseudoxanthoma elasticum and vitamin K-dependent coagulation factor deficiency: Compound heterozygosity for mutations in the GGCX gene. Am. J. Pathol. 2009, 174, 534–540. [Google Scholar] [CrossRef] [Green Version]
- Moreau, M.E.; Garbacki, N.; Molinaro, G.; Brown, N.J.; Marceau, F.; Adam, A. The Kallikrein-Kinin System: Current and Future Pharmacological Targets. J. Pharmacol. Sci. 2005, 99, 6–38. [Google Scholar] [CrossRef] [Green Version]
- Carretero, O.A.; Scicli, A.G. The renal kallikrein-kinin system in human and in experimental hypertension. Klin. Wochenschr. 1978, 56 (Suppl. 1), 113–125. [Google Scholar] [CrossRef]
- Madeddu, P.; Emanueli, C.; El-Dahr, S. Mechanisms of disease: The tissue kallikrein-kinin system in hypertension and vascular remodeling. Nat. Clin. Pract. Nephrol. 2007, 3, 208–221. [Google Scholar] [CrossRef]
- Woodley-Miller, C.; Chao, J.; Chao, L. Restriction fragment length polymorphisms mapped in spontaneously hypertensive rats using kallikrein probes. J. Hypertens. 1989, 7, 865–871. [Google Scholar] [CrossRef]
- Madeddu, P.; Varoni, M.V.; Demontis, M.P.; Chao, J.; Simson, J.A.; Glorioso, N.; Anania, V. Kallikrein-kinin system and blood pressure sensitivity to salt. Hypertension 1997, 29, 471–477. [Google Scholar] [CrossRef] [Green Version]
- Hua, H.; Zhou, S.; Liu, Y.; Wang, Z.; Wan, C.; Li, H.; Chen, C.; Li, G.; Zeng, C.; Chen, L.; et al. Relationship between the regulatory region polymorphism of human tissue kallikrein gene and essential hypertension. J. Hum. Hypertens. 2005, 19, 715–721. [Google Scholar] [CrossRef] [Green Version]
- Alexander-Curtis, M.; Pauls, R.; Chao, J.; Volpi, J.J.; Bath, P.M.; Verdoorn, T.A. Human tissue kallikrein in the treatment of acute ischemic stroke. Ther. Adv. Neurol. Disord. 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Anthony, T.G.; McDaniel, B.J.; Byerley, R.L.; McGrath, B.C.; Cavener, D.R.; McNurlan, M.A.; Wek, R.C. Preservation of liver protein synthesis during dietary leucine deprivation occurs at the expense of skeletal muscle mass in mice deleted for eIF2 kinase GCN2. J. Biol. Chem. 2004, 279, 36553–36561. [Google Scholar] [CrossRef] [Green Version]
- Waypa, G.B.; Osborne, S.W.; Marks, J.D.; Berkelhamer, S.K.; Kondapalli, J.; Schumacker, P.T. Sirtuin 3 deficiency does not augment hypoxia-induced pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 2013, 49, 885–891. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Kim, I.-K.; Yang, J.M.; Lee, E.; Koh, B.I.; Song, S.; Park, J.; Lee, S.; Choi, C.; Kim, J.W.; et al. SoxF Transcription Factors Are Positive Feedback Regulators of VEGF Signaling. Circ. Res. 2016, 119, 839–852. [Google Scholar] [CrossRef] [Green Version]
- Varshney, G.K.; Carrington, B.; Pei, W.; Bishop, K.; Chen, Z.; Fan, C.; Xu, L.; Jones, M.; LaFave, M.C.; Ledin, J.; et al. A high-throughput functional genomics workflow based on CRISPR/Cas9-mediated targeted mutagenesis in zebrafish. Nat. Protoc. 2016, 11, 2357–2375. [Google Scholar] [CrossRef]
- Vickovic, S.; Eraslan, G.; Salmén, F.; Klughammer, J.; Stenbeck, L.; Schapiro, D.; Äijö, T.; Bonneau, R.; Bergenstråhle, L.; Navarro, J.F.; et al. High-definition spatial transcriptomics for in situ tissue profiling. Nat. Methods 2019, 16, 987–990. [Google Scholar] [CrossRef]
- Grunewald, J.; Eklund, A. Sex-Specific Manifestations of Löfgren’s Syndrome. Am. J. Respir. Crit. Care Med. 2007, 175, 40–44. [Google Scholar] [CrossRef] [Green Version]
- Landini, S.; Mazzinghi, B.; Becherucci, F.; Allinovi, M.; Provenzano, A.; Palazzo, V.; Ravaglia, F.; Artuso, R.; Bosi, E.; Stagi, S.; et al. Reverse Phenotyping after Whole-Exome Sequencing in Steroid-Resistant Nephrotic Syndrome. Clin. J. Am. Soc. Nephrol. 2020, 15, 89–100. [Google Scholar] [CrossRef]
- Nelson, M.R.; Tipney, H.; Painter, J.L.; Shen, J.; Nicoletti, P.; Shen, Y.; Floratos, A.; Sham, P.C.; Li, M.J.; Wang, J.; et al. The support of human genetic evidence for approved drug indications. Nat. Genet. 2015, 47, 856–860. [Google Scholar] [CrossRef]
- Whicher, D.; Philbin, S.; Aronson, N. An overview of the impact of rare disease characteristics on research methodology. Orphanet J. Rare Dis. 2018, 13, 14. [Google Scholar] [CrossRef]
- UK Biobank. Available online: https://www.ukbiobank.ac.uk (accessed on 17 October 2020).
- Welcome to eMerge > Collaborate. Available online: https://emerge-network.org (accessed on 17 October 2020).
- National Institutes of Health (NIH)—All of Us. Available online: https://www.nih.gov/precision-medicine-initiative-cohort-program (accessed on 17 October 2020).
