Genotypes Predispose Phenotypes—Clinical Features and Genetic Spectrum of ABCA4-Associated Retinal Dystrophies

 , and
, and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Subjects and Clinical Evaluation
2.2. Next-Generation Sequencing
2.3. Classification System
2.4. Lesion Area Measurement with Fundus Autofluorescence Examination (FAF)
2.5. Statistical Analysis
3. Results
3.1. Demographics
3.2. Clinical Presentations of ABCA4-Associated Stargardt Disease 1
3.3. Foveal Sparing Phenotype of Stargardt Disease 1
3.4. Clinical Presentations of ABCA4-Associated Retinitis Pigmentosa
3.5. Cone-Rod Dystrophy
3.6. Genetic Spectrum of ABCA4-Associated Retinal Dystrophies in Taiwanese
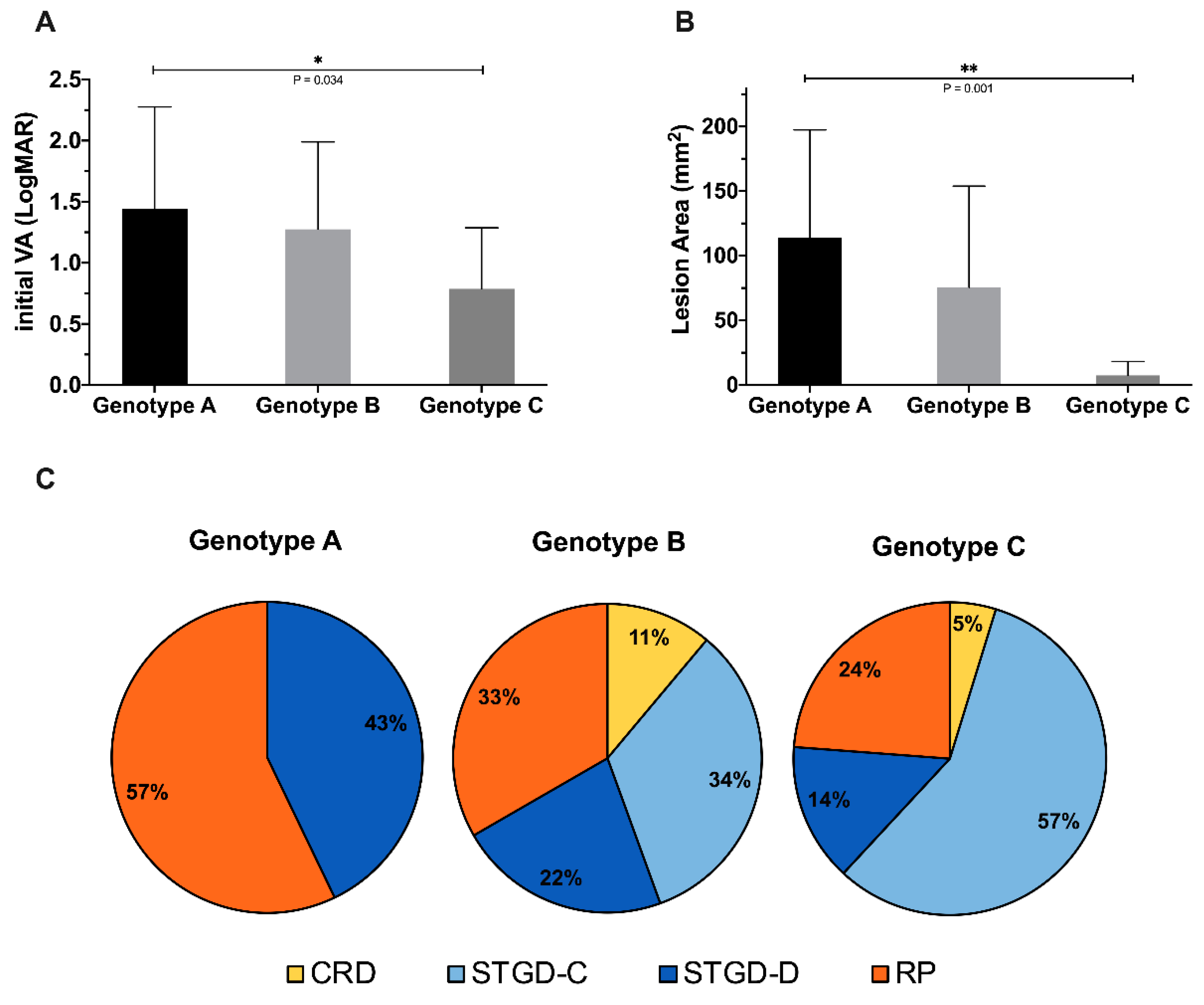
3.7. Genotype–Phenotype Correlation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Trezza, A.; Bernini, A.; Langella, A.; Ascher, D.B.; Pires, D.E.V.; Sodi, A.; Passerini, I.; Pelo, E.; Rizzo, S.; Niccolai, N.; et al. A Computational Approach From Gene to Structure Analysis of the Human ABCA4 Transporter Involved in Genetic Retinal Diseases. Investig. Opthalmol. Vis. Sci. 2017, 58, 5320–5328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molday, R.S.; Zhong, M.; Quazi, F. The role of the photoreceptor ABC transporter ABCA4 in lipid transport and Stargardt macular degeneration. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2009, 1791, 573–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsybovsky, Y.; Molday, R.S.; Palczewski, K. The ATP-Binding Cassette Transporter ABCA4: Structural and Functional Properties and Role in Retinal Disease. Adv. Exp. Med. Biol. 2010, 703, 105–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.S.; Joo, K.; Seong, M.-W.; Kim, M.J.; Park, K.H.; Park, S.S.; Woo, S.J. Genetic Mutation Profiles in Korean Patients with Inherited Retinal Diseases. J. Korean Med. Sci. 2019, 34, e161. [Google Scholar] [CrossRef]
- Allikmets, R.; Singh, N.; Sun, H.; Shroyer, N.F.; Hutchinson, A.; Chidambaram, A.; Gerrard, B.; Baird, L.; Stauffer, D.; Peiffer, A.; et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat. Genet. 1997, 15, 236–246. [Google Scholar] [CrossRef]
- Tanna, P.; Strauss, R.W.; Fujinami, K.; Michaelides, M. Stargardt disease: Clinical features, molecular genetics, animal models and therapeutic options. Br. J. Ophthalmol. 2017, 101, 25–30. [Google Scholar] [CrossRef] [Green Version]
- Mullins, R.F.; Kuehn, M.H.; Radu, R.A.; Enriquez, G.S.; East, J.S.; Schindler, E.I.; Travis, G.H.; Stone, E.A. Autosomal Recessive Retinitis Pigmentosa Due ToABCA4Mutations: Clinical, Pathologic, and Molecular Characterization. Investig. Opthalmol. Vis. Sci. 2012, 53, 1883–1894. [Google Scholar] [CrossRef] [Green Version]
