Unusual Longevity of Edwards Syndrome: A Case Report

 , ,
, , {kind=link}
Abstract
:1. Introduction
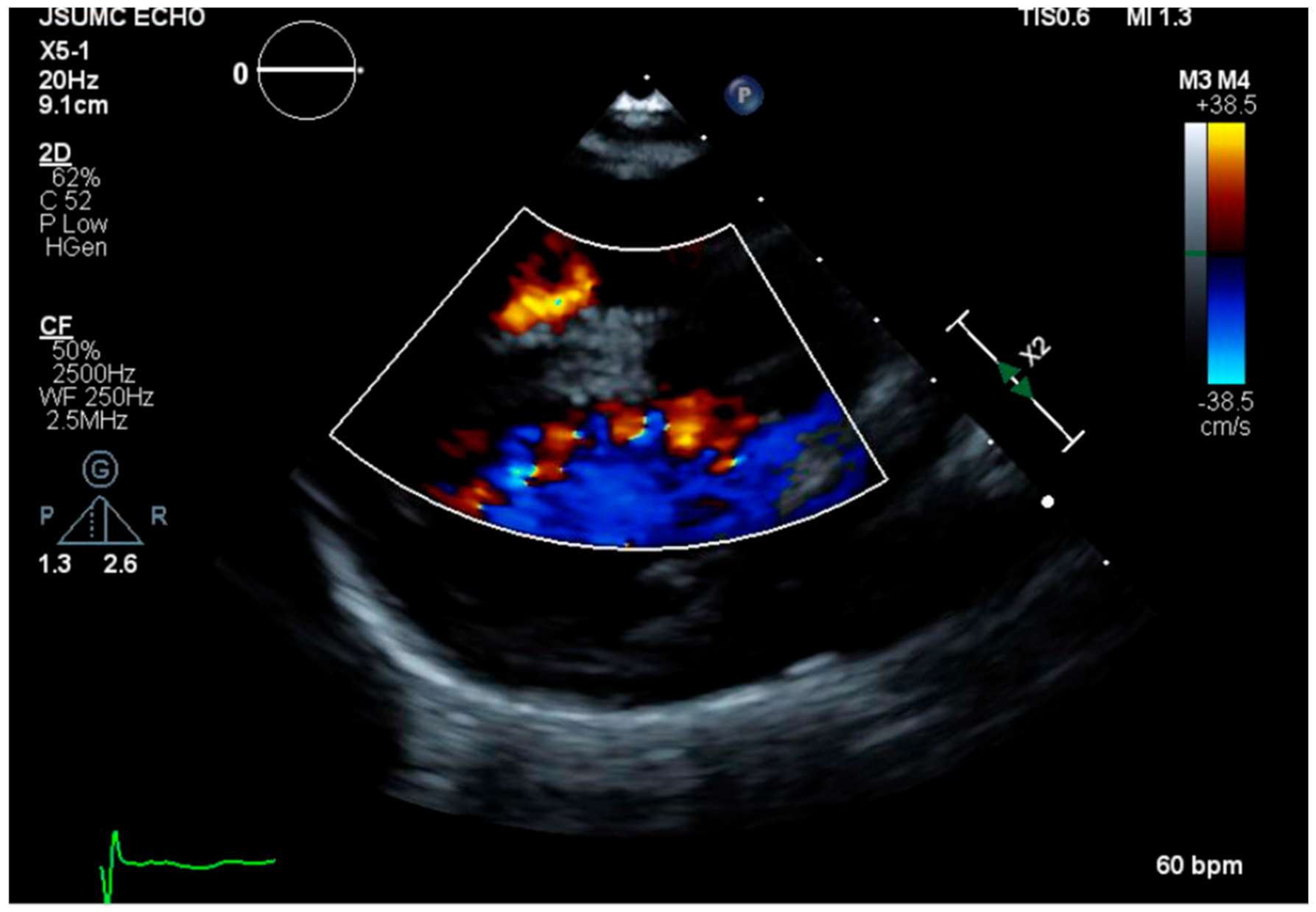
2. Case Presentation
3. Discussion
4. Conclusions
Author Contributions
Funding
Statement of Consent
Conflicts of Interest
References
- Edwards, J.H.; Harnden, D.G.; Cameron, A.H.; Crosse, V.M.; Wolff, O.H. A new trisomic syndrome. Lancet 1960, 1, 787–790. [Google Scholar] [CrossRef]
- Imataka, G.; Suzumura, H.; Arisaka, O. Clinical features and survival in individuals with trisomy 18: A retrospective one-center study of 44 patients who received intensive care treatments. Mol. Med. Rep. 2016, 13, 2457–2466. [Google Scholar] [CrossRef] [Green Version]
- Cereda, A.; Carey, J.C. The trisomy 18 syndrome. Orphanet. J. Rare Dis. 2012, 7, 81. [Google Scholar] [CrossRef] [Green Version]
- Baty, B.J.; Blackburn, B.L.; Carey, J.C. Natural history of trisomy 18 and trisomy 13: I. Growth, physical assessment, medical histories, survival, and recurrence risk. Am. J. Med. Genet. 1994, 49, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Root, S.; Carey, J.C. Survival in trisomy 18. Am. J. Med. Genet. 1994, 49, 170–174. [Google Scholar] [CrossRef] [PubMed]
- López-Ríos, V.; Grajales-Marín, E.; Gómez-Zambrano, V.; Barrios-Arroyave, F.A. Prolonged survival in Edwards syndrome with congenital heart disease: A case report and literature review. Medwave 2020, 20, e8015. [Google Scholar] [CrossRef] [PubMed]
- Moyano, D.; Huggon, I.C.; Allan, L.D. Fetal echocardiography in trisomy 18. Arch. Dis. Child Fetal Neonatal Ed. 2005, 90, F520–F522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bussmann, N.; Cunningham, K.; Green, A.; Ryan, C.A. Phenotypic extremes in liveborn monozygotic twins with mosaic Edwards syndrome. BMJ Case Rep. 2015, 2015, bcr2015211587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, M.; Robinson, B.W.; Moore, J.W. Trisomy 18 in a 20-year-old woman. Am. J. Med. Genet. 2002, 112, 397–399. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.; Field, B.; Learoyd, B.M. Trisomy 18 at age 21 years. Am. J. Med. Genet. 