An Update on the Lithogenic Mechanisms of Cholecystokinin a Receptor (CCKAR), an Important Gallstone Gene for Lith13




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
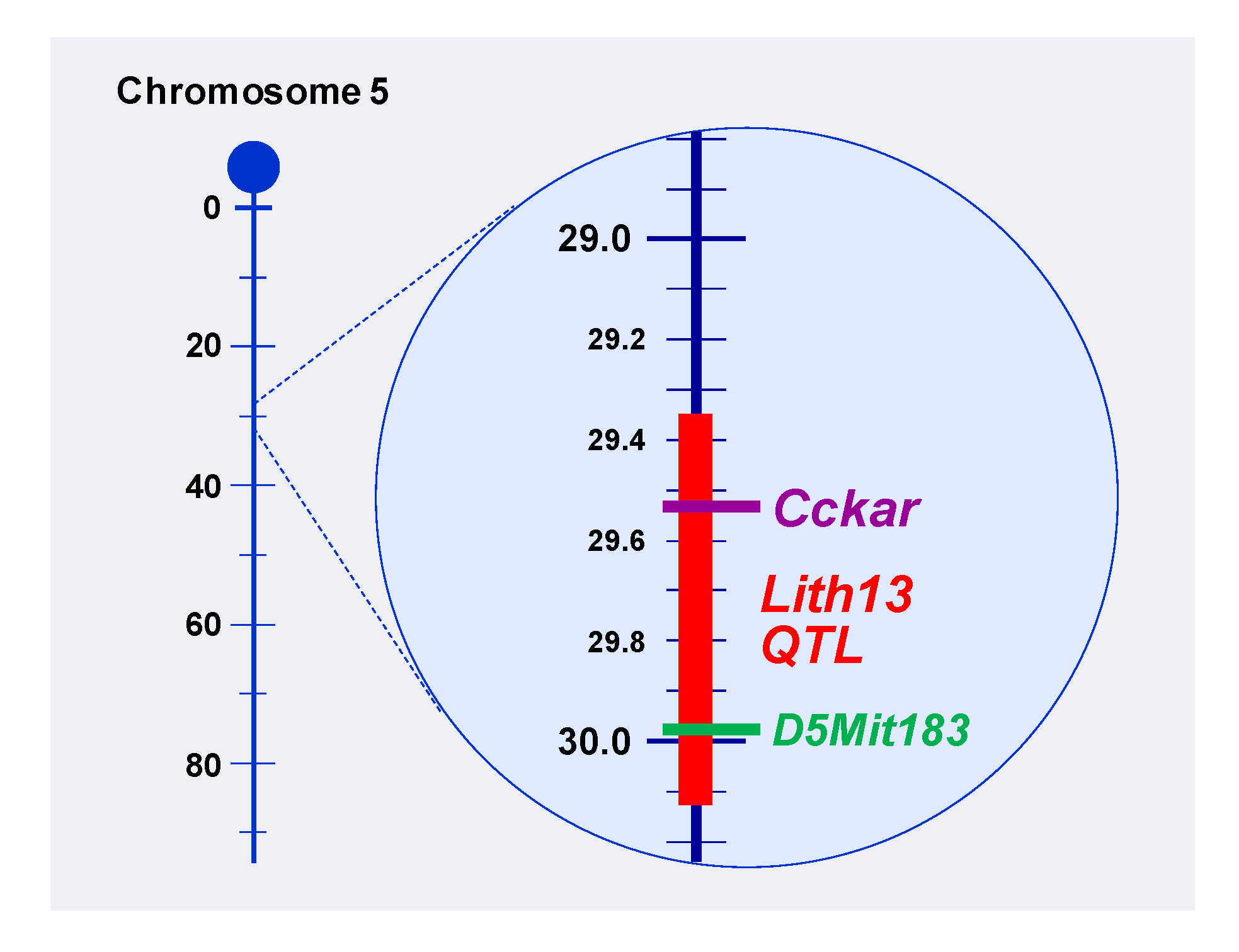
2. Identification of Lith13 in Mice
3. Regulation of Gallbladder and Gastrointestinal Motility, as well as Pancreatic Secretion by CCK and CCKAR
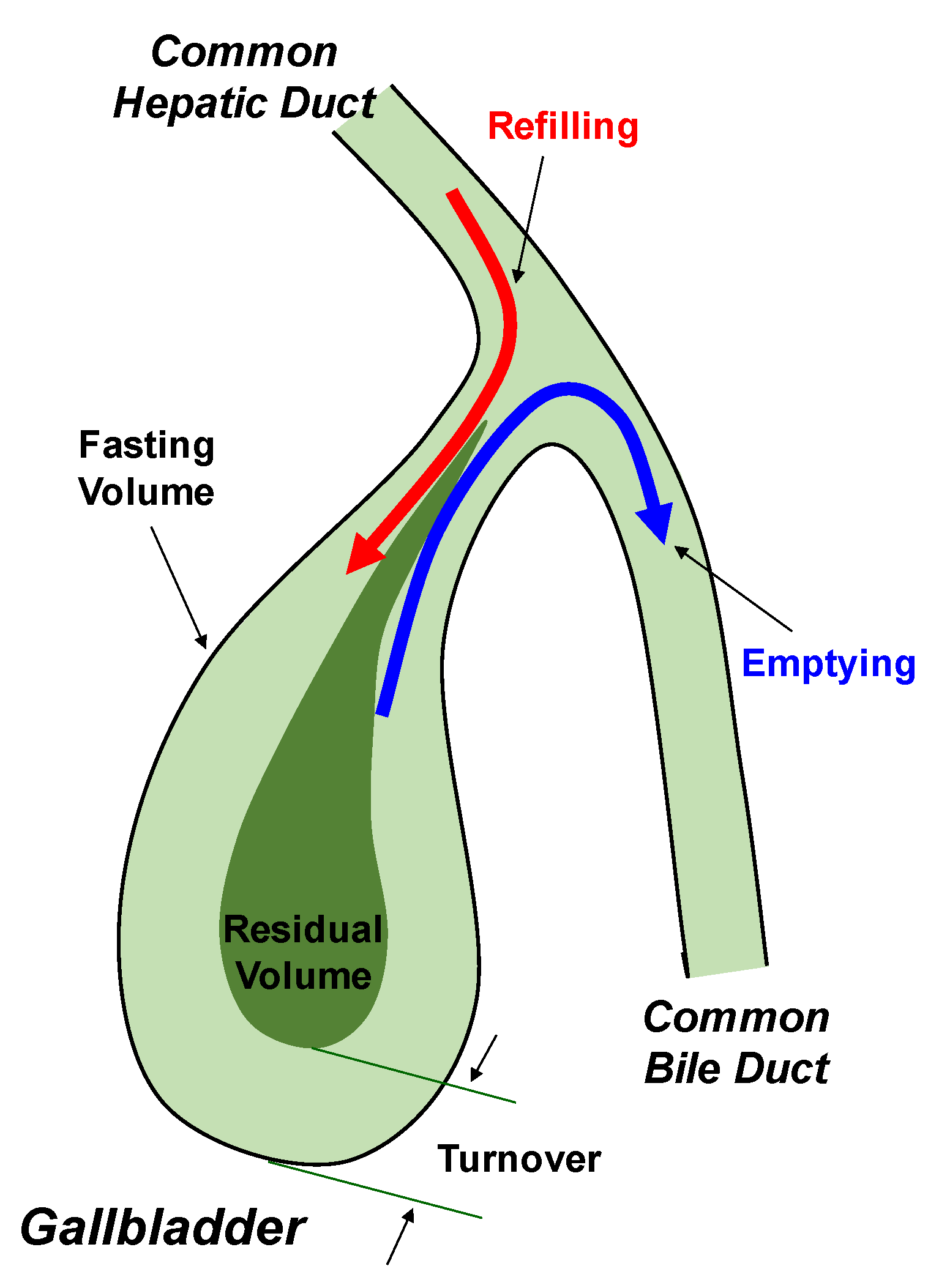
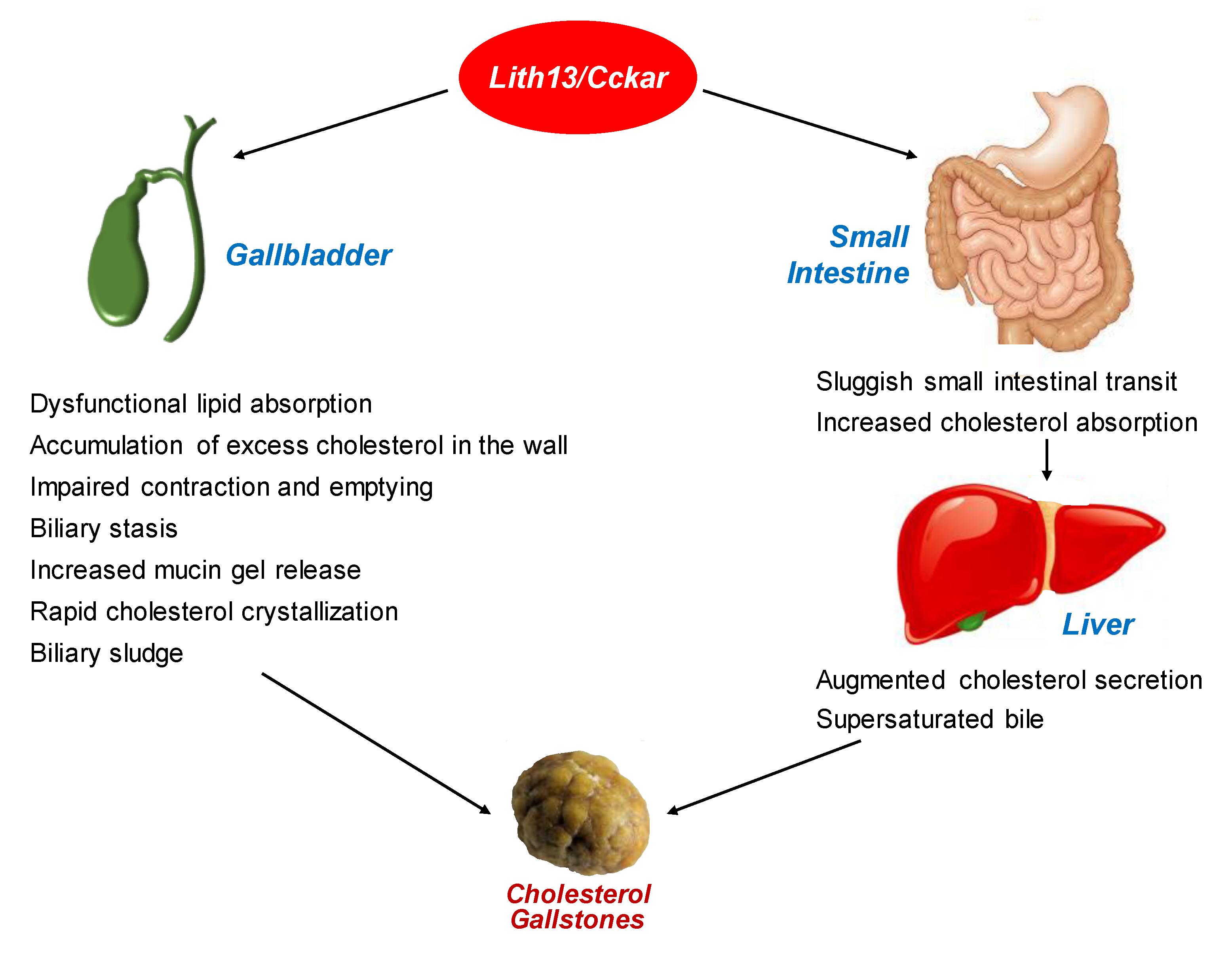
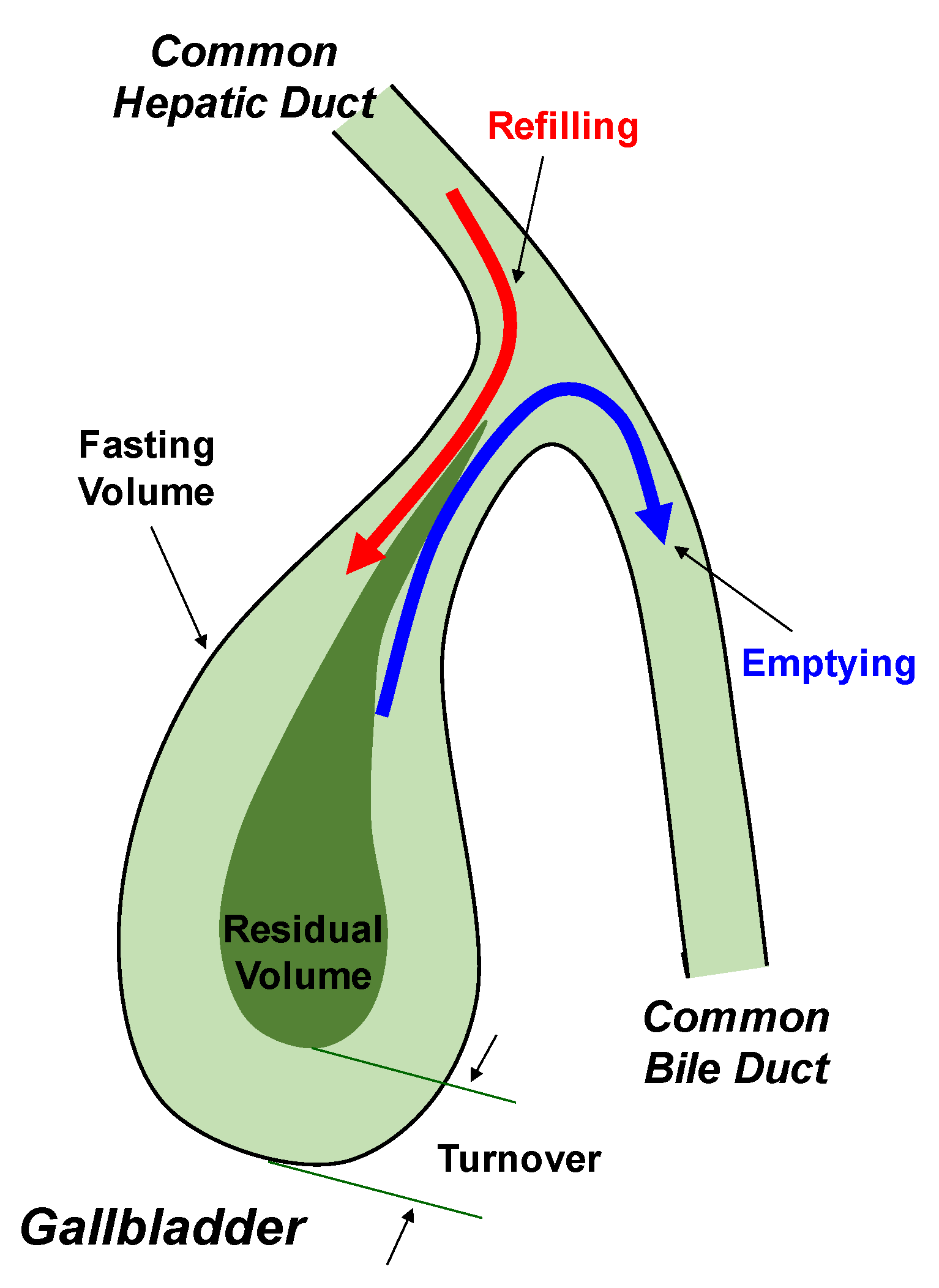
4. Role of Lith13 in Impairing Gallbladder Motility
5. Effect of Lith13 on Delaying Intestinal Transit Time
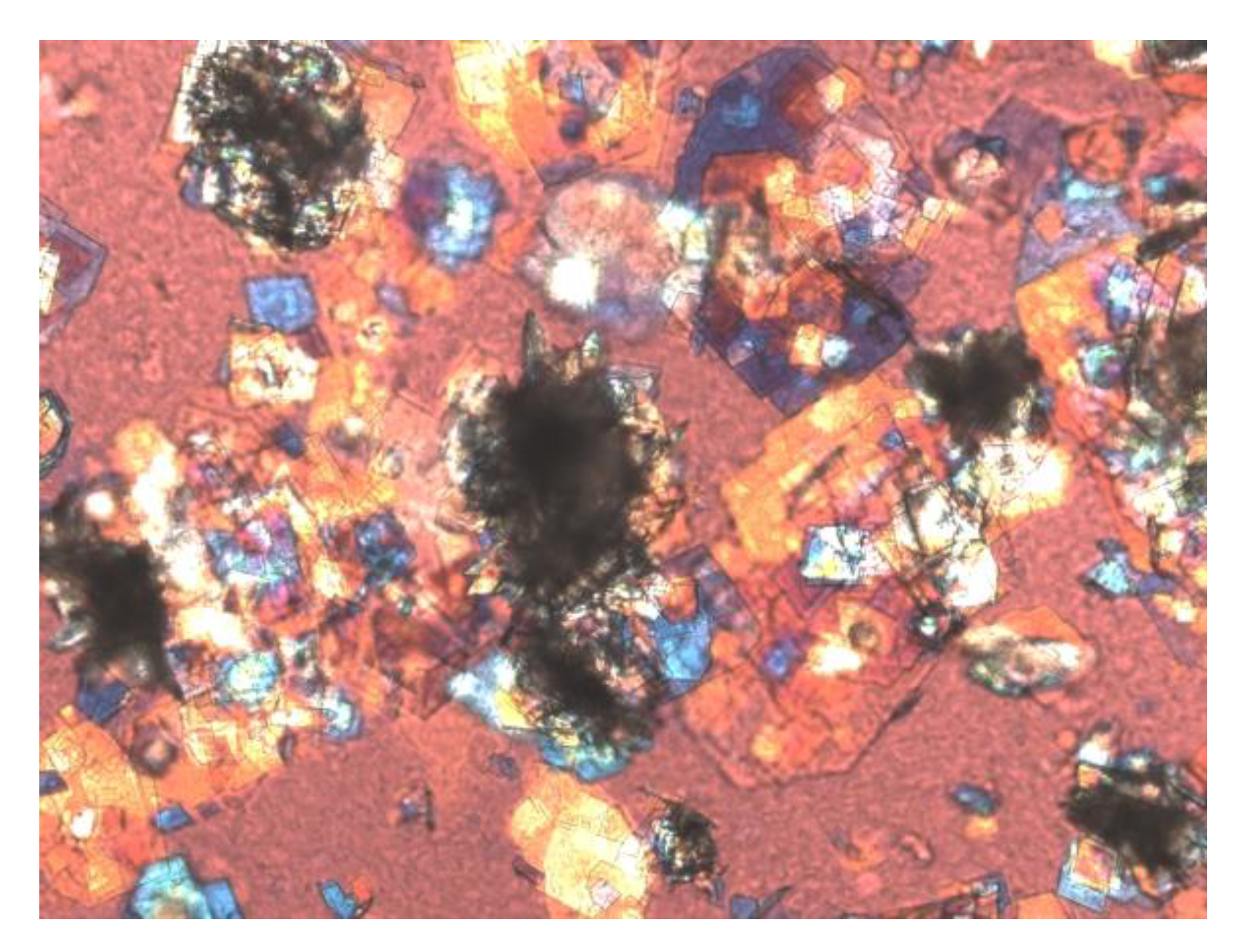
6. Lithogenic Mechanisms Underlying the Role of Lith13 in Cholesterol Crystallization and Gallstone Formation in Mice
7. Effect of LITH13 on the Pathogenesis and Pathophysiology of Cholesterol Gallstone Formation in Humans
8. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, H.H.; Portincasa, P.; Afdhal, N.H.; Wang, D.Q. Lith genes and genetic analysis of cholesterol gallstone formation. Gastroenterol. Clin. N. Am. 2010, 39, 185–207. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Q.; Portincasa, P.; Wang, H.H. Bile formation and pathophysiology of gallstones. In Encyclopedia of Gastroenterology, 2nd ed.; Kuipers, E.J., Ed.; Elsevier: New York, NY, USA, 2020; Volume 1, pp. 287–306. [Google Scholar]
- Wang, D.Q.; Afdhal, N.H. Gallstone Disease. In Sleisenger and Fordtran’s Gastrointestinal and Liver Disease, 11th ed.; Feldman, M., Friedman, L.S., Brandt, L.J., Chung, R.T., Rubin, D.T., Wilcox, C.M., Eds.; Elsevier: Philadelphia, PA, USA, 2020; Volume 1, pp. 1016–1046. [Google Scholar]
- Wang, H.H.; Portincasa, P.; de Bari, O.; Liu, K.J.; Garruti, G.; Neuschwander-Tetri, B.A.; Wang, D.Q. Prevention of cholesterol gallstones by inhibiting hepatic biosynthesis and intestinal absorption of cholesterol. Eur. J. Clin. Investig. 2013, 43, 413–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.Q.; Cohen, D.E.; Carey, M.C. Biliary lipids and cholesterol gallstone disease. J. Lipid Res. 2009, 50, S406–S411. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.Q.; Portincasa, P. Gallstones: Recent Advances in Epidemiology, Pathogenesis, Diagnosis and Management; Nova Biomedical: New York, NY, USA, 2017; pp. 1–676. [Google Scholar]
- Wang, D.Q.; Neuschwander-Tetri, B.A.; Portincasa, P. The Biliary System, 1st ed.; Morgan & Claypool Life Sciences: Princeton, NJ, USA, 2012; pp. 1–146. [Google Scholar]
- Meilstrup, J.W.; Hopper, K.D.; Thieme, G.A. Imaging of gallbladder variants. AJR Am. J. Roentgenol. 1991, 157, 1205–1208. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Q. Regulation of intestinal cholesterol absorption. Annu. Rev. Physiol. 2007, 69, 221–248. [Google Scholar] [CrossRef] [PubMed]
- Lammert, F.; Gurusamy, K.; Ko, C.W.; Miquel, J.F.; Mendez-Sanchez, N.; Portincasa, P.; van Erpecum, K.J.; van Laarhoven, C.J.; Wang, D.Q. Gallstones. Nat. Rev. Dis. Primers 2016, 2, 16024. [Google Scholar] [CrossRef] [PubMed]
- Jazrawi, R.P.; Pazzi, P.; Petroni, M.L.; Prandini, N.; Paul, C.; Adam, J.A.; Gullini, S.; Northfield, T.C. Postprandial gallbladder motor function: Refilling and turnover of bile in health and in cholelithiasis. Gastroenterology 1995, 109, 582–591. [Google Scholar] [CrossRef]
- LaMorte, W.W.; Schoetz, D.J., Jr.; Birkett, D.H.; Williams, L.F., Jr. The role of the gallbladder in the pathogenesis of cholesterol gallstones. Gastroenterology 1979, 77, 580–592. [Google Scholar] [CrossRef]
- Howard, P.J.; Murphy, G.M.; Dowling, R.H. Gall bladder emptying patterns in response to a normal meal in healthy subjects and patients with gall stones: Ultrasound study. Gut 1991, 32, 1406–1411. [Google Scholar] [CrossRef] [Green Version]
- Shaffer, E.A.; McOrmond, P.; Duggan, H. Quantitative cholescintigraphy: Assessment of gallbladder filling and emptying and duodenogastric reflux. Gastroenterology 1980, 79, 899–906. [Google Scholar] [CrossRef]
- Portincasa, P.; Moschetta, A.; Palasciano, G. Cholesterol gallstone disease. Lancet 2006, 368, 230–239. [Google Scholar] [CrossRef]
- Portincasa, P.; Di Ciaula, A.; Baldassarre, G.; Palmieri, V.; Gentile, A.; Cimmino, A.; Palasciano, G. Gallbladder motor function in gallstone patients: Sonographic and in vitro studies on the role of gallstones, smooth muscle function and gallbladder wall inflammation. J. Hepatol. 1994, 21, 430–440. [Google Scholar] [CrossRef]
- Low-Beer, T.S.; Harvey, R.F.; Davies, E.R.; Read, A.F. Abnormalities of serum cholecystokinin and gallbladder emptying in celiac disease. N. Engl. J. Med. 1975, 292, 961–963. [Google Scholar] [CrossRef] [PubMed]
- Maton, P.N.; Selden, A.C.; Fitzpatrick, M.L.; Chadwick, V.S. Defective gallbladder emptying and cholecystokinin release in celiac disease. Reversal by gluten-free diet. Gastroenterology 1985, 88, 391–396. [Google Scholar] [CrossRef]
- Hopman, W.P.; Rosenbusch, G.; Hectors, M.P.; Jansen, J.B. Effect of predigested fat on intestinal stimulation of plasma cholecystokinin and gall bladder motility in coeliac disease. Gut 1995, 36, 17–21. [Google Scholar] [CrossRef] [Green Version]