- Polubriaginof, F.C.G.; Vanguri, R.; Quinnies, K.; Belbin, G.M.; Yahi, A.; Salmasian, H.; Lorberbaum, T.; Nwankwo, V.; Li, L.; Shervey, M.M.; et al. Disease Heritability Inferred from Familial Relationships Reported in Medical Records. Cell 2018, 173, 1692–1704. [Google Scholar] [CrossRef] [Green Version]
- Firth, H.V.; Richards, S.M.; Bevan, A.P.; Clayton, S.; Corpas, M.; Rajan, D.; Van Vooren, S.; Moreau, Y.; Pettett, R.M.; Carter, N.P. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 2009, 84, 524–533. [Google Scholar] [CrossRef] [Green Version]
- Philippakis, A.A.; Azzariti, D.R.; Beltran, S.; Brookes, A.J.; Brownstein, C.A.; Brudno, M.; Brunner, H.G.; Buske, O.J.; Carey, K.; Doll, C.; et al. The Matchmaker Exchange: A platform for rare disease gene discovery. Hum. Mutat. 2015, 36, 915–921. [Google Scholar] [CrossRef]
- Boycott, K.M.; Rath, A.; Chong, J.X.; Hartley, T.; Alkuraya, F.S.; Baynam, G.; Brookes, A.J.; Brudno, M.; Carracedo, A.; den Dunnen, J.T.; et al. International Cooperation to Enable the Diagnosis of All Rare Genetic Diseases. Am. J. Hum. Genet. 2017, 100, 695–705. [Google Scholar] [CrossRef] [Green Version]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef]
- Freeman, P.J.; Hart, R.K.; Gretton, L.J.; Brookes, A.J.; Dalgleish, R. VariantValidator: Accurate validation, mapping, and formatting of sequence variation descriptions. Hum. Mutat. 2018, 39, 61–68. [Google Scholar] [CrossRef] [Green Version]
- Corbin, L.J.; Tan, V.Y.; Hughes, D.A.; Wade, K.H.; Paul, D.S.; Tansey, K.E.; Butcher, F.; Dudbridge, F.; Howson, J.M.; Jallow, M.W.; et al. Formalising recall by genotype as an efficient approach to detailed phenotyping and causal inference. Nat. Commun. 2018, 9, 711. [Google Scholar] [CrossRef] [Green Version]
- Koolen, D.A.; Kramer, J.M.; Neveling, K.; Nillesen, W.M.; Moore-Barton, H.L.; Elmslie, F.V.; Toutain, A.; Amiel, J.; Malan, V.; Tsai, A.C.-H.; et al. Mutations in the chromatin modifier gene KANSL1 cause the 17q21.31 microdeletion syndrome. Nat. Genet. 2012, 44, 639–641. [Google Scholar] [CrossRef]
- Potocki, L.; Chen, K.-S.; Park, S.-S.; Osterholm, D.E.; Withers, M.A.; Kimonis, V.; Summers, A.M.; Meschino, W.S.; Anyane-Yeboa, K.; Kashork, C.D.; et al. Molecular mechanism for duplication 17p11.2—The homologous recombination reciprocal of the Smith-Magenis microdeletion. Nat. Genet. 2000, 24, 84–87. [Google Scholar] [CrossRef]
- Potocki, L.; Bi, W.; Treadwell-Deering, D.; Carvalho, C.M.B.; Eifert, A.; Friedman, E.M.; Glaze, D.; Krull, K.; Lee, J.A.; Lewis, R.A.; et al. Characterization of Potocki-Lupski syndrome (dup(17)(p11.2p11.2)) and delineation of a dosage-sensitive critical interval that can convey an autism phenotype. Am. J. Hum. Genet. 2007, 80, 633–649. [Google Scholar] [CrossRef] [Green Version]
- Ghigna, M.-R.; Guignabert, C.; Montani, D.; Girerd, B.; Jaïs, X.; Savale, L.; Hervé, P.; Thomas de Montpréville, V.; Mercier, O.; Sitbon, O.; et al. BMPR2 mutation status influences bronchial vascular changes in pulmonary arterial hypertension. Eur. Respir. J. 2016, 48, 1668–1681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Austin, E.D.; Phillips, J.A.; Cogan, J.D.; Hamid, R.; Yu, C.; Stanton, K.C.; Phillips, C.A.; Wheeler, L.A.; Robbins, I.M.; Newman, J.H.; et al. Truncating and missense BMPR2 mutations differentially affect the severity of heritable pulmonary arterial hypertension. Respir. Res. 2009, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girerd, B.; Coulet, F.; Jaïs, X.; Eyries, M.; Van Der Bruggen, C.; De Man, F.; Houweling, A.; Dorfmüller, P.; Savale, L.; Sitbon, O.; et al. Characteristics of pulmonary arterial hypertension in affected carriers of a mutation located in the cytoplasmic tail of bone morphogenetic protein receptor type 2. Chest 2015, 147, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Ballif, B.C.; Theisen, A.; Rosenfeld, J.A.; Traylor, R.N.; Gastier-Foster, J.; Thrush, D.L.; Astbury, C.; Bartholomew, D.; McBride, K.L.; Pyatt, R.E.; et al. Identification of a recurrent microdeletion at 17q23.1q23.2 flanked by segmental duplications associated with heart defects and limb abnormalities. Am. J. Hum. Genet. 2010, 86, 454–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nimmakayalu, M.; Major, H.; Sheffield, V.; Solomon, D.H.; Smith, R.J.; Patil, S.R.; Shchelochkov, O.A. Microdeletion of 17q22q23.2 encompassing TBX2 and TBX4 in a patient with congenital microcephaly, thyroid duct cyst, sensorineural hearing loss, and pulmonary hypertension. Am. J. Med Genet. Part A 2011, 155, 418–423. [Google Scholar] [CrossRef]
- German, K.; Deutsch, G.H.; Freed, A.S.; Dipple, K.M.; Chabra, S.; Bennett, J.T. Identification of a deletion containing TBX4 in a neonate with acinar dysplasia by rapid exome sequencing. Am. J. Med Genet. Part A 2019, 179, 842–845. [Google Scholar] [CrossRef]
- Suhrie, K.; Pajor, N.M.; Ahlfeld, S.K.; Dawson, D.B.; Dufendach, K.R.; Kitzmiller, J.A.; Leino, D.; Lombardo, R.C.; Smolarek, T.A.; Rathbun, P.A.; et al. Neonatal Lung Disease Associated with TBX4 Mutations. J. Pediatr. 2019, 206, 286–292.e1. [Google Scholar] [CrossRef]