- Maugeri, A.; Klevering, B.J.; Rohrschneider, K.; Blankenagel, A.; Brunner, H.G.; Deutman, A.F.; Hoyng, C.B.; Cremers, F.P.M. Mutations in the ABCA4 (ABCR) Gene Are the Major Cause of Autosomal Recessive Cone-Rod Dystrophy. Am. J. Hum. Genet. 2000, 67, 960–966. [Google Scholar] [CrossRef] [Green Version]
- De La Paz, M.A.; Guy, V.K.; Abou-Donia, S.; Heinis, R.; Bracken, B.; Vance, J.M.; Gilbert, J.R.; Gass, J.M.; Haines, J.L.; Pericak-Vance, M.A. Analysis of the stargardt disease gene (ABCR) in age-related macular degeneration. Ophthalmology 1999, 106, 1531–1536. [Google Scholar] [CrossRef]
- Fritsche, L.G.; Fleckenstein, M.; Fiebig, B.S.; Schmitz-Valckenberg, S.; Bindewald-Wittich, A.; Keilhauer, C.N.; Renner, A.B.; Mackensen, F.; Mößner, A.; Pauleikhoff, D.; et al. A Subgroup of Age-Related Macular Degeneration is Associated with Mono-Allelic Sequence Variants in theABCA4Gene. Investig. Opthalmol. Vis. Sci. 2012, 53, 2112–2299. [Google Scholar] [CrossRef] [Green Version]
- Fujinami, K.; Zernant, J.; Chana, R.K.; Wright, G.A.; Tsunoda, K.; Ozawa, Y.; Tsubota, K.; Robson, A.G.; Holder, G.E.; Allikmets, R.; et al. Clinical and Molecular Characteristics of Childhood-Onset Stargardt Disease. Ophthalmology 2015, 122, 326–334. [Google Scholar] [CrossRef] [Green Version]
- Sheffield, V.C.; Stone, E.M. Genomics and the Eye. N. Engl. J. Med. 2011, 364, 1932–1942. [Google Scholar] [CrossRef]
- Lee, W.; Schuerch, K.; Zernant, J.; Collison, F.T.; Bearelly, S.; Fishman, G.A.; Tsang, S.H.; Sparrow, J.R.; Allikmets, R. Genotypic spectrum and phenotype correlations of ABCA4-associated disease in patients of south Asian descent. Eur. J. Hum. Genet. 2017, 25, 735–743. [Google Scholar] [CrossRef]
- Utz, V.M.; Coussa, R.G.; Marino, M.J.; Chappelow, A.V.; Pauer, G.J.; Hagstrom, S.A.; Traboulsi, E.I. Predictors of visual acuity and genotype-phenotype correlates in a cohort of patients with Stargardt disease. Br. J. Ophthalmol. 2014, 98, 513–518. [Google Scholar] [CrossRef]
- Klevering, B.J.; Deutman, A.F.; Maugeri, A.; Cremers, F.P.M.; Hoyng, C.B. The spectrum of retinal phenotypes caused by mutations in the ABCA4 gene. Graefe’s Arch. Clin. Exp. Ophthalmol. 2005, 243, 90–100. [Google Scholar] [CrossRef]
- Yuan, E. The Republic of China Yearbook 2014; Executive Yuan: Taipei, Taiwan, 2014. [Google Scholar]
- Chen, C.-H.; Yang, J.-H.; Chiang, C.W.; Hsiung, C.-N.; Wu, P.-E.; Chang, L.-C.; Chu, H.-W.; Chang, J.; Yuan-Tsong, C.; Yang, S.-L.; et al. Population structure of Han Chinese in the modern Taiwanese population based on 10,000 participants in the Taiwan Biobank project. Hum. Mol. Genet. 2016, 25, 5321–5331. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Kong, X.; Fujinami, K.; Strauss, R.W.; Munoz, B.; West, S.K.; Cideciyan, A.V.; Michaelides, M.; Ahmed, M.; Ervin, A.-M.; Schönbach, E.; et al. Visual Acuity Change Over 24 Months and Its Association With Foveal Phenotype and Genotype in Individuals With Stargardt Disease: ProgStar Study Report No. 10. JAMA Ophthalmol. 2018, 136, 920–928. [Google Scholar] [CrossRef]