- Fraquelli, M.; Bardella, M.T.; Peracchi, M.; Cesana, B.M.; Bianchi, P.A.; Conte, D. Gallbladder emptying and somatostatin and cholecystokinin plasma levels in celiac disease. Am. J. Gastroenterol. 1999, 94, 1866–1870. [Google Scholar] [CrossRef]
- Brown, A.M.; Bradshaw, M.J.; Richardson, R.; Wheeler, J.G.; Harvey, R.F. Pathogenesis of the impaired gall bladder contraction of coeliac disease. Gut 1987, 28, 1426–1432. [Google Scholar] [CrossRef] [Green Version]
- Sjolund, K.; Alumets, J.; Berg, N.O.; Hakanson, R.; Sundler, F. Duodenal endocrine cells in adult coeliac disease. Gut 1979, 20, 547–552. [Google Scholar] [CrossRef] [Green Version]
- Low-Beer, T.S.; Heaton, K.W.; Heaton, S.T.; Read, A.E. Gallbladder inertia and sluggish enterohepatic circulation of bile-salts in coeliac disease. Lancet 1971, 1, 991–994. [Google Scholar] [CrossRef]
- Wang, H.H.; Liu, M.; Li, X.; Portincasa, P.; Wang, D.Q. Impaired intestinal cholecystokinin secretion, a fascinating but overlooked link between coeliac disease and cholesterol gallstone disease. Eur. J. Clin. Investig. 2017, 47, 328–333. [Google Scholar] [CrossRef] [Green Version]
- Portincasa, P.; Di Ciaula, A.; Wang, H.H.; Palasciano, G.; van Erpecum, K.J.; Moschetta, A.; Wang, D.Q. Coordinate regulation of gallbladder motor function in the gut-liver axis. Hepatology 2008, 47, 2112–2126. [Google Scholar] [CrossRef] [PubMed]
- Chandra, R.; Liddle, R.A. Cholecystokinin. Curr. Opin. Endocrinol. Diabetesand Obes. 2007, 14, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Grider, J.R. Role of cholecystokinin in the regulation of gastrointestinal motility. J. Nutr. 1994, 124 (Suppl. 8), 1334S–1339S. [Google Scholar] [CrossRef] [Green Version]
- Schjoldager, B.T. Role of CCK in gallbladder function. Ann. N. Y. Acad. Sci. 1994, 713, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Q.; Afdhal, N.H. Genetic analysis of cholesterol gallstone formation: Searching for Lith (gallstone) genes. Curr. Gastroenterol. Rep. 2004, 6, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Lyons, M.A.; Wittenburg, H. Cholesterol gallstone susceptibility loci: A mouse map, candidate gene evaluation, and guide to human LITH genes. Gastroenterology 2006, 131, 1943–1970. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Q.; Schmitz, F.; Kopin, A.S.; Carey, M.C. Targeted disruption of the murine cholecystokinin-1 receptor promotes intestinal cholesterol absorption and susceptibility to cholesterol cholelithiasis. J. Clin. Investig. 2004, 114, 521–528. [Google Scholar] [CrossRef] [Green Version]
- Miller, L.J.; Holicky, E.L.; Ulrich, C.D.; Wieben, E.D. Abnormal processing of the human cholecystokinin receptor gene in association with gallstones and obesity. Gastroenterology 1995, 109, 1375–1380. [Google Scholar] [CrossRef]
- Nardone, G.; Ferber, I.A.; Miller, L.J. The integrity of the cholecystokinin receptor gene in gallbladder disease and obesity. Hepatology 1995, 22, 1751–1753. [Google Scholar]
- Schneider, H.; Sanger, P.; Hanisch, E. In vitro effects of cholecystokinin fragments on human gallbladders. Evidence for an altered CCK-receptor structure in a subgroup of patients with gallstones. J. Hepatol. 1997, 26, 1063–1068. [Google Scholar] [CrossRef]
- Ivy, A.C.; Oldberg, E. A hormone mechanism for gallbladder contraction and evacuation. Am. J. Physiol. 1928, 86, 559–613. [Google Scholar] [CrossRef]
- Liddle, R.A. Cholecystokinin. In Gut Peptides, 1st ed.; Walsh, J.H., Dockray, G.J., Eds.; Raven: New York, NY, USA, 1994; pp. 175–216. [Google Scholar]
- Wank, S.A.; Harkins, R.; Jensen, R.T.; Shapira, H.; de Weerth, A.; Slattery, T. Purification, molecular cloning, and functional expression of the cholecystokinin receptor from rat pancreas. Proc. Natl. Acad. Sci. USA 1992, 89, 3125–3129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopin, A.S.; Lee, Y.M.; McBride, E.W.; Miller, L.J.; Lu, M.; Lin, H.Y.; Kolakowski, L.F., Jr.; Beinborn, M. Expression cloning and characterization of the canine parietal cell gastrin receptor. Proc. Natl. Acad. Sci. USA 1992, 89, 3605–3609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiegelman, B.M.; Flier, J.S. Obesity and the regulation of energy balance. Cell 2001, 104, 531–543. [Google Scholar] [CrossRef] [Green Version]
- Badman, M.K.; Flier, J.S. The gut and energy balance: Visceral allies in the obesity wars. Science 2005, 307, 1909–1914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guyenet, S.J.; Schwartz, M.W. Clinical review: Regulation of food intake, energy balance, and body fat mass: Implications for the pathogenesis and treatment of obesity. J. Clin. Endocrinol. Metab. 2012, 97, 745–755. [Google Scholar] [CrossRef] [Green Version]
- Cheung, G.W.; Kokorovic, A.; Lam, C.K.; Chari, M.; Lam, T.K. Intestinal cholecystokinin controls glucose production through a neuronal network. Cell Metab. 2009, 10, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Heijboer, A.C.; Pijl, H.; Van den Hoek, A.M.; Havekes, L.M.; Romijn, J.A.; Corssmit, E.P. Gut-brain axis: Regulation of glucose metabolism. J. Neuroendocrinol. 2006, 18, 883–894. [Google Scholar] [CrossRef]
- Harro, J. CCK and NPY as anti-anxiety treatment targets: Promises, pitfalls, and strategies. Amino Acids 2006, 31, 215–230. [Google Scholar] [CrossRef]
- Wang, T.Y.; Portincasa, P.; Liu, M.; Tso, P.; Wang, D.Q. Mouse models of gallstone disease. Curr. Opin. Gastroenterol. 2018, 34, 59–70. [Google Scholar] [CrossRef]
- Liddle, R.A. Cholecystokinin cells. Annu. Rev. Physiol. 1997, 59, 221–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liddle, R.A. Gastrointestinal Hormones and Neurotransmitters. In Sleisenger and Fordtran’s Gastrointestinal and Liver Disease, 9th ed.; Feldman, M., Friedman, L.S., Brandt, L., Eds.; Elsevier Saunders: Philadelphia, PA, USA, 2010; pp. 3–19. [Google Scholar]
- Martinez, M.A.; Lajas, A.I.; Yago, M.D.; Redondo, P.C.; Granados, M.P.; Gonzalez, A.; Rosado, J.A.; Martinez-Victoria, E.; Manas, M.; Pariente, J.A. Dietary virgin olive oil enhances secretagogue-evoked calcium signaling in rat pancreatic acinar cells. Nutrition 2004, 20, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Liddle, R.A. Regulation of cholecystokinin secretion by intraluminal releasing factors. Am. J. Physiol. 1995, 269, G319–G327. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chandra, R.; Samsa, L.A.; Gooch, B.; Fee, B.E.; Cook, J.M.; Vigna, S.R.; Grant, A.O.; Liddle, R.A. Amino acids stimulate cholecystokinin release through the Ca2+-sensing receptor. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G528–G537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Prpic, V.; Green, G.M.; Reeve, J.R., Jr.; Liddle, R.A. Luminal CCK-releasing factor stimulates CCK release from human intestinal endocrine and STC-1 cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, G16–G22. [Google Scholar] [CrossRef]
- Ohta, H.; Guan, D.; Tawil, T.; Liddle, R.A.; Green, G.M. Regulation of plasma cholecystokinin levels by bile and bile acids in the rat. Gastroenterology 1990, 99, 819–825. [Google Scholar] [CrossRef]
- Liddle, R.A.; Goldfine, I.D.; Rosen, M.S.; Taplitz, R.A.; Williams, J.A. Cholecystokinin bioactivity in human plasma. Molecular forms, responses to feeding, and relationship to gallbladder contraction. J. Clin. Investig. 1985, 75, 1144–1152. [Google Scholar] [CrossRef]
- Maton, P.N.; Selden, A.C.; Chadwick, V.S. Differential distribution of molecular forms of cholecystokinin in human and porcine small intestinal mucosa. Regul. Pept. 1984, 8, 9–19. [Google Scholar] [CrossRef]
- Noble, F.; Roques, B.P. CCK-B receptor: Chemistry, molecular biology, biochemistry and pharmacology. Prog. Neurobiol. 1999, 58, 349–379. [Google Scholar] [CrossRef]
- Maton, P.N.; Selden, A.C.; FitzPatrick, M.L.; Chadwick, V.S. Infusion of cholecystokinin octapeptide in man: Relation between plasma cholecystokinin concentrations and gallbladder emptying rates. Eur. J. Clin. Investig. 1984, 14, 37–41. [Google Scholar] [CrossRef]
- Rehfeld, J.F.; Friis-Hansen, L.; Goetze, J.P.; Hansen, T.V. The biology of cholecystokinin and gastrin peptides. Curr. Top. Med. Chem. 2007, 7, 1154–1165. [Google Scholar] [CrossRef] [PubMed]
- Rehfeld, J.F.; Larsson, L.I.; Goltermann, N.R.; Schwartz, T.W.; Holst, J.J.; Jensen, S.L.; Morley, J.S. Neural regulation of pancreatic hormone secretion by the C-terminal tetrapeptide of CCK. Nature 1980, 284, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Carey, M.C.; Hernell, O. Digestion and absorption of fat. Semin. Gastrointest. Dis. 1992, 3, 189–208. [Google Scholar]
- Hofmann, A.F.; Hagey, L.R. Bile acids: Chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell. Mol. Life Sci. 2008, 65, 2461–2483. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, A.F. Bile Acids and the Enterohepatic Circulation. In The Liver: Biology and Pathobiology, 5th ed.; Arias, I.M., Alter, H.J., Boyer, J.L., Cohen, D.E., Fausto, N., Shafritz, D.A., Wolkoff, A.W., Eds.; Wiley-Blackwell: West Sussex, UK, 2009; pp. 290–304. [Google Scholar]
- Hay, D.W.; Carey, M.C. Chemical species of lipids in bile. Hepatology 1990, 12, 6S–S14; discussion 14S–S16. [Google Scholar] [PubMed]
- Wang, H.H.; Liu, M.; Portincasa, P.; Tso, P.; Wang, D.Q. Lack of endogenous cholecystokinin promotes cholelithogenesis in mice. Neurogastroenterol. Motil. Off. J. Eur. Gastrointest. Motil. Soc. 2016, 28, 364–375. [Google Scholar] [CrossRef] [Green Version]
- de Leon, M.P.; Iori, R.; Barbolini, G.; Pompei, G.; Zaniol, P.; Carulli, N. Influence of small-bowel transit time on dietary cholesterol absorption in human beings. N. Engl. J. Med. 1982, 307, 102–103. [Google Scholar] [CrossRef]
- Chandra, R.; Liddle, R.A. Recent advances in pancreatic endocrine and exocrine secretion. Curr. Opin. Gastroenterol. 2011, 27, 439–443. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Nylander, A.G.; Rehfeld, J.F.; Axelson, J.; Ihse, I.; Hakanson, R. Does vagotomy affect the growth of the pancreas in the rat? Scand. J. Gastroenterol. 1992, 27, 606–608. [Google Scholar] [CrossRef]
- Chu, M.; Borch, K.; Lilja, I.; Blomqvist, L.; Rehfeld, J.F.; Ihse, I. Endogenous hypercholecystokininemia model in the hamster: Trophic effect on the exocrine pancreas. Pancreas 1992, 7, 220–225. [Google Scholar] [CrossRef]
- Shetzline, M.A.; Liddle, R.A. Neurohumoral control of the exocrine pancreas. Curr. Opin. Gastroenterol. 1999, 15, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Portincasa, P.; Di Ciaula, A.; de Bari, O.; Garruti, G.; Palmieri, V.O.; Wang, D.Q. Management of gallstones and its related complications. Expert Rev. Gastroenterol. Hepatol. 2016, 10, 93–112. [Google Scholar] [CrossRef] [PubMed]
- Portincasa, P.; Di Ciaula, A.; Vendemiale, G.; Palmieri, V.; Moschetta, A.; Vanberge-Henegouwen, G.P.; Palasciano, G. Gallbladder motility and cholesterol crystallization in bile from patients with pigment and cholesterol gallstones. Eur. J. Clin. Investig. 2000, 30, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Portincasa, P.; Wang, D.Q. Gallstones. In Yamada’s Atlas of Gastroenterology, 5th ed.; Podolsky, D.K., Camilleri, M., Fitz, J.G., Kalloo, A.N., Shanahan, F., Wang, T.C., Eds.; Wiley-Blackwell: Hoboken, NJ, USA, 2016; pp. 335–353. [Google Scholar]
- Wang, H.H.; Liu, M.; Clegg, D.J.; Portincasa, P.; Wang, D.Q. New insights into the molecular mechanisms underlying effects of estrogen on cholesterol gallstone formation. Biochim. Et Biophys. Acta 2009, 1791, 1037–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bari, O.; Wang, T.Y.; Liu, M.; Paik, C.N.; Portincasa, P.; Wang, D.Q. Cholesterol cholelithiasis in pregnant women: Pathogenesis, prevention and treatment. Ann. Hepatol. 2014, 13, 728–745. [Google Scholar] [CrossRef]
- Ko, C.W.; Beresford, S.A.; Schulte, S.J.; Matsumoto, A.M.; Lee, S.P. Incidence, natural history, and risk factors for biliary sludge and stones during pregnancy. Hepatology 2005, 41, 359–365. [Google Scholar] [CrossRef]
- Ko, C.W. Risk factors for gallstone-related hospitalization during pregnancy and the postpartum. Am. J. Gastroenterol. 2006, 101, 2263–2268. [Google Scholar] [CrossRef]
- Spengler, U.; Sackmann, M.; Sauerbruch, T.; Holl, J.; Paumgartner, G. Gallbladder motility before and after extracorporeal shock-wave lithotripsy. Gastroenterology 1989, 96, 860–863. [Google Scholar] [CrossRef]
- Festi, D.; Frabboni, R.; Bazzoli, F.; Sangermano, A.; Ronchi, M.; Rossi, L.; Parini, P.; Orsini, M.; Primerano, A.M.; Mazzella, G.; et al. Gallbladder motility in cholesterol gallstone disease. Effect of ursodeoxycholic acid administration and gallstone dissolution. Gastroenterology 1990, 99, 1779–1785. [Google Scholar] [CrossRef]
- Abdou, M.S.; Strichartz, S.D.; Abedin, M.Z.; Roslyn, J.J. Biliary lipids alter ion transport during cholesterol gallstone formation. J. Surg. Res. 1988, 44, 672–679. [Google Scholar] [CrossRef]
- Abedin, M.Z.; Strichartz, S.D.; Festekdjian, S.; Roslyn, J.J. Increased biliary calcium in cholesterol and pigment gallstone disease: The role of altered bile acid composition. Lipids 1989, 24, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Cates, J.A.; Abedin, M.Z.; Saunders-Kirkwood, K.D.; Moser, A.J.; Giurgiu, D.I.; Roslyn, J.J. Protein kinase C regulates prairie dog gallbladder ion transport. Surgery 1995, 117, 206–212. [Google Scholar] [CrossRef]
- Behar, J.; Lee, K.Y.; Thompson, W.R.; Biancani, P. Gallbladder contraction in patients with pigment and cholesterol stones. Gastroenterology 1989, 97, 1479–1484. [Google Scholar] [CrossRef]
- Behar, J.; Rhim, B.Y.; Thompson, W.; Biancani, P. Inositol trisphosphate restores impaired human gallbladder motility associated with cholesterol stones. Gastroenterology 1993, 104, 563–568. [Google Scholar] [CrossRef]
- Xiao, Z.; Schmitz, F.; Pricolo, V.E.; Biancani, P.; Behar, J. Role of caveolae in the pathogenesis of cholesterol-induced gallbladder muscle hypomotility. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G1641–G1649. [Google Scholar] [CrossRef]
- Cong, P.; Xiao, Z.L.; Biancani, P.; Behar, J. Prostaglandins mediate tonic contraction of the guinea pig and human gallbladder. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G409–G418. [Google Scholar] [CrossRef] [Green Version]
- Conter, R.L.; Roslyn, J.J.; Porter-Fink, V.; DenBesten, L. Gallbladder absorption increases during early cholesterol gallstone formation. Am. J. Surg. 1986, 151, 184–191. [Google Scholar] [CrossRef]
- Roslyn, J.J.; Doty, J.; Pitt, H.A.; Conter, R.L.; Den Besten, L. Enhanced gallbladder absorption during gallstone formation: The roles of cholesterol saturated bile and gallbladder stasis. Am. J. Med. Sci. 1986, 292, 75–80. [Google Scholar] [CrossRef]
- Giurgiu, D.I.; Saunders-Kirkwood, K.D.; Roslyn, J.J.; Abedin, M.Z. Sequential changes in biliary lipids and gallbladder ion transport during gallstone formation. Ann. Surg. 1997, 225, 382–390. [Google Scholar] [CrossRef]
- Behar, J. Clinical aspects of gallbladder motor function and dysfunction. Curr. Gastroenterol. Rep. 1999, 1, 91–94. [Google Scholar] [CrossRef]
- Behar, J.; Corazziari, E.; Guelrud, M.; Hogan, W.; Sherman, S.; Toouli, J. Functional gallbladder and sphincter of oddi disorders. Gastroenterology 2006, 130, 1498–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cong, P.; Pricolo, V.; Biancani, P.; Behar, J. Effects of cholesterol on CCK-1 receptors and caveolin-3 proteins recycling in human gallbladder muscle. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G742–G750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feeley, T.M.; Clanachan, A.S.; Scott, G.W. Contractility of human gallbladder muscle in vitro. Aliment. Pharmacol. Ther. 1987, 1, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Chen, Q.; Biancani, P.; Behar, J. Membrane cholesterol alters gallbladder muscle contractility in prairie dogs. Am. J. Physiol. 1996, 271, G56–G61. [Google Scholar] [CrossRef]
- Pauletzki, J.; Cicala, M.; Holl, J.; Sauerbruch, T.; Schafmayer, A.; Paumgartner, G. Correlation between gall bladder fasting volume and postprandial emptying in patients with gall stones and healthy controls. Gut 1993, 34, 1443–1447. [Google Scholar] [CrossRef] [Green Version]
- Kishk, S.M.; Darweesh, R.M.; Dodds, W.J.; Lawson, T.L.; Stewart, E.T.; Kern, M.K.; Hassanein, E.H. Sonographic evaluation of resting gallbladder volume and postprandial emptying in patients with gallstones. AJR Am. J. Roentgenol. 1987, 148, 875–879. [Google Scholar] [CrossRef]
- Fisher, R.S.; Stelzer, F.; Rock, E.; Malmud, L.S. Abnormal gallbladder emptying in patients with gallstones. Dig. Dis. Sci. 1982, 27, 1019–1024. [Google Scholar] [CrossRef]
- Guarino, M.P.; Cong, P.; Cicala, M.; Alloni, R.; Carotti, S.; Behar, J. Ursodeoxycholic acid improves muscle contractility and inflammation in symptomatic gallbladders with cholesterol gallstones. Gut 2007, 56, 815–820. [Google Scholar] [CrossRef]
- Desai, A.J.; Miller, L.J. Sensitivity of cholecystokinin receptors to membrane cholesterol content. Front. Endocrinol. 2012, 3, 123. [Google Scholar] [CrossRef] [Green Version]
- Potter, R.M.; Harikumar, K.G.; Wu, S.V.; Miller, L.J. Differential sensitivity of types 1 and 2 cholecystokinin receptors to membrane cholesterol. J. Lipid Res. 2012, 53, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Harikumar, K.G.; Potter, R.M.; Patil, A.; Echeveste, V.; Miller, L.J. Membrane cholesterol affects stimulus-activity coupling in type 1, but not type 2, CCK receptors: Use of cell lines with elevated cholesterol. Lipids 2013, 48, 231–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Chitinavis, V.; Xiao, Z.; Yu, P.; Oh, S.; Biancani, P.; Behar, J. Impaired G protein function in gallbladder muscle from progesterone-treated guinea pigs. Am. J. Physiol. 1998, 274, G283–G289. [Google Scholar] [CrossRef]
- Harikumar, K.G.; Puri, V.; Singh, R.D.; Hanada, K.; Pagano, R.E.; Miller, L.J. Differential effects of modification of membrane cholesterol and sphingolipids on the conformation, function, and trafficking of the G protein-coupled cholecystokinin receptor. J. Biol. Chem. 2005, 280, 2176–2185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Amaral, J.; Biancani, P.; Behar, J. Excess membrane cholesterol alters human gallbladder muscle contractility and membrane fluidity. Gastroenterology 1999, 116, 678–685. [Google Scholar] [CrossRef]
- Yu, P.; Chen, Q.; Harnett, K.M.; Amaral, J.; Biancani, P.; Behar, J. Direct G protein activation reverses impaired CCK signaling in human gallbladders with cholesterol stones. Am. J. Physiol. 1995, 269, G659–G665. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.L.; Chen, Q.; Amaral, J.; Biancani, P.; Behar, J. Defect of receptor-G protein coupling in human gallbladder with cholesterol stones. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 278, G251–G258. [Google Scholar] [CrossRef]
- Xiao, Z.L.; Chen, Q.; Biancani, P.; Behar, J. Mechanisms of gallbladder hypomotility in pregnant guinea pigs. Gastroenterology 1999, 116, 411–419. [Google Scholar] [CrossRef]
- Yu, P.; Chen, Q.; Xiao, Z.; Harnett, K.; Biancani, P.; Behar, J. Signal transduction pathways mediating CCK-induced gallbladder muscle contraction. Am. J. Physiol. 1998, 275, G203–G211. [Google Scholar] [CrossRef]
- Yu, P.; De Petris, G.; Biancani, P.; Amaral, J.; Behar, J. Cholecystokinin-coupled intracellular signaling in human gallbladder muscle. Gastroenterology 1994, 106, 763–770. [Google Scholar] [CrossRef]
- Xiao, Z.L.; Chen, Q.; Amaral, J.; Biancani, P.; Jensen, R.T.; Behar, J. CCK receptor dysfunction in muscle membranes from human gallbladders with cholesterol stones. Am. J. Physiol. 1999, 276, G1401–G1407. [Google Scholar] [CrossRef]
- Scott, A.J. Epithelial cell proliferation in diverse models of experimental cholelithiasis. Gut 1978, 19, 558–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pemsingh, R.S.; MacPherson, B.R.; Scott, G.W. Morphological observations on the gallbladder of ground squirrels fed a lithogenic diet. J. Pathol. 1987, 152, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.L.; Biancani, P.; Carey, M.C.; Behar, J. Hydrophilic but not hydrophobic bile acids prevent gallbladder muscle dysfunction in acute cholecystitis. Hepatology 2003, 37, 1442–1450. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Q.; Zhang, L.; Wang, H.H. High cholesterol absorption efficiency and rapid biliary secretion of chylomicron remnant cholesterol enhance cholelithogenesis in gallstone-susceptible mice. Biochim. Biophys. Acta 2005, 1733, 90–99. [Google Scholar] [CrossRef]
- Wang, D.Q.; Paigen, B.; Carey, M.C. Genetic factors at the enterocyte level account for variations in intestinal cholesterol absorption efficiency among inbred strains of mice. J. Lipid Res. 2001, 42, 1820–1830. [Google Scholar]
- Wang, D.Q.; Tazuma, S.; Cohen, D.E.; Carey, M.C. Feeding natural hydrophilic bile acids inhibits intestinal cholesterol absorption: Studies in the gallstone-susceptible mouse. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 285, G494–G502. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.Q.; Lammert, F.; Cohen, D.E.; Paigen, B.; Carey, M.C. Cholic acid aids absorption, biliary secretion, and phase transitions of cholesterol in murine cholelithogenesis. Am. J. Physiol. 1999, 276, G751–G760. [Google Scholar] [CrossRef]
- Krawczyk, M.; Lutjohann, D.; Schirin-Sokhan, R.; Villarroel, L.; Nervi, F.; Pimentel, F.; Lammert, F.; Miquel, J.F. Phytosterol and cholesterol precursor levels indicate increased cholesterol excretion and biosynthesis in gallstone disease. Hepatology 2012, 55, 1507–1517. [Google Scholar] [CrossRef]
- Thomas, L.A.; Veysey, M.J.; Bathgate, T.; King, A.; French, G.; Smeeton, N.C.; Murphy, G.M.; Dowling, R.H. Mechanism for the transit-induced increase in colonic deoxycholic acid formation in cholesterol cholelithiasis. Gastroenterology 2000, 119, 806–815. [Google Scholar] [CrossRef]
- Dowling, R.H.; Veysey, M.J.; Pereira, S.P.; Hussaini, S.H.; Thomas, L.A.; Wass, J.A.; Murphy, G.M. Role of intestinal transit in the pathogenesis of gallbladder stones. Can. J. Gastroenterol. J. Can. De Gastroenterol. 1997, 11, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Hussaini, S.H.; Pereira, S.P.; Veysey, M.J.; Kennedy, C.; Jenkins, P.; Murphy, G.M.; Wass, J.A.; Dowling, R.H. Roles of gall bladder emptying and intestinal transit in the pathogenesis of octreotide induced gall bladder stones. Gut 1996, 38, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Hussaini, S.H.; Pereira, S.P.; Dowling, R.H.; Wass, J.A. Slow intestinal transit and gallstone formation. Lancet 1993, 341, 638. [Google Scholar] [CrossRef]
- Hussaini, S.H.; Pereira, S.P.; Murphy, G.M.; Dowling, R.H. Deoxycholic acid influences cholesterol solubilization and microcrystal nucleation time in gallbladder bile. Hepatology 1995, 22, 1735–1744. [Google Scholar] [PubMed]
- Thomas, L.A.; Veysey, M.J.; Murphy, G.M.; Russell-Jones, D.; French, G.L.; Wass, J.A.; Dowling, R.H. Octreotide induced prolongation of colonic transit increases faecal anaerobic bacteria, bile acid metabolising enzymes, and serum deoxycholic acid in patients with acromegaly. Gut 2005, 54, 630–635. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.Q.; Paigen, B.; Carey, M.C. Phenotypic characterization of Lith genes that determine susceptibility to cholesterol cholelithiasis in inbred mice: Physical-chemistry of gallbladder bile. J. Lipid Res. 1997, 38, 1395–1411. [Google Scholar]
- Wang, D.Q.; Lammert, F.; Paigen, B.; Carey, M.C. Phenotypic characterization of Lith genes that determine susceptibility to cholesterol cholelithiasis in inbred mice: Pathophysiology of biliary lipid secretion. J. Lipid Res. 1999, 40, 2066–2079. [Google Scholar]
- Lammert, F.; Wang, D.Q.; Paigen, B.; Carey, M.C. Phenotypic characterization of Lith genes that determine susceptibility to cholesterol cholelithiasis in inbred mice: Integrated activities of hepatic lipid regulatory enzymes. J. Lipid Res. 1999, 40, 2080–2090. [Google Scholar]
- Eckhardt, E.R.; Wang, D.Q.; Donovan, J.M.; Carey, M.C. Dietary sphingomyelin suppresses intestinal cholesterol absorption by decreasing thermodynamic activity of cholesterol monomers. Gastroenterology 2002, 122, 948–956. [Google Scholar] [CrossRef]
- Heaton, K.W.; Emmett, P.M.; Symes, C.L.; Braddon, F.E. An explanation for gallstones in normal-weight women: Slow intestinal transit. Lancet 1993, 341, 8–10. [Google Scholar] [CrossRef]
- Thomas, L.A.; Veysey, M.J.; French, G.; Hylemon, P.B.; Murphy, G.M.; Dowling, R.H. Bile acid metabolism by fresh human colonic contents: A comparison of caecal versus faecal samples. Gut 2001, 49, 835–842. [Google Scholar] [CrossRef]
- Shoda, J.; He, B.F.; Tanaka, N.; Matsuzaki, Y.; Osuga, T.; Yamamori, S.; Miyazaki, H.; Sjovall, J. Increase of deoxycholate in supersaturated bile of patients with cholesterol gallstone disease and its correlation with de novo syntheses of cholesterol and bile acids in liver, gallbladder emptying, and small intestinal transit. Hepatology 1995, 21, 1291–1302. [Google Scholar] [CrossRef]
- Azzaroli, F.; Mazzella, G.; Mazzeo, C.; Simoni, P.; Festi, D.; Colecchia, A.; Montagnani, M.; Martino, C.; Villanova, N.; Roda, A.; et al. Sluggish small bowel motility is involved in determining increased biliary deoxycholic acid in cholesterol gallstone patients. Am. J. Gastroenterol. 1999, 94, 2453–2459. [Google Scholar] [CrossRef] [PubMed]
- Marcus, S.N.; Heaton, K.W. Intestinal transit, deoxycholic acid and the cholesterol saturation of bile--three inter-related factors. Gut 1986, 27, 550–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.H.; Portincasa, P.; Wang, D.Q. The cholecystokinin-1 receptor antagonist devazepide increases cholesterol cholelithogenesis in mice. Eur. J. Clin. Investig. 2016, 46, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.H.; Portincasa, P.; Liu, M.; Tso, P.; Samuelson, L.C.; Wang, D.Q. Effect of gallbladder hypomotility on cholesterol crystallization and growth in CCK-deficient mice. Biochim. Et Biophys. Acta 2010, 1801, 138–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.H.; Portincasa, P.; Wang, D.Q. Update on the molecular mechanisms underlying the effect of cholecystokinin and cholecystokinin-1 receptor on the formation of cholesterol gallstones. Curr. Med. Chem. 2019, 26, 3407–3423. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.P.; LaMont, J.T.; Carey, M.C. Role of gallbladder mucus hypersecretion in the evolution of cholesterol gallstones. J. Clin. Investig. 1981, 67, 1712–1723. [Google Scholar] [CrossRef] [Green Version]
- Holzbach, R.T.; Corbusier, C.; Marsh, M.; Naito, H.K. The process of cholesterol cholelithiasis induced by diet in the prairie dog: A physicochemical characterization. J. Lab. Clin. Med. 1976, 87, 987–998. [Google Scholar]
- Mazer, N.A.; Carey, M.C. Quasi-elastic light-scattering studies of aqueous biliary lipid systems. Cholesterol solubilization and precipitation in model bile solutions. Biochemistry 1983, 22, 426–442. [Google Scholar] [CrossRef]
- Halpern, Z.; Dudley, M.A.; Lynn, M.P.; Nader, J.M.; Breuer, A.C.; Holzbach, R.T. Vesicle aggregation in model systems of supersaturated bile: Relation to crystal nucleation and lipid composition of the vesicular phase. J. Lipid Res. 1986, 27, 295–306. [Google Scholar]
- Halpern, Z.; Dudley, M.A.; Kibe, A.; Lynn, M.P.; Breuer, A.C.; Holzbach, R.T. Rapid vesicle formation and aggregation in abnormal human biles. A time-lapse video-enhanced contrast microscopy study. Gastroenterology 1986, 90, 875–885. [Google Scholar] [CrossRef]
- Konikoff, F.M.; Chung, D.S.; Donovan, J.M.; Small, D.M.; Carey, M.C. Filamentous, helical, and tubular microstructures during cholesterol crystallization from bile. Evidence that cholesterol does not nucleate classic monohydrate plates. J. Clin. Investig. 1992, 90, 1155–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.Q.; Carey, M.C. Complete mapping of crystallization pathways during cholesterol precipitation from model bile: Influence of physical-chemical variables of pathophysiologic relevance and identification of a stable liquid crystalline state in cold, dilute and hydrophilic bile salt-containing systems. J. Lipid Res. 1996, 37, 606–630. [Google Scholar] [PubMed]
- Wang, D.Q.; Carey, M.C. Characterization of crystallization pathways during cholesterol precipitation from human gallbladder biles: Identical pathways to corresponding model biles with three predominating sequences. J. Lipid Res. 1996, 37, 2539–2549. [Google Scholar] [PubMed]
- Toor, E.W.; Evans, D.F.; Cussler, E.L. Cholesterol monohydrate growth in model bile solutions. Proc. Natl. Acad. Sci. USA 1978, 75, 6230–6234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portincasa, P.; Venneman, N.G.; Moschetta, A.; van den Berg, A.; Palasciano, G.; vanBerge-Henegouwen, G.P.; van Erpecum, K.J. Quantitation of cholesterol crystallization from supersaturated model bile. J Lipid Res. 2002, 43, 604–610. [Google Scholar] [PubMed]
- Corradini, S.G.; Elisei, W.; Giovannelli, L.; Ripani, C.; Della Guardia, P.; Corsi, A.; Cantafora, A.; Capocaccia, L.; Ziparo, V.; Stipa, V.; et al. Impaired human gallbladder lipid absorption in cholesterol gallstone disease and its effect on cholesterol solubility in bile. Gastroenterology 2000, 118, 912–920. [Google Scholar] [CrossRef]
- Ginanni Corradini, S.; Ripani, C.; Della Guardia, P.; Giovannelli, L.; Elisei, W.; Cantafora, A.; Codacci Pisanelli, M.; Tebala, G.D.; Nuzzo, G.; Corsi, A.; et al. The human gallbladder increases cholesterol solubility in bile by differential lipid absorption: A study using a new in vitro model of isolated intra-arterially perfused gallbladder. Hepatology 1998, 28, 314–322. [Google Scholar] [CrossRef]
- Ginanni Corradini, S.; Yamashita, G.; Nuutinen, H.; Chernosky, A.; Williams, C.; Hays, L.; Shiffman, M.L.; Walsh, R.M.; Svanvik, J.; Della Guardia, P.; et al. Human gallbladder mucosal function: Effects on intraluminal fluid and lipid composition in health and disease. Dig. Dis. Sci. 1998, 43, 335–343. [Google Scholar] [CrossRef]
- van Erpecum, K.J.; van Berge Henegouwen, G.P.; Stolk, M.F.; Hopman, W.P.; Jansen, J.B.; Lamers, C.B. Effects of ursodeoxycholic acid on gallbladder contraction and cholecystokinin release in gallstone patients and normal subjects. Gastroenterology 1990, 99, 836–842. [Google Scholar] [CrossRef]
- Forgacs, I.C.; Maisey, M.N.; Murphy, G.M.; Dowling, R.H. Influence of gallstones and ursodeoxycholic acid therapy on gallbladder emptying. Gastroenterology 1984, 87, 299–307. [Google Scholar] [CrossRef]
- Sylwestrowicz, T.A.; Shaffer, E.A. Gallbladder function during gallstone dissolution. Effect of bile acid therapy in patients with gallstones. Gastroenterology 1988, 95, 740–748. [Google Scholar] [CrossRef]
- Hussaini, S.H.; Pereira, S.P.; Murphy, G.M.; Kennedy, C.; Wass, J.A.; Besser, G.M.; Dowling, R.H. Composition of gall bladder stones associated with octreotide: Response to oral ursodeoxycholic acid. Gut 1995, 36, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Holzbach, R.T. Gallbladder stasis: Consequence of long-term parenteral hyperalimentation and risk factor for cholelithiasis. Gastroenterology 1983, 84, 1055–1058. [Google Scholar] [CrossRef]
- Roslyn, J.J.; Pitt, H.A.; Mann, L.L.; Ament, M.E.; DenBesten, L. Gallbladder disease in patients on long-term parenteral nutrition. Gastroenterology 1983, 84, 148–154. [Google Scholar] [CrossRef]
- Cano, N.; Cicero, F.; Ranieri, F.; Martin, J.; di Costanzo, J. Ultrasonographic study of gallbladder motility during total parenteral nutrition. Gastroenterology 1986, 91, 313–317. [Google Scholar] [CrossRef]
- Nakano, K.; Chijiiwa, K.; Noshiro, H.; Hirota, I.; Yamasaki, T. Human gallbladder bile becomes lithogenic during short-term intravenous hyperalimentation. J. Surg. Res. 1992, 53, 396–401. [Google Scholar] [CrossRef]
- Angelico, M.; De Santis, A.; Capocaccia, L. Biliary sludge: A critical update. J. Clin. Gastroenterol. 1990, 12, 656–662. [Google Scholar] [CrossRef]
- Messing, B.; Bories, C.; Kunstlinger, F.; Bernier, J.J. Does total parenteral nutrition induce gallbladder sludge formation and lithiasis? Gastroenterology 1983, 84, 1012–1019. [Google Scholar] [CrossRef]
- Pazzi, P.; Gamberini, S.; Buldrini, P.; Gullini, S. Biliary sludge: The sluggish gallbladder. Dig. Liver Dis. Off. J. Ital. Soc. Gastroenterol. Ital. Assoc. Study Liver 2003, 35 (Suppl. S3), S39–S45. [Google Scholar] [CrossRef]
- Sitzmann, J.V.; Pitt, H.A.; Steinborn, P.A.; Pasha, Z.R.; Sanders, R.C. Cholecystokinin prevents parenteral nutrition induced biliary sludge in humans. Surg. Gynecol. Obstet. 1990, 170, 25–31. [Google Scholar] [PubMed]
- Green, P.H.; Cellier, C. Celiac disease. N. Engl. J. Med. 2007, 357, 1731–1743. [Google Scholar] [CrossRef] [PubMed]
- Calam, J.; Ellis, A.; Dockray, G.J. Identification and measurement of molecular variants of cholecystokinin in duodenal mucosa and plasma. Diminished concentrations in patients with celiac disease. J. Clin. Investig. 1982, 69, 218–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masclee, A.A.; Jansen, J.B.; Driessen, W.M.; Geuskens, L.M.; Lamers, C.B. Gallbladder sensitivity to cholecystokinin in coeliac disease. Correlation of gallbladder contraction with plasma cholecystokinin-like immunoreactivity during infusion of cerulein. Scand. J. Gastroenterol. 1991, 26, 1279–1284. [Google Scholar] [CrossRef] [PubMed]
- Lebwohl, B.; Green, P.H.R. Celiac Disease. In Sleisenger and Fordtran’s Gastrointestinal and Liver Disease, 11th ed.; Feldman, M., Friedman, L.S., Brandt, L.J., Chung, R.T., Rubin, D.T., Wilcox, C.M., Eds.; Elsevier Saunders: Philadelphia, PA, USA, 2020; Volume 2, pp. 1736–1755. [Google Scholar]
- Wang, H.H.; Li, T.; Portincasa, P.; Ford, D.A.; Neuschwander-Tetri, B.A.; Tso, P.; Wang, D.Q. New insights into the role of Lith genes in the formation of cholesterol-supersaturated bile. Liver Res. 2017, 1, 42–53. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Wang, D.Q.; Portincasa, P. An update on the pathogenesis of cholesterol gallstone disease. Curr. Opin. Gastroenterol. 2018, 34, 71–80. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.H.; Portincasa, P.; Liu, M.; Tso, P.; Wang, D.Q.-H. An Update on the Lithogenic Mechanisms of Cholecystokinin a Receptor (CCKAR), an Important Gallstone Gene for Lith13. Genes 2020, 11, 1438. https://doi.org/10.3390/genes11121438
Wang HH, Portincasa P, Liu M, Tso P, Wang DQ-H. An Update on the Lithogenic Mechanisms of Cholecystokinin a Receptor (CCKAR), an Important Gallstone Gene for Lith13. Genes. 2020; 11(12):1438. https://doi.org/10.3390/genes11121438
Chicago/Turabian StyleWang, Helen H., Piero Portincasa, Min Liu, Patrick Tso, and David Q.-H. Wang. 2020. "An Update on the Lithogenic Mechanisms of Cholecystokinin a Receptor (CCKAR), an Important Gallstone Gene for Lith13" Genes 11, no. 12: 1438. https://doi.org/10.3390/genes11121438
APA StyleWang, H. H., Portincasa, P., Liu, M., Tso, P., & Wang, D. Q.-H. (2020). An Update on the Lithogenic Mechanisms of Cholecystokinin a Receptor (CCKAR), an Important Gallstone Gene for Lith13. Genes, 11(12), 1438. https://doi.org/10.3390/genes11121438