1989, 34, 338–339. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.K.; Savva, G.M. The risk of fetal loss following a prenatal diagnosis of trisomy 13 or trisomy 18. Am. J. Med. Genet. A 2008, 146, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.K.; Kochilas, L.K.; Catton, K.G.; Moller, J.H.; Setty, S.P. Long-Term Outcomes of Children With Trisomy 13 and 18 After Congenital Heart Disease Interventions. Ann. Thorac. Surg. 2017, 103, 1941–1949. [Google Scholar] [CrossRef] [PubMed]
- Tucker, M.E.; Garringer, H.J.; Weaver, D.D. Phenotypic spectrum of mosaic trisomy 18: Two new patients, a literature review, and counseling issues. Am. J. Med. Genet. A 2007, 143, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Imataka, G.; Yamanouchi, H.; Arisaka, O. Dandy-Walker syndrome and chromosomal abnormalities. Congenit. Anom. 2007, 47, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Van den Veyver, I.B. Recent advances in prenatal genetic screening and testing. F1000Research 2016, 5, 2591. [Google Scholar] [CrossRef]
- Batees, H.; Altirkawi, K.A. Trisomy 18 syndrome: Towards a balanced approach. Sudan. J. Paediatr. 2014, 14, 76–84. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alshami, A.; Douedi, S.; Guida, M.; Ajam, F.; Desai, D.; Zales, V.; Calderon, D.M. Unusual Longevity of Edwards Syndrome: A Case Report. Genes 2020, 11, 1466. https://doi.org/10.3390/genes11121466
Alshami A, Douedi S, Guida M, Ajam F, Desai D, Zales V, Calderon DM. Unusual Longevity of Edwards Syndrome: A Case Report. Genes. 2020; 11(12):1466. https://doi.org/10.3390/genes11121466
Chicago/Turabian StyleAlshami, Abbas, Steven Douedi, Melissa Guida, Firas Ajam, Dhaval Desai, Vincent Zales, and Dawn M Calderon. 2020. "Unusual Longevity of Edwards Syndrome: A Case Report" Genes 11, no. 12: 1466. https://doi.org/10.3390/genes11121466
APA StyleAlshami, A., Douedi, S., Guida, M., Ajam, F., Desai, D., Zales, V., & Calderon, D. M. (2020). Unusual Longevity of Edwards Syndrome: A Case Report. Genes, 11(12), 1466. https://doi.org/10.3390/genes11121466