- Schmitz-Valckenberg, S.; Brinkmann, C.K.; Alten, F.; Herrmann, P.; Stratmann, N.K.; Göbel, A.P.; Fleckenstein, M.; Diller, M.; Jaffe, G.J.; Holz, F.G. Semiautomated Image Processing Method for Identification and Quantification of Geographic Atrophy in Age-Related Macular Degeneration. Investig. Opthalmol. Vis. Sci. 2011, 52, 7640–7646. [Google Scholar] [CrossRef] [Green Version]
- Strauss, R.W.; Ho, A.; Munoz, B.; Cideciyan, A.V.; Sahel, J.A.; Sunness, J.S.; Birch, D.G.; Bernstein, P.S.; Michaelides, M.; Traboulsi, E.I.; et al. The natural history of the progression of atrophy secondary to Stargardt disease (progstar) studies: Design and baseline characteristics: Progstar report no. 1. Ophthalmology 2016, 123, 817–828. [Google Scholar] [CrossRef]
- Strauss, R.W.; Muñoz, B.; Ho, A.; Jha, A.; Michaelides, M.; Cideciyan, A.V.; Audo, I.; Birch, D.G.; Hariri, A.H.; Nittala, M.G.; et al. Progression of Stargardt Disease as Determined by Fundus Autofluorescence in the Retrospective Progression of Stargardt Disease Study (ProgStar Report No. 9). JAMA Ophthalmol. 2017, 135, 1232–1241. [Google Scholar] [CrossRef]
- Chen, Z.-J.; Lin, K.-H.; Lee, S.-H.; Shen, R.-J.; Feng, Z.-K.; Wang, X.-F.; Ms, X.H.; Huang, Z.-Q.; Jin, Z.-B. Mutation spectrum and genotype-phenotype correlation of inherited retinal dystrophy in Taiwan. Clin. Exp. Ophthalmol. 2020, 48, 486–499. [Google Scholar] [CrossRef]
- Huang, X.-F.; Huang, F.; Wu, K.-C.; Wu, J.; Chen, J.; Pang, C.-P.; Lu, F.; Qu, J.; Jin, Z.-B. Genotype–phenotype correlation and mutation spectrum in a large cohort of patients with inherited retinal dystrophy revealed by next-generation sequencing. Genet. Med. 2015, 17, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Fujinami, K.; Lois, N.; Mukherjee, R.; McBain, V.A.; Tsunoda, K.; Tsubota, K.; Stone, E.M.; Fitzke, F.W.; Bunce, C.; Moore, A.T.; et al. A Longitudinal Study of Stargardt Disease: Quantitative Assessment of Fundus Autofluorescence, Progression, and Genotype Correlations. Investig. Opthalmol. Vis. Sci. 2013, 54, 8181–8190. [Google Scholar] [CrossRef] [Green Version]
- Sparrow, J.R.; Marsiglia, M.; Allikmets, R.; Tsang, S.; Lee, W.; Duncker, T.; Zernant, J. Flecks in Recessive Stargardt Disease: Short-Wavelength Autofluorescence, Near-Infrared Autofluorescence, and Optical Coherence Tomography. Investig. Opthalmol. Vis. Sci. 2015, 56, 5029–5039. [Google Scholar] [CrossRef] [Green Version]
- Kong, X.; West, S.K.; Strauss, R.W.; Munoz, B.; Cideciyan, A.V.; Michaelides, M.; Ho, A.; Ahmed, M.; Schönbach, E.M.; Cheetham, J.K.; et al. Progression of Visual Acuity and Fundus Autofluorescence in Recent-Onset Stargardt Disease: ProgStar Study Report #4. Ophthalmol. Retin. 2017, 1, 514–523. [Google Scholar] [CrossRef]