- Iannuzzi, M.C.; Baughman, R.P. Reverse phenotyping in sarcoidosis. Am. J. Respir. Crit. Care Med. 2007, 175, 4–5. [Google Scholar] [CrossRef]
- Stram, M.; Gigliotti, T.; Hartman, D.; Pitkus, A.; Huff, S.M.; Riben, M.; Henricks, W.H.; Farahani, N.; Pantanowitz, L. Logical Observation Identifiers Names and Codes for Laboratorians. Arch. Pathol. Lab. Med. 2020, 144, 229–239. [Google Scholar] [CrossRef] [Green Version]
- Grubert, F.; Zaugg, J.B.; Kasowski, M.; Ursu, O.; Spacek, D.V.; Martin, A.R.; Greenside, P.; Srivas, R.; Phanstiel, D.H.; Pekowska, A.; et al. Genetic Control of Chromatin States in Humans Involves Local and Distal Chromosomal Interactions. Cell 2015, 162, 1051–1065. [Google Scholar] [CrossRef] [Green Version]
- Perenthaler, E.; Yousefi, S.; Niggl, E.; Barakat, T.S. Beyond the Exome: The Non-coding Genome and Enhancers in Neurodevelopmental Disorders and Malformations of Cortical Development. Front. Cell. Neurosci. 2019, 13, 352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes-Palomares, A.; Gu, M.; Grubert, F.; Berest, I.; Sa, S.; Kasowski, M.; Arnold, C.; Shuai, M.; Srivas, R.; Miao, S.; et al. Remodeling of active endothelial enhancers is associated with aberrant gene-regulatory networks in pulmonary arterial hypertension. Nat. Commun. 2020, 11, 1673. [Google Scholar] [CrossRef] [PubMed]
- Machado, R.D.; James, V.; Southwood, M.; Harrison, R.E.; Atkinson, C.; Stewart, S.; Morrell, N.W.; Trembath, R.C.; Aldred, M.A. Investigation of second genetic hits at the BMPR2 locus as a modulator of disease progression in familial pulmonary arterial hypertension. Circulation 2005, 111, 607–613. [Google Scholar] [CrossRef] [Green Version]
- Aldred, M.A.; Comhair, S.A.; Varella-Garcia, M.; Asosingh, K.; Xu, W.; Noon, G.P.; Thistlethwaite, P.A.; Tuder, R.M.; Erzurum, S.C.; Geraci, M.W.; et al. Somatic Chromosome Abnormalities in the Lungs of Patients with Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2010, 182, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Drake, K.M.; Comhair, S.A.; Erzurum, S.C.; Tuder, R.M.; Aldred, M.A. Endothelial chromosome 13 deletion in congenital heart disease-associated pulmonary arterial hypertension dysregulates SMAD9 signaling. Am. J. Respir. Crit. Care Med. 2015, 191, 850–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Best, D.H.; Vaughn, C.; McDonald, J.; Damjanovich, K.; Runo, J.R.; Chibuk, J.M.; Bayrak-Toydemir, P. Mosaic ACVRL1 and ENG mutations in hereditary haemorrhagic telangiectasia patients. J. Med Genet. 2011, 48, 358–360. [Google Scholar] [CrossRef] [PubMed]
- Eyries, M.; Coulet, F.; Girerd, B.; Montani, D.; Humbert, M.; Lacombe, P.; Chinet, T.; Gouya, L.; Roume, J.; Axford, M.M.; et al. ACVRL1 germinal mosaic with two mutant alleles in hereditary hemorrhagic telangiectasia associated with pulmonary arterial hypertension. Clin. Genet. 2012, 82, 173–179. [Google Scholar] [CrossRef]
- McDonald, J.; Gedge, F.; Burdette, A.; Carlisle, J.; Bukjiok, C.J.; Fox, M.; Bayrak-Toydemir, P. Multiple sequence variants in hereditary hemorrhagic telangiectasia cases: Illustration of complexity in molecular diagnostic interpretation. J. Mol. Diagn. 2009, 11, 569–575. [Google Scholar] [CrossRef] [Green Version]
- Culley, M.K.; Chan, S.Y. Mitochondrial metabolism in pulmonary hypertension: Beyond mountains there are mountains. J. Clin. Investig. 2018, 128, 3704–3715. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, C.J.; Ghataorhe, P.; Wharton, J.; Rue-Albrecht, K.C.; Hadinnapola, C.; Watson, G.; Bleda, M.; Haimel, M.; Coghlan, G.; Corris, P.A.; et al. Plasma Metabolomics Implicates Modified Transfer RNAs and Altered Bioenergetics in the Outcomes of Pulmonary Arterial Hypertension. Circulation 2017, 135, 460–475. [Google Scholar] [CrossRef] [PubMed]
- Farha, S.; Hu, B.; Comhair, S.; Zein, J.; Dweik, R.; Erzurum, S.C.; Aldred, M.A. Mitochondrial Haplogroups and Risk of Pulmonary Arterial Hypertension. PLoS ONE 2016, 11, e0156042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bris, C.; Goudenege, D.; Desquiret-Dumas, V.; Charif, M.; Colin, E.; Bonneau, D.; Amati-Bonneau, P.; Lenaers, G.; Reynier, P.; Procaccio, V. Bioinformatics Tools and Databases to Assess the Pathogenicity of Mitochondrial DNA Variants in the Field of Next Generation Sequencing. Front. Genet. 2018, 9, 632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, J.T.; Pai, A.A.; Pickrell, J.K.; Gaffney, D.J.; Pique-Regi, R.; Degner, J.F.; Gilad, Y.; Pritchard, J.K. DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biol. 2011, 12, R10. [Google Scholar] [CrossRef] [Green Version]