- Van Huet, R.A.C.; Bax, N.; Haaften, S.C.W.-V.; Muhamad, M.; Zonneveld-Vrieling, M.N.; Hoefsloot, L.H.; Cremers, F.P.M.; Boon, C.J.F.; Klevering, B.J.; Hoyng, C.B. Foveal Sparing in Stargardt Disease. Investig. Opthalmol. Vis. Sci. 2014, 55, 7467–7478. [Google Scholar] [CrossRef] [Green Version]
- Fujinami, K.; Sergouniotis, P.I.; Davidson, A.E.; Wright, G.; Chana, R.K.; Tsunoda, K.; Tsubota, K.; Egan, C.A.; Robson, A.G.; Moore, A.T.; et al. Clinical and Molecular Analysis of Stargardt Disease With Preserved Foveal Structure and Function. Am. J. Ophthalmol. 2013, 156, 487–501. [Google Scholar] [CrossRef]
- Lambertus, S.; Lindner, M.; Bax, N.M.; Mauschitz, M.M.; Nadal, J.; Schmid, M.; Schmitz-Valckenberg, S.; Hollander, A.I.D.; Weber, B.H.F.; Holz, F.G.; et al. Progression of Late-Onset Stargardt Disease. Investig. Opthalmol. Vis. Sci. 2016, 57, 5186–5191. [Google Scholar] [CrossRef]
- Sayegh, R.G.; Sacu, S.; Dunavölgyi, R.; Kroh, M.E.; Roberts, P.; Mitsch, C.; Montuoro, A.; Ehrenmüller, M.; Schmidt-Erfurth, U. Geographic Atrophy and Foveal-Sparing Changes Related to Visual Acuity in Patients With Dry Age-Related Macular Degeneration Over Time. Am. J. Ophthalmol. 2017, 179, 118–128. [Google Scholar] [CrossRef] [Green Version]
- Bax, N.M.; Valkenburg, D.; Lambertus, S.; Klevering, B.J.; Boon, C.J.F.; Holz, F.G.; Cremers, F.P.M.; Fleckenstein, M.; Hoyng, C.B.; Lindner, M.; et al. Foveal Sparing in Central Retinal Dystrophies. Investig. Opthalmol. Vis. Sci. 2019, 60, 3456–3467. [Google Scholar] [CrossRef] [Green Version]
- Genead, M.A.; Fishman, G.A.; Stone, E.M.; Allikmets, R. The Natural History of Stargardt Disease with Specific Sequence Mutation in the ABCA4 Gene. Investig. Opthalmol. Vis. Sci. 2009, 50, 5867–5871. [Google Scholar] [CrossRef] [Green Version]
- Cideciyan, A.V.; Swider, M.; Schwartz, S.B.; Stone, E.M.; Jacobson, S.G. Predicting Progression ofABCA4-Associated Retinal Degenerations Based on Longitudinal Measurements of the Leading Disease Front. Investig. Opthalmol. Vis. Sci. 2015, 56, 5946–5955. [Google Scholar] [CrossRef] [Green Version]
- Han, Z.; Conley, S.M.; Naash, M.I. Gene therapy for Stargardt disease associated with ABCA4 gene. Adv. Exp. Med. Biol. 2014, 801, 719–724. [Google Scholar]
- Kong, J.; Kim, S.-R.; Binley, K.; Pata, I.; Doi, K.; Mannik, J.; Zernant-Rajang, J.; Kan, O.; Iqball, S.; Naylor, S.A.; et al. Correction of the disease phenotype in the mouse model of Stargardt disease by lentiviral gene therapy. Gene Ther. 2008, 15, 1311–1320. [Google Scholar] [CrossRef] [Green Version]
- Travis, G.H.; Golczak, M.; Moise, A.R.; Palczewski, K. Diseases Caused by Defects in the Visual Cycle: Retinoids as Potential Therapeutic Agents. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 469–512. [Google Scholar] [CrossRef] [Green Version]
- Charbel Issa, P.; Barnard, A.R.; Herrmann, P.; Washington, I.; MacLaren, R.E. Rescue of the Stargardt phenotype in Abca4 knockout mice through inhibition of vitamin A dimerization. Proc. Natl. Acad. Sci. USA 2015, 112, 8415–8420. [Google Scholar] [CrossRef] [Green Version]
- Ortube, M.C.; Strom, S.P.; Nelson, S.F.; Nusinowitz, S.; Martinez, A.; Gorin, M.B. Whole exome sequencing detects homozygosity for ABCA4 p.Arg602Trp missense mutation in a pediatric patient with rapidly progressive retinal dystrophy. BMC Med. Genet. 2014, 15, 11. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, T.; Klie, F.; Garred, P.; Schwartz, M. N965S is a common ABCA4 variant in Stargardt-related retinopathies in the Danish population. Mol. Vis. 2007, 13, 1962–1969. [Google Scholar]
- Hu, F.-Y.; Li, J.-K.; Gao, F.-J.; Qi, Y.-H.; Xu, P.; Zhang, Y.-J.; Wang, D.-D.; Wang, L.-S.; Li, W.; Wang, M.; et al. ABCA4 Gene Screening in a Chinese Cohort With Stargardt Disease: Identification of 37 Novel Variants. Front. Genet. 2019, 10, 773. [Google Scholar] [CrossRef]
- Jiang, F.; Pan, Z.; Xu, K.; Tian, L.; Xie, Y.; Zhang, X.; Chen, J.; Dong, B.; Li, Y. Screening of ABCA4 Gene in a Chinese Cohort With Stargardt Disease or Cone-Rod Dystrophy With a Report on 85 Novel Mutations. Investig. Opthalmol. Vis. Sci. 2016, 57, 145–152. [Google Scholar] [CrossRef] [Green Version]
- Roberts, L.; Nossek, C.A.; Greenberg, L.J.; Ramesar, R.S. Stargardt macular dystrophy: Common ABCA4 mutations in South Africa—Establishment of a rapid genetic test and relating risk to patients. Mol. Vis. 2012, 18, 280–289. [Google Scholar]
- Del Pozo-Valero, M.; Riveiro-Alvarez, R.; Blanco-Kelly, F.; Aguirre-Lamban, J.; Martin-Merida, I.; Iancu, I.F.; Swafiri, S.; Lorda-Sanchez, I.; Rodriguez-Pinilla, E.; Trujillo-Tiebas, M.J.; et al. Genotype-phenotype correlations in a Spanish cohort of 506 families with bi-allelic ABCA4 pathogenic variants. Am. J. Ophthalmol. 2020, 219, 195–204. [Google Scholar] [CrossRef]
- September, A.V.; Vorster, A.A.; Ramesar, R.; Greenberg, L.J. Mutation spectrum and founder chromosomes for the ABCA4 gene in South African patients with Stargardt disease. Investig. Opthalmol. Vis. Sci. 2004, 45, 1705–1711. [Google Scholar] [CrossRef] [Green Version]
- Cremers, F.P.M.; Van De Pol, D.J.R.; Van Driel, M.; Hollander, A.I.D.; Van Haren, F.J.J.; Knoers, N.V.A.M.; Tijmes, N.; Bergen, A.A.B.; Rohrschneider, K.; Blankenagel, A.; et al. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt’s disease gene ABCR. Hum. Mol. Genet. 1998, 7, 355–362. [Google Scholar] [CrossRef] [Green Version]
- Joo, K.; Seong, M.-W.; Park, K.H.; Park, S.S.; Woo, S.J. Genotypic profile and phenotype correlations of ABCA4-associated retinopathy in Koreans. Mol. Vis. 2019, 25, 679–690. [Google Scholar]
- Riveiro-Alvarez, R.; Lopez-Martinez, M.-A.; Zernant, J.; Aguirre-Lamban, J.; Cantalapiedra, D.; Avila-Fernandez, A.; Gimenez, A.; Lopez-Molina, M.-I.; Garcia-Sandoval, B.; Blanco-Kelly, F.; et al. Outcome of ABCA4 disease-associated alleles in autosomal recessive retinal dystrophies: Retrospective analysis in 420 Spanish families. Ophthalmology 2013, 120, 2332–2337. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stargardt Disease 1 | Retinitis Pigmentosa | p-Value | ||
|---|---|---|---|---|
| Central Type | Dispersed Type | |||
| Patients (n = 35) | 15 | 8 | 12 | |
| Male (n) | 7 (46.66%) | 3 (37.5%) | 4 (33.33%) | |
| Female (n) | 8 (53.33%) | 5 (62.5%) | 8 (66.67%) | |
| Age at first visit, mean ± SD (years) | 27.07 ± 10.21 | 24.88 ± 20.57 | 44.33 ± 9.39 | p = 0.038 |
| 0–19 y (n) | 7 (46.66%) | 5 (62.5%) | 1 (8.33%) | |
| 20–29 y (n) | 3 (20%) | 0 | 1 (8.33%) | |
| 30–39 y (n) | 1 (6.67%) | 0 | 2 (16.67%) | |
| 40–49 y (n) | 1 (6.67%) | 1 (12.5%) | 3 (25%) | |
| 50–59 y (n) | 2 (13.33%) | 1 (12.5%) | 4 (33.33%) | |
| >60 y (n) | 1 (6.67%) | 1 (12.5%) | 1 (8.33%) | |
| Age at symptom onset, mean ± SD (years) | 18.73 ± 6.05 | 15.14 ± 15.21 | 14.91 ± 7.83 | p = 0.695 |
| 0–19 y (n) | 9 (60%) | 6 (75%) | 9 (75%) | |
| 20–29 y (n) | 4 (26.67%) | 1 (12.5%) | 2 (16.67%) | |
| 30–39 y (n) | 0 | 0 | 0 | |
| >40 y (n) | 2 (13.33%) | 1 (12.5%) | 1 (8.33%) | |
| Visual acuity at first visit, mean ± SD (LogMAR) | 0.67 ± 0.16 | 0.77 ± 0.4 | 1.63 ± 0.46 | p < 0.001 |
| Visual acuity at recent visit, mean ± SD (LogMAR) | 0.98 ± 0.28 | 0.79 ± 0.73 | 1.8 ± 1.04 | p = 0.033 |
| Follow-up period (months) | 64.5 ± 56.55 | 73.5 ± 73.4 | 49 ± 37.86 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sung, Y.-C.; Yang, C.-H.; Yang, C.-M.; Lin, C.-W.; Huang, D.-S.; Huang, Y.-S.; Hu, F.-R.; Chen, P.-L.; Chen, T.-C. Genotypes Predispose Phenotypes—Clinical Features and Genetic Spectrum of ABCA4-Associated Retinal Dystrophies. Genes 2020, 11, 1421. https://doi.org/10.3390/genes11121421
Sung Y-C, Yang C-H, Yang C-M, Lin C-W, Huang D-S, Huang Y-S, Hu F-R, Chen P-L, Chen T-C. Genotypes Predispose Phenotypes—Clinical Features and Genetic Spectrum of ABCA4-Associated Retinal Dystrophies. Genes. 2020; 11(12):1421. https://doi.org/10.3390/genes11121421
Chicago/Turabian StyleSung, Yu-Chi, Chang-Hao Yang, Chung-May Yang, Chao-Wen Lin, Ding-Siang Huang, Yu-Shu Huang, Fung-Rong Hu, Pei-Lung Chen, and Ta-Ching Chen. 2020. "Genotypes Predispose Phenotypes—Clinical Features and Genetic Spectrum of ABCA4-Associated Retinal Dystrophies" Genes 11, no. 12: 1421. https://doi.org/10.3390/genes11121421