- Cheung, W.A.; Shao, X.; Morin, A.; Siroux, V.; Kwan, T.; Ge, B.; Aïssi, D.; Chen, L.; Vasquez, L.; Allum, F.; et al. Correction to: Functional variation in allelic methylomes underscores a strong genetic contribution and reveals novel epigenetic alterations in the human epigenome. Genome Biol. 2019, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archer, S.L.; Marsboom, G.; Kim, G.H.; Zhang, H.J.; Toth, P.T.; Svensson, E.C.; Dyck, J.R.B.; Gomberg-Maitland, M.; Thébaud, B.; Husain, A.N.; et al. Epigenetic Attenuation of Mitochondrial Superoxide Dismutase 2 in Pulmonary Arterial Hypertension. Circulation 2010, 121, 2661–2671. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, S.; Michelakis, E.D.; Porter, C.J.; Andrade-Navarro, M.A.; Thébaud, B.; Bonnet, S.; Haromy, A.; Harry, G.; Moudgil, R.; Sean McMurtry, M.; et al. An Abnormal Mitochondrial–Hypoxia Inducible Factor-1α–Kv Channel Pathway Disrupts Oxygen Sensing and Triggers Pulmonary Arterial Hypertension in Fawn Hooded Rats. Circulation 2006, 113, 2630–2641. [Google Scholar] [CrossRef] [Green Version]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Chen, C.-N.; Hajji, N.; Oliver, E.; Cotroneo, E.; Wharton, J.; Wang, D.; Li, M.; McKinsey, T.A.; Stenmark, K.R.; et al. Histone deacetylation inhibition in pulmonary hypertension: Therapeutic potential of valproic acid and suberoylanilide hydroxamic acid. Circulation 2012, 126, 455–467. [Google Scholar] [CrossRef]
- Hon, C.-C.; Ramilowski, J.A.; Harshbarger, J.; Bertin, N.; Rackham, O.J.L.; Gough, J.; Denisenko, E.; Schmeier, S.; Poulsen, T.M.; Severin, J.; et al. An atlas of human long non-coding RNAs with accurate 5’ ends. Nature 2017, 543, 199–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- St Laurent, G.; Wahlestedt, C.; Kapranov, P. The Landscape of long noncoding RNA classification. Trends Genet. 2015, 31, 239–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, H.; Xu, X.; Yan, C.; Shi, Y.; Hu, Y.; Dong, L.; Ying, S.; Ying, K.; Zhang, R. LncRNA H19 promotes the proliferation of pulmonary artery smooth muscle cells through AT1R via sponging let-7b in monocrotaline-induced pulmonary arterial hypertension. Respir. Res. 2018, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jandl, K.; Thekkekara Puthenparampil, H.; Marsh, L.M.; Hoffmann, J.; Wilhelm, J.; Veith, C.; Sinn, K.; Klepetko, W.; Olschewski, H.; Olschewski, A.; et al. Long non-coding RNAs influence the transcriptome in pulmonary arterial hypertension: The role of PAXIP1-AS1. J. Pathol. 2019, 247, 357–370. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Xu, H.; Wu, B.; Jiang, S.; Pan, H.; Wang, R.; Chen, J. Long non‑coding RNA MALAT1 sponges miR‑124‑3p.1/KLF5 to promote pulmonary vascular remodeling and cell cycle progression of pulmonary artery hypertension. Int. J. Mol. Med. 2019, 44, 871–884. [Google Scholar] [CrossRef] [Green Version]
- Leung, A.; Trac, C.; Jin, W.; Lanting, L.; Akbany, A.; Sætrom, P.; Schones, D.E.; Natarajan, R. Novel long noncoding RNAs are regulated by angiotensin II in vascular smooth muscle cells. Circ. Res. 2013, 113, 266–278. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Liang, H.; Shen, L.; Guan, Z.; Meng, X. LncRNA Tug1 involves in the pulmonary vascular remodeling in mice with hypoxic pulmonary hypertension via the microRNA-374c-mediated Foxc1. Life Sci. 2019, 237, 116769. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, Y.; Yan, L.; Wang, S.; Zhang, M.; Ma, C.; Zheng, X.; Chen, H.; Zhu, D. Long noncoding RNA Hoxaas3 contributes to hypoxia-induced pulmonary artery smooth muscle cell proliferation. Cardiovasc. Res. 2019, 115, 647–657. [Google Scholar] [CrossRef]
- Xing, Y.; Zheng, X.; Fu, Y.; Qi, J.; Li, M.; Ma, M.; Wang, S.; Li, S.; Zhu, D. Long Noncoding RNA-Maternally Expressed Gene 3 Contributes to Hypoxic Pulmonary Hypertension. Mol. Ther. 2019, 27, 2166–2181. [Google Scholar] [CrossRef]
- Zhu, T.-T.; Sun, R.-L.; Yin, Y.-L.; Quan, J.-P.; Song, P.; Xu, J.; Zhang, M.-X.; Li, P. Long noncoding RNA UCA1 promotes the proliferation of hypoxic human pulmonary artery smooth muscle cells. Pflugers Arch. 2019, 471, 347–355. [Google Scholar] [CrossRef]
- Sun, Z.; Nie, X.; Sun, S.; Dong, S.; Yuan, C.; Li, Y.; Xiao, B.; Jie, D.; Liu, Y. Long Non-Coding RNA MEG3 Downregulation Triggers Human Pulmonary Artery Smooth Muscle Cell Proliferation and Migration via the p53 Signaling Pathway. Cell. Physiol. Biochem. 2017, 42, 2569–2581. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Gong, Y.; Yan, G.; Wang, D.; Qiao, Y.; Wang, Q.; Liu, B.; Hou, J.; Li, R.; Tang, C. Down-regulation of lncRNA MEG3 promotes hypoxia-induced human pulmonary artery smooth muscle cell proliferation and migration via repressing PTEN by sponging miR-21. Biochem. Biophys. Res. Commun. 2018, 495, 2125–2132. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Chen, Z.; Chen, Y.; Lv, H.; Lu, H.; Yan, F.; Li, L.; Zhang, W.; Shi, J. Long non-coding RNA CASC2 suppresses pulmonary artery smooth muscle cell proliferation and phenotypic switch in hypoxia-induced pulmonary hypertension. Respir. Res. 2019, 20, 53. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Guo, J.; Cui, X.; Dai, Y.; Tang, Z.; Qu, J.; Usha Raj, J.; Hu, Q.; Gou, D. The Long Noncoding RNA LnRPT Is Regulated by PDGF-BB and Modulates the Proliferation of Pulmonary Artery Smooth Muscle Cells. Am. J. Respir. Cell Mol. Biol. 2018, 58, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Zhang, Y.; Tang, Y.; Li, Q. MALAT1 Modulates TGF-β1-Induced Endothelial-to-Mesenchymal Transition through Downregulation of miR-145. Cell. Physiol. Biochem. 2017, 42, 357–372. [Google Scholar] [CrossRef]
- Neumann, P.; Jaé, N.; Knau, A.; Glaser, S.F.; Fouani, Y.; Rossbach, O.; Krüger, M.; John, D.; Bindereif, A.; Grote, P.; et al. The lncRNA GATA6-AS epigenetically regulates endothelial gene expression via interaction with LOXL2. Nat. Commun. 2018, 9, 237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.; Louie, J.; Weisman, A.; Sheu-Gruttadauria, J.; Davis-Dusenbery, B.N.; Lagna, G.; Hata, A. Inhibition of microRNA-302 (miR-302) by bone morphogenetic protein 4 (BMP4) facilitates the BMP signaling pathway. J. Biol. Chem. 2012, 287, 38656–38664. [Google Scholar] [CrossRef] [Green Version]
- Pullamsetti, S.S.; Doebele, C.; Fischer, A.; Savai, R.; Kojonazarov, B.; Dahal, B.K.; Ghofrani, H.A.; Weissmann, N.; Grimminger, F.; Bonauer, A.; et al. Inhibition of microRNA-17 improves lung and heart function in experimental pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2012, 185, 409–419. [Google Scholar] [CrossRef]
- Brock, M.; Samillan, V.J.; Trenkmann, M.; Schwarzwald, C.; Ulrich, S.; Gay, R.E.; Gassmann, M.; Ostergaard, L.; Gay, S.; Speich, R.; et al. AntagomiR directed against miR-20a restores functional BMPR2 signalling and prevents vascular remodelling in hypoxia-induced pulmonary hypertension. Eur. Heart J. 2014, 35, 3203–3211. [Google Scholar] [CrossRef] [Green Version]
- Caruso, P.; MacLean, M.R.; Khanin, R.; McClure, J.; Soon, E.; Southgate, M.; MacDonald, R.A.; Greig, J.A.; Robertson, K.E.; Masson, R.; et al. Dynamic Changes in Lung MicroRNA Profiles During the Development of Pulmonary Hypertension due to Chronic Hypoxia and Monocrotaline. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 716–723. [Google Scholar] [CrossRef] [Green Version]
- Uchida, S.; Dimmeler, S. Long Noncoding RNAs in Cardiovascular Diseases. Circ. Res. 2015, 116, 737–750. [Google Scholar] [CrossRef] [Green Version]
- Thum, T.; Condorelli, G. Long Noncoding RNAs and MicroRNAs in Cardiovascular Pathophysiology. Circ. Res. 2015, 116, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Chinnappan, M.; Mohan, A.; Agarwal, S.; Dalvi, P.; Dhillon, N.K. Network of MicroRNAs Mediate Translational Repression of Bone Morphogenetic Protein Receptor-2: Involvement in HIV-Associated Pulmonary Vascular Remodeling. J. Am. Heart Assoc. 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Durrington, H.J.; Upton, P.D.; Hoer, S.; Boname, J.; Dunmore, B.J.; Yang, J.; Crilley, T.K.; Butler, L.M.; Blackbourn, D.J.; Nash, G.B.; et al. Identification of a lysosomal pathway regulating degradation of the bone morphogenetic protein receptor type II. J. Biol. Chem. 2010, 285, 37641–37649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalvi, P.; O’Brien-Ladner, A.; Dhillon, N.K. Downregulation of bone morphogenetic protein receptor axis during HIV-1 and cocaine-mediated pulmonary smooth muscle hyperplasia: Implications for HIV-related pulmonary arterial hypertension. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2585–2595. [Google Scholar] [CrossRef] [Green Version]
- Mansoor, J.K.; Schelegle, E.S.; Davis, C.E.; Walby, W.F.; Zhao, W.; Aksenov, A.A.; Pasamontes, A.; Figueroa, J.; Allen, R. Analysis of volatile compounds in exhaled breath condensate in patients with severe pulmonary arterial hypertension. PLoS ONE 2014, 9, e95331. [Google Scholar] [CrossRef] [PubMed]
- Hakim, M.; Broza, Y.Y.; Barash, O.; Peled, N.; Phillips, M.; Amann, A.; Haick, H. Volatile organic compounds of lung cancer and possible biochemical pathways. Chem. Rev. 2012, 112, 5949–5966. [Google Scholar] [CrossRef] [PubMed]
- Montani, D.; Lau, E.M.; Descatha, A.; Jaïs, X.; Savale, L.; Andujar, P.; Bensefa-Colas, L.; Girerd, B.; Zendah, I.; Le Pavec, J.; et al. Occupational exposure to organic solvents: A risk factor for pulmonary veno-occlusive disease. Eur. Respir. J. 2015, 46, 1721–1731. [Google Scholar] [CrossRef] [Green Version]
- Barker, D. Infant Mortality, Childhood Nutrition, and Ischaemic Heart Disease in England and Wales. Lancet 1986, 327, 1077–1081. [Google Scholar] [CrossRef]
- Barker, D.J.P.; Osmond, C.; Winter, P.D.; Margetts, B.; Simmonds, S.J. Weight in Infancy and Death from Ischaemic Heart Disease. Lancet 1989, 334, 577–580. [Google Scholar] [CrossRef]
- Hales, C.N.; Barker, D.J.; Clark, P.M.; Cox, L.J.; Fall, C.; Osmond, C.; Winter, P.D. Fetal and infant growth and impaired glucose tolerance at age 64. BMJ 1991, 303, 1019–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stern, D.A.; Morgan, W.J.; Wright, A.L.; Guerra, S.; Martinez, F.D. Poor airway function in early infancy and lung function by age 22 years: A non-selective longitudinal cohort study. Lancet 2007, 370, 758–764. [Google Scholar] [CrossRef] [Green Version]
- Berry, C.E.; Billheimer, D.; Jenkins, I.C.; Lu, Z.J.; Stern, D.A.; Gerald, L.B.; Carr, T.F.; Guerra, S.; Morgan, W.J.; Wright, A.L.; et al. A Distinct Low Lung Function Trajectory from Childhood to the Fourth Decade of Life. Am. J. Respir. Crit. Care Med. 2016, 194, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Mann, S.L.; Wadsworth, M.E.; Colley, J.R. Accumulation of factors influencing respiratory illness in members of a national birth cohort and their offspring. J. Epidemiol. Community Health 1992, 46, 286–292. [Google Scholar] [CrossRef] [Green Version]
- Grigoriadis, S.; Vonderporten, E.H.; Mamisashvili, L.; Tomlinson, G.; Dennis, C.-L.; Koren, G.; Steiner, M.; Mousmanis, P.; Cheung, A.; Ross, L.E. Prenatal exposure to antidepressants and persistent pulmonary hypertension of the newborn: Systematic review and meta-analysis. BMJ 2014, 348, f6932. [Google Scholar] [CrossRef] [Green Version]
- Marques, F.Z. Missing Heritability of Hypertension and Our Microbiome. Circulation 2018, 138, 1381–1383. [Google Scholar] [CrossRef]
- Ranchoux, B.; Bigorgne, A.; Hautefort, A.; Girerd, B.; Sitbon, O.; Montani, D.; Humbert, M.; Tcherakian, C.; Perros, F. Gut-Lung Connection in Pulmonary Arterial Hypertension. Am. J. Respir. Cell Mol. Biol. 2017, 56, 402–405. [Google Scholar] [CrossRef]
- Sofianopoulou, E.; Kaptoge, S.; Gräf, S.; Hadinnapola, C.; Treacy, C.M.; Church, C.; Coghlan, G.; Gibbs, J.S.R.; Haimel, M.; Howard, L.S.; et al. Traffic exposures, air pollution and outcomes in pulmonary arterial hypertension: A UK cohort study analysis. Eur. Respir. J. 2019, 53. [Google Scholar] [CrossRef]
- Gazzo, A.; Raimondi, D.; Daneels, D.; Moreau, Y.; Smits, G.; Van Dooren, S.; Lenaerts, T. Understanding mutational effects in digenic diseases. Nucleic Acids Res. 2017, 45, e140. [Google Scholar] [CrossRef] [Green Version]
- Eichstaedt, C.A.; Song, J.; Benjamin, N.; Harutyunova, S.; Fischer, C.; Grünig, E.; Hinderhofer, K. EIF2AK4 mutation as “second hit” in hereditary pulmonary arterial hypertension. Respir. Res. 2016, 17, 141. [Google Scholar] [CrossRef] [Green Version]
- Pousada, G.; Baloira, A.; Valverde, D. Pulmonary arterial hypertension associated with several mutations. Eur. Respir. J. 2016, 48, PA2478. [Google Scholar]
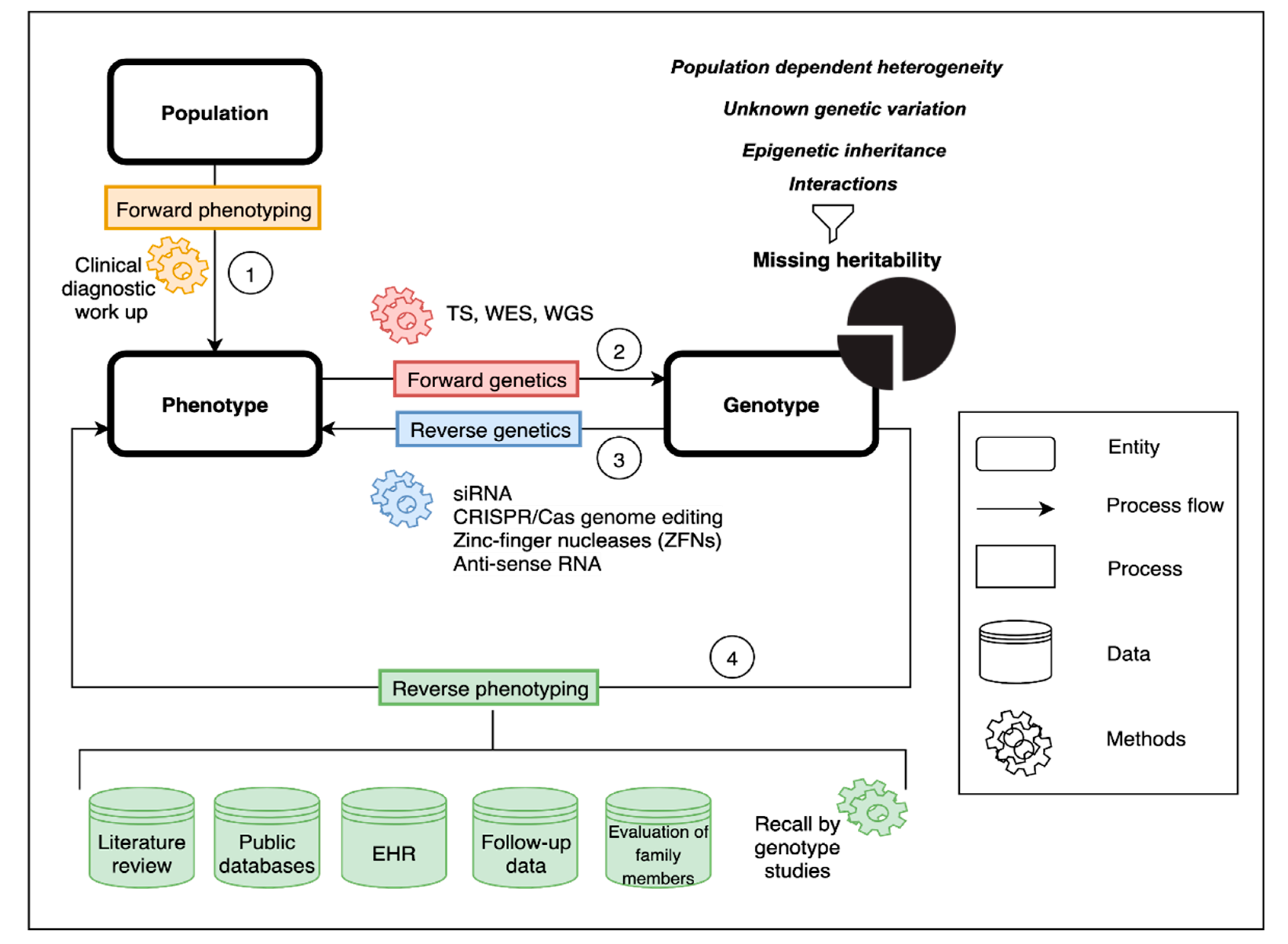

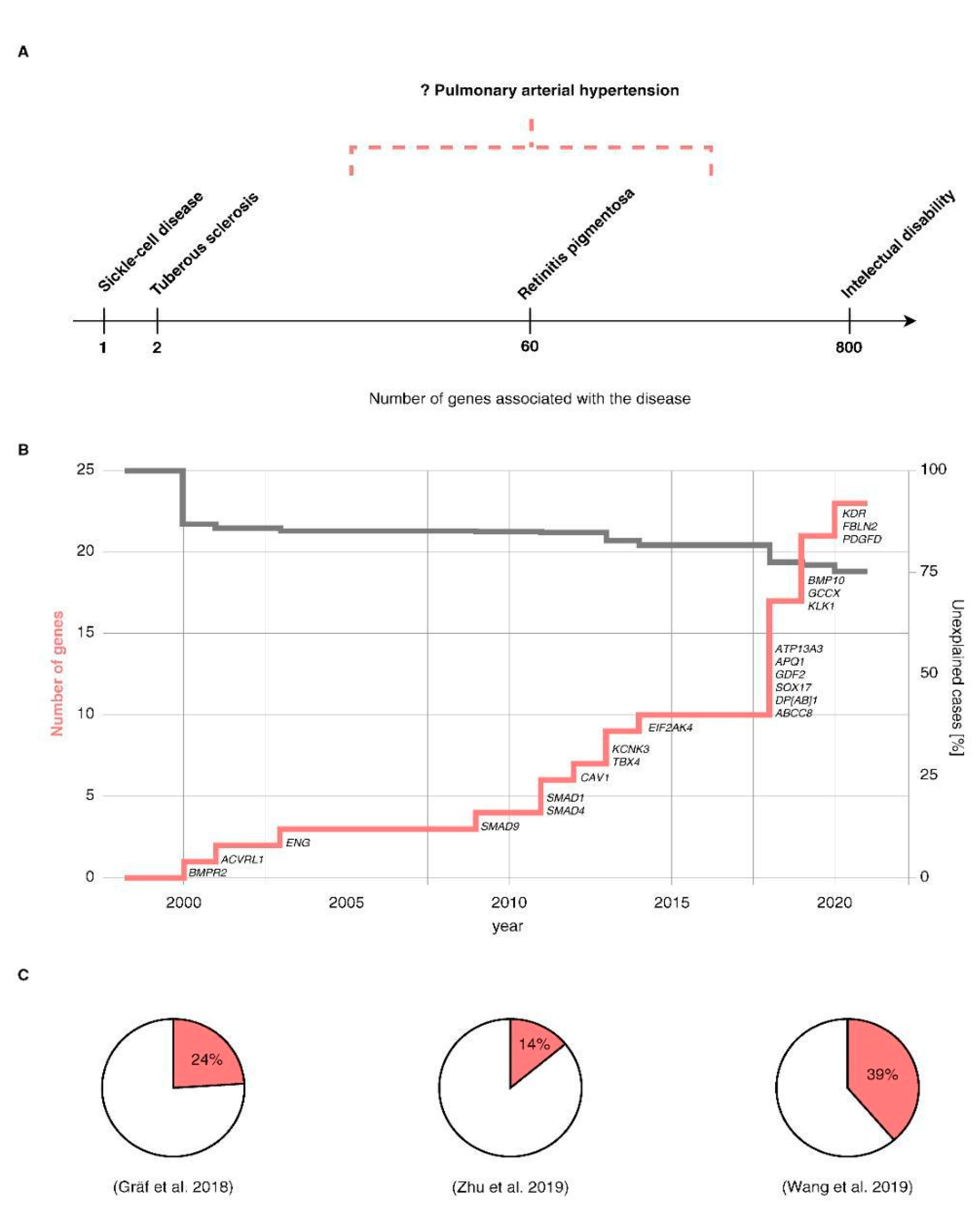
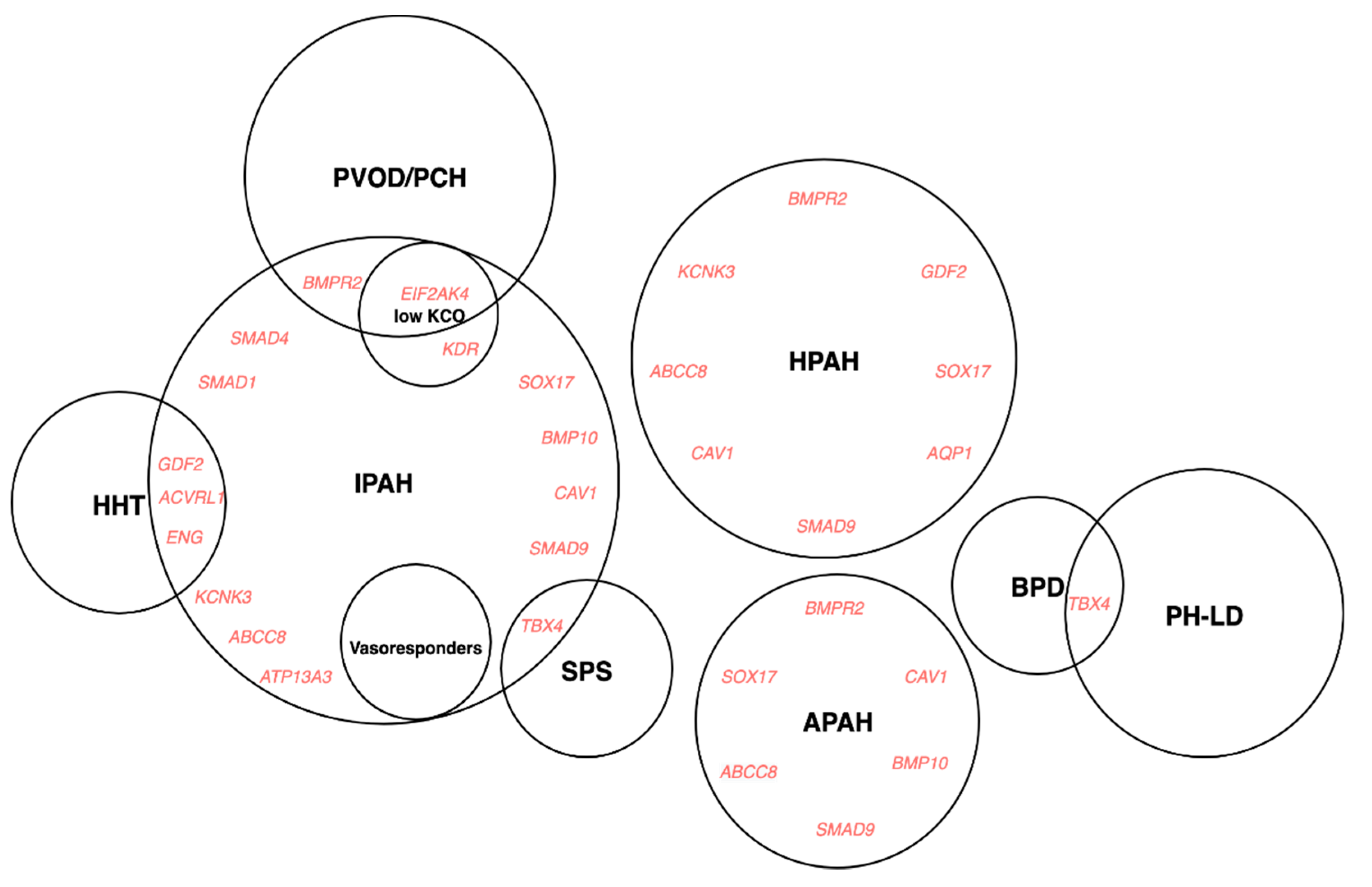

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WSPH Proceedings and ERS/ESC Guidelines | Definition of Group 1 | Comments | Changes to the Classification |
|---|---|---|---|
| 1st WSPH, Geneva, 1973 [12] | No haemodynamic definition mentioned | ||
| 2nd WSPH, Evian, 1998 [13] | No haemodynamic definition mentioned, but RHC recommended for diagnosis | Introduction of the terms primary (PPH) and secondary (related to other conditions) pulmonary hypertension, recognition of familial forms of PH | |
| 3rd WSPH, Venice, 2003 [14] | mPAP(R) > 25 mmHg; mPAP(E) > 30 mmHg; PAWP < 15 mmHg; PVR > 3 WU | Abandonment of the term primary pulmonary hypertension, the introduction of terms idiopathic and familial PAH as well as associated PAH, BMPR2 and ACVRL1 implicated in the pathogenesis of PAH | |
| 4th WSPH, Dana Point, 2008 [5] | mPAP(R) ≥ 25 mmHg; PAWP ≤ 15 mmHg | Exercise-induced PH removed from the definition as although (R) mPAP has been shown to be stable across age groups, (E) mPAP increases with age hence based on the available data it was not possible to define a cutoff | Introduction of the terms idiopathic (no family history, no precipitating risk factor) and hereditary (encompassing familial cases with or without identified germline mutations and PAH). Inclusion of PH associated with Schistosomiasis and PH associated with chronic hemolytic anaemia to Group 1 |
| ERS/ESC Guidelines, 2009 [15] | mPAP(R) ≥ 25 mmHg; PAWP ≤ 15 mmHg; CO normal or reduced | No definition of PH on exercise | |
| 5th WSPH, Nice, 2013 [16] | mPAP(R) ≥ 25 mmHg; PAWP ≤ 15 mmHg; PVR > 3 WU | Introduction of PVR to the definition, a recommendation to report PVR in WU; fluid challenge may be helpful to unmask occult LV diastolic dysfunction | SMAD9, CAV1 and KCNK3 included as risk genes for HPAH |
| ERS/ESC Guidelines, 2015 [17] | mPAP(R) ≥ 25 mmHg; PAWP ≤ 15 mmHg | The clinical significance of a mPAP between 21 and 24 mmHg is unclear | Group 1′ PVOD/PCH has been expanded and includes idiopathic, heritable, drug-, toxin- and radiation-induced and associated forms; PPHN includes a heterogeneous group of conditions that may differ from classical PAH. As a consequence, PPHN has been sub-categorised as group I′′. |
| 6th WSPH, Nice, 2018 [18] | mPAP(R) ≥ 20 mmHg; PAWP ≤ 15 mmHg; PVR ≥ 3 WU | PVR ≥ 3WU should be used as a diagnostic criterion for all forms of PH | PAH long-term responses to calcium channel blockers established as a subtype of Group 1; PAH with overt features of venous/capillaries (PVOD/PCH) involvement established as a subtype of Group 1 |
| Study (Reference) | Genes | Study Design | Sample | Ethnicity | Method | Reference Genome |
|---|---|---|---|---|---|---|
| Lane et al. 2000, [101] | BMPR2 | Case-level data | Cases: n = 8 PPH kindreds for candidate gene mutational analysis | Not stated | TS | H.sapiens mRNA for BMPR-II: Genbank Z48923 |
| Thomson 2000, [102] | BMPR2 | Case-level data | Cases: n = 50 PPH | Not stated | TS | Not stated |
| Trembath et al. 2001, [103] | ACVRL1 | Case-level data | Cases: 5 kindreds plus 1 individual patient with HHT, including n = 10 cases with PH | Not stated | TS | Not stated |
| Chaouat 2004, [104] | ENG | Case-level data | Case: n=1 HHT, PPH with history of anorexigen use | Not stated | TS | Not stated |
| Harrison et al. 2005, [105] | ACVRL1, ENG | Case-level data | Cases: n = 18 I/APAH | Not stated | TS | Not stated |
| Shintani et al. 2009, [106] | SMAD9 (SMAD8) | Case-level data | Cases: n = 23 IPAH | Japanese | TS | Not stated |
| Nasim et al. 2011, [54] | SMAD1, SMAD4, SMAD9 | Case-level data | Cases: n = 324 IPAH/APAH/CTEPH; Controls: n = 1584 | European & Japanese | TS | Not stated |
| Austin et al. 2012, [74] | CAV1 | Case-level data | Cases: 3-generation family, 6 with PAH; Additional cohort: n = 260 unrelated I/HPAH cases; Controls: n = 1000 | European | WES | GRCh37 |
| Ma et al. 2013, [107] | KCNK3 | Case-level data | Cases: Family in which multiple members had PAH | Not stated | WES | GRCh37 |
| Kerstjens-Frederikse et al. 2013, [108] | TBX4 | Case-level data | Cases: n = 20 childhood-onset I/HPAH; n = 49 adult-onset I/HPAH; n = 23 SPS | Not stated | TS | Not stated |
| Eyries et al. 2014, [109] | EIF2AK4 | Case-level data | Cases: n = 13 PVOD families | Not stated | WES | GRCh37 |
| Best et al. 2017, [110] | EIF2AK4 | Case-level data | Cases: n = 81 I/HPAH | Not stated | TS | Not stated |
| Hadinnapola et al. 2017, [111] | EIF2AK4 | Case-control data | Cases: n = 880 I/FPAH, PVOD/PCH; Controls: n = 7134 non-PAH controls and their relatives recruited to NBR | European: 84.6% | WGS | GRCh37 |
| Gräf et al. 2018, [24] | GDF2, SOX17, ATP13A3, AQP1 | Case-control data | Cases: n = 1048 I/F/PAH, PVOD/PCH; Controls: n = 7979 non-PAH controls and their relatives recruited to NBR | European: 84.6% | WGS | GRCh37 |
| Zhu et al. 2018, [75] | SOX17 | Case-control data | Cases: n = 256 I/FPAH-CHD; Additional cohort: n = 413 I/FPAH screened for rare variants in SOX17; Controls: n = 7509 gnomAD | Not stated | WES | GRCh37 |
| Hiraide et al. 2018, [112] | SOX17 | Case-level data | Cases: n = 12 IPAH and 12 family members; Additional cohort: n = 128 I/HPAH screened for SOX17 mutations | Japanese: 100% | WES | Not stated |
| Bohnen et al. 2018, [113] | ABCC8 | Case-control data | Cases: n = 913; Controls: n = 33,369 European adults from ExAC & n = 49,630 Europeans from the Regeneron-Geisinger DiscovEHR study | Not stated | WES, WGS | GRCh37 |
| Wang et al. 2019, [26] | GDF2 | Case-control data | Cases: n = 331 IPAH; Controls: n = 10,508 from available reference data sets | East Asian: 100% | WES, WGS | GRCh37 |
| Eyries et al. 2019, [114] | BMP10 | Case-level data | Cases: n = 268 I/HPAH, PVOD/PCH | European: > 90% | TS | GRCh37 |
| Hodgson et al. 2020, [115] | BMP10 | Case-level data | Cases: n = 1048 I/FPAH, PVOD/PCH | European: 84.6% | WGS | GRCh37 |
| Zhu et al. 2019, [25] | KLK1, GCCX | Case-control data | Cases: n = 2572 Group 1 PAH; Controls: n = 12,771 | European: 72% | WES | GRCh38 |
| Rhodes et al. 2019, [116] | HLA-DPA1/DPB1, SOX17 enhancer | Case-control data | Cases: n = 2085 cases; Controls: n = 9659 | European: 100% | WGS | GRCh37 |
| Swietlik et al. 2019, [30] | KDR | Case-control data | Cases: n = 1122 PAH; Controls: n = 11,889 non-PAH NBR | European: 84% | WGS | GRCh37 |
| Eyries et al. 2020, [117] | KDR | Case-level data | Cases: n = 311 unrelated PAH | Not stated | TS | Not stated |
| Potus et al. 2020, [55] | TET2 | Case-control data | Cases: n = 2572; Controls: n = 7509 non-Finnish European subjects from gnomAD | European: 72% | WES | GRCh38 |
| Zhu et al. 2020, [118] | FBLN2, PDGFD | Case-control data | Cases: n = 4175; Controls: n = 18,819 from SPARK and gnomAD cohorts | European: 54.5% | WES | GRCh38 |
| Forward Genetics | Reverse Genetics | |||||
|---|---|---|---|---|---|---|
| Gene | Case-Control Data | Case-Level Data | Segregation Data | Functional Aberration | Disease Model | Rescue |
| MOI: Autosomal Dominant | ||||||
| BMPR2 | (+) [24,25] | (+) [101,114] | (+) [101,122] | (+) [123,124,125,126,127,128,129,130] | Animal: (+) [123,126,128,129,130,131] Cell culture: (+) [124,129,130] | (+) [128,129,130,131] (+) |
| ACVRL1 | (+) [24] | (+) [25,103,105,114] | (+) [103] | |||
| ENG | (−) [24,25] | (+) [24,75,104] | (+) [104] | Animal: (−) [132] | ||
| SMAD9 | (−) [24,25] | (+) [24,25,75,106,114] | (+) [54,106,133] | Animal: (+) [134] Cell culture: (+) [54,106,133] | (+) [133,135] | |
| SMAD1 | (−) [24,25] | (+) [54], (−) [24,25] | (+) [54,136] | Animal: (+) [136] Cell culture: (+) [54] | ||
| SMAD4 | (−) [24,25] | (+) [25,54], (−) [24] | (+) [54], (−) [54] | Cell culture: (+) [54] | ||
| CAV1 | (+) [74] | (+) [25,75], (−) [24] | (+) [137] | Animal: (+) [138,139] Cell culture: (+) [137] | (+) [137] | |
| TBX4 | (+) [24,140] | (+) [75,108,114,141,142] | (+) [143,144] | |||
| KCNK3 | (+) [24,25,107] | (+) [107] | (+) [107,145] | Animal: (+) [146] Cell culture: (+) [107,145] | (+) [107] | |
| ATP13A3 | (+) [24] | (+) [147,148] | Animal: (+) [147,148] | |||
| AQP1 | (+) [24] | (+) [149,150] (−) [149] | Animal: (+) [149,151] Cell culture: (+) [150] | (+) [149] | ||
| GDF2 | (+) [24,25,26] | (+) [114] | (+) [115] | (+) [26,115] | Animal: (−) [152] Cell culture: (+) [26,115] | |
| SOX17 | (+) [24,75] | (+) [112] | (+) [153,154] | Animal: (+) [153] Cell culture: (+) [154] | ||
| ABCC8 | (+) [113] | (+) [113] | (+) [113] | Cell culture: (+) [113] | (+) [113] | |
| BMP10 | (+) [114,115] | |||||
| GGCX | (+) [25] | |||||
| KLK1 | (+) [25] | |||||
| KDR | (+) [30] | (+) [30,117] | (+) [155] | Animal: (+) [155] | ||
| FBLN2 | (+) [118] | |||||
| PDGFD | (+) [118] | |||||
| TET2 | (+) [55] | (+) [55] | Animal: (+) [55] | (+) [55] | ||
| BMPR1A | (+) [25,75] | (+) [156,157,158,159] | Animal: (+) [156,157,159] | (+) [156] | ||
| BMPR1B | (+) [24,25,75] | (+) [160] | Cell culture: (+) [160] | |||
| TOPBP1 | (+) [24] | |||||
| THBS1 | (+) [161] | Cell culture: (+) [161] | (+) [161] | |||
| KCNA5 | (+) [162] | (+) [163] | Cell culture: (+) [163] | (+) [163] | ||
| MOI: Autosomal Recessive | ||||||
| EIF2AK4 | (+) [111] | (+) [114] | (+) [109,110] | (+) [109] | Animal: (+) [164] | |
| Common Variation | ||||||
| enhancer near SOX17 | (+) [116] | (+) [116] | (+) [116] | |||
| locus within HLA-DPA1/DPB1 | (+) [116] | |||||
| CBLN2 | (+) [165] (−) [116] | |||||
| SIRT3 | (+) [166] | (+) [166] | (+) [166] | Animal: (+) [166,167] | ||
| UCP2 | (+) [168] | Animal: (+) [167,169] | ||||
| EDN1 | (+) [170] | (+) [171] | ||||
| AGTR1 | (+) [34] | |||||
| TOPBP1 | (+) [172] | (+) [172] | Cell culture: (+) [172] | |||
| Endostatin | (+) [173] | |||||
| TRPC6 | (+) [174] | Cell culture: (+) [174] | ||||
| Gene | Condition | Study Design | Data Sources | Results | References |
|---|---|---|---|---|---|
| HLA-DRB1*0301/DQB1*0201 | Sarcoidosis | Mix of retrospective and prospective cases with acute onset sarcoidosis | Health records | Patients with acute onset sarcoidosis carrying HLA-DRB1*0301/DQB1*0201 genotype have good prognosis, manifestations of Lofgren’s syndrome differ between man and woman. | [278,302] |
| BMPR2 | PAH | Retrospective analysis of 169 PAH patients | WES & Health records | Patients with missense mutations that escape nonsense-mediated decay have more severe disease than those with truncating mutations. | [296] |
| BMPR2 | PAH | Retrospective analysis of 171 patients | Missense variants in the cytoplasmic tail appear to confer less severe phenotype than other BMPR2 variants with a later age of onset, milder haemodynamics, and more vasoreactivity. | [297] | |
| BMPR2 | PAH | A large individual participant data meta-analysis | Literature review | Patients harbouring deleterious BMPR2 mutations have earlier disease onset, worse haemodynamics, are less likely to respond to NO challenge and have lower survival when compared to those without BMPR2 mutations. | [180] |
| BMPR2 | PAH | Retrospective analysis of 44 PAH patients who underwent lung transplantation between 2005 and 2014 | Histology, immunohistochemistry, morphometry of explanted lungs; French registry database | BMPR2 mutation carriers are more prone to haemoptysis; haemoptysis is closely correlated to bronchial arterial remodelling and angiogenesis; pronounced changes in the systemic vasculature correlate with increased pulmonary venous remodelling, creating a distinctive profile in PAH patients harbouring a BMPR2 mutation. | [295] |
| multiple genes | Nephrotic syndrome | A retrospective analysis of all patients diagnosed with nephrotic syndrome between 2000 and 2018 | WES & personalised diagnostic workflow | Reverse phenotyping after WES increased the diagnostic accuracy in patients referred with the diagnosis of steroid-resistant nephrotic syndrome. | [279] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Swietlik, E.M.; Prapa, M.; Martin, J.M.; Pandya, D.; Auckland, K.; Morrell, N.W.; Gräf, S. ‘There and Back Again’—Forward Genetics and Reverse Phenotyping in Pulmonary Arterial Hypertension. Genes 2020, 11, 1408. https://doi.org/10.3390/genes11121408
Swietlik EM, Prapa M, Martin JM, Pandya D, Auckland K, Morrell NW, Gräf S. ‘There and Back Again’—Forward Genetics and Reverse Phenotyping in Pulmonary Arterial Hypertension. Genes. 2020; 11(12):1408. https://doi.org/10.3390/genes11121408
Chicago/Turabian StyleSwietlik, Emilia M., Matina Prapa, Jennifer M. Martin, Divya Pandya, Kathryn Auckland, Nicholas W. Morrell, and Stefan Gräf. 2020. "‘There and Back Again’—Forward Genetics and Reverse Phenotyping in Pulmonary Arterial Hypertension" Genes 11, no. 12: 1408. https://doi.org/10.3390/genes11121408
APA StyleSwietlik, E. M., Prapa, M., Martin, J. M., Pandya, D., Auckland, K., Morrell, N. W., & Gräf, S. (2020). ‘There and Back Again’—Forward Genetics and Reverse Phenotyping in Pulmonary Arterial Hypertension. Genes, 11(12), 1408. https://doi.org/10.3390/genes